

Abstract
Diastereoselectivity in control
The combined C—H functionalization/Cope rearrangement (CHCR) is a highly diastereoselective process that typically proceeds through a chair transition state. A recent computational study of a model system for the CHCR reaction revealed that a boat transition state was only slightly less favored than a chair transition state. Guided by these computational results, this study describes the design of substrates that would react by means of a boat transition state. The resulting C—H functionalization products are the opposite diastereomeric series to what had been previously obtained with this chemistry.
Keywords: carbenoids, diastereoselectivity, C-H functionalization, Cope rearrangement, rhodium
Developing practical methods for C—H functionalization has attracted considerable attention from the synthetic community.[1] One of the major challenges in this field is to achieve transformations that are not only site selective, but also stereoselective.[2] One highly stereoselective intermolecular C—H functionalization method is the combined C—H functionalization/Cope rearrangement (CHCR) between allylic C—H bonds and vinylcarbenoids.[3] This transformation can generate two new stereocenters. When chiral dirhodium catalysts such as Rh2(S-DOSP)4[4] are used, the products are formed essentially as single diastereomers and in the majority of cases with >97% ee. This method has been developed into a powerful protocol for the synthesis of natural products and pharmaceutical targets.[3] In all of the studies reported to date, the stereochemistry is consistent with a reaction occurring on the s-cis conformation of the vinylcarbenoid and proceeding through a chair transition state as illustrated in [Eq. (1)].
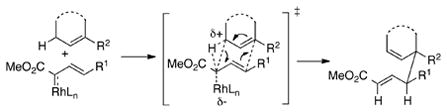 |
(1) |
Recently, we have completed a detailed computational study of the CHCR reaction.[5] The reaction was shown to be an asynchronous process, involving an initial hydride transfer event followed by carbon-carbon bond formation. Even though all the previously reported examples of the CHCR reactions are highly diastereoselective, the calculations showed that different product outcomes are possible, depending on whether the s-cis or s-trans con of the vinylcarbenoids[6] are involved and whether the reaction proceeded through a chair or a boat transition state. Furthermore, the calculations on a model system showed that the transition states for other products were energetically accessible. In particular the s-cis chair transition state was only 2 kcal/mole more stable than the s-cis boat transition state. Inspired by the computational studies, this paper is directed towards switching the diastereoselectivity of the CHCR reaction by forcing the reaction to proceed through the s-cis boat transition state B instead of the s-cis chair transition state A (Figure 1).
Figure 1.

The chair and boat transition states for the CHCR reaction.
In order to limit the number of potential transition states available for the CHCR reaction, the study described herein was conducted with β-siloxyvinyldiazoacetates. The carbenoid derived from E-vinyldiazoacetates has little preference for the s-trans over the s-cis configuration,[5] whereas the internal substituent in the vinylcarbenoid derived from the β-siloxyvinyldiazoacetate strongly prefers the s-cis configuration.[5] In the s-trans configuration, the siloxy group would be pointing towards the “wall” of the catalyst (Figure 2).
Figure 2.

The s-cis and s-trans configurations of the rhodium carbenoid derived from 1.
Previous studies have shown that Rh2(S-PTAD)4 (Figure 3) is the optimum chiral catalyst for asymmetric reactions with siloxyvinyldiazoacetate 1.[7] In order to test a baseline substrate, the Rh2(S-PTAD)4 catalyzed reaction of diazoacetate 1 with the siloxycyclohexene 2a was examined [Eq. (2)]. Characterizable material was obtained by hydrolysis of the silyl enol ether of the crude product followed by conversion of the β-keto ester to the β-keto-α-diazoacetate 3a in 74% yield for the three-step sequence.[8] The β-keto-α-diazoacetate 3a was formed as a single diastereomer with 89% ee. The reaction with the bulky siloxycyclohexene 2b selectively afforded the diazoacetate 3b with even higher enantioselectivity (97% ee). The relative and absolute configuration of product 3b was unambiguously determined using X-ray crystallography.[9]
Figure 3.

Structures of Rh2(S-DOSP)4 and Rh2(S-PTAD)4.
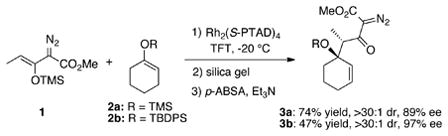 |
(2) |
The observed stereochemistry is consistent with the previously published examples of the CHCR reaction and would occur in a reaction proceeding through a chair transition state.[3e] An examination of the two possible transition states reveals that in the boat transition state C the remainder of the cyclohexyl ring would be pointing towards the “wall” of the catalyst, and therefore, it would be reasonable to propose that this arrangement would be unfavorable (Figure 4).
Figure 4.

s-Cis/boat transition state model for reaction of 1 with 2.
We envisioned that a possible way to limit the steric influence of the ring would be to use a smaller ring size. Indeed, when the reaction was repeated with the siloxycyclopentene 4, two diastereomers of the CHCR product 5 were produced in a 4/1 ratio [Eq. (3)]. This is the first example of a CHCR reaction generating a mixture of diastereomeric products.
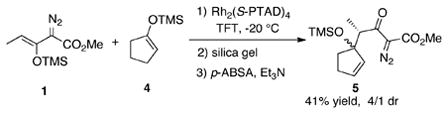 |
(3) |
Further evaluation of the proposed transition states D and E related to the formation of 5, suggested that the cyclopentyl ring could be incorporated into the boat transition state E (Figure 5). Furthermore, it became evident that a 2-substituent on the cyclopentenyl ring would cause the chair transition state D to be destabilized. If this proved to be the case, then the opposite diastereomeric series of products would become accessible.
Figure 5.

Transition state models for reaction of 1 with cyclopentenes.
The Rh2(S-PTAD)4 catalyzed decomposition of siloxydiazoacetate 1 in the presence of 1,2-disubstituted cyclopentenyl derivatives afforded the β-keto-α-diazoacetates 6–11 as summarized in Table 1. In all cases, a single CHCR product was produced with excellent diastereoselectivity (dr >30 : 1) and enantioselectivity (>97% ee). In the case of the unsymmetrical cyclopentene substrates, the resulting products, 6, 7 and 9, are derived from site selective C—H functionalization initiated at the methylene group allylic to the siloxy group. The relative and absolute configuration of 7 was unambiguously assigned by X-ray crystallography. The stereochemical configurations of products 9 and 10 were also unambiguously confirmed by X-ray crystallography of products derived from them (see supporting information). In each case, the relative configuration was consistent with a reaction proceeding through a boat transition state, and is opposite to the products 3a and 3b derived from the cyclohexene derivatives 2a and 2b. The structures of 6, 8 and 11 were tentatively assigned by assuming they are formed through a similar boat transition state.
Table 1.
The CHCR reactions with cyclopentenyl derivatives
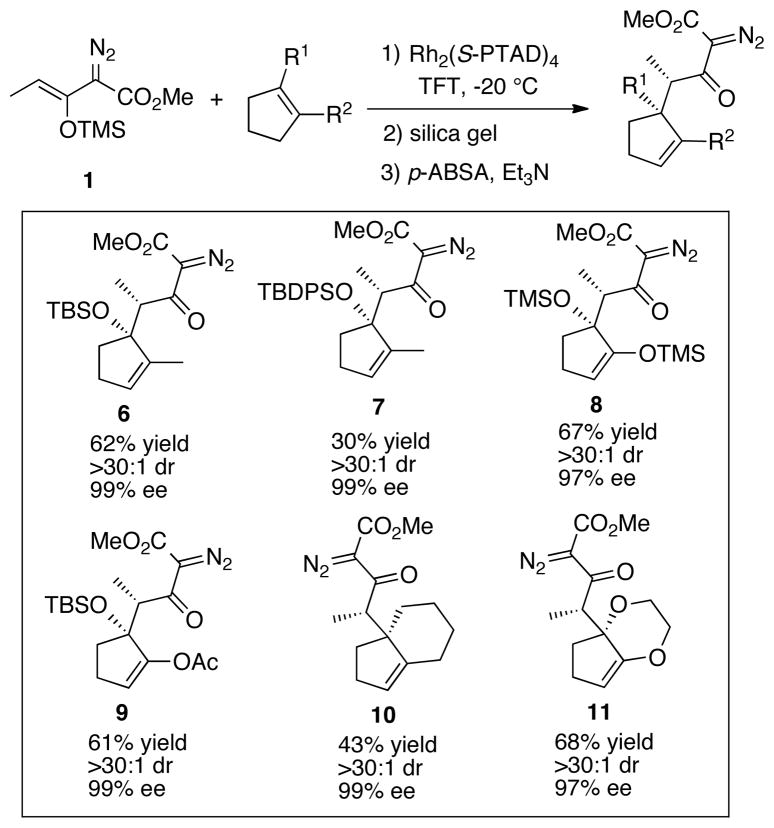
|
Normally, the CHCR reaction is influenced by the presence of other stereogenic centers in the substrate and high levels of enantiomeric differentiation have been reported.[3a–d] Consequently, we explored if a desymmetrization would be feasible in a CHCR reaction. The reaction with cyclopentene 12 successfully generated product 13 as a single diastereomer with extremely high enantioselectivity [Eq. (4)]. This represents the first example of desymmetrization in the CHCR reaction. The relative configuration of 13 inside the ring was assigned by nOe studies and was consistent with the outcome predicted by a boat transition state model (see SI), while the stereochemistry in the chain was tentatively assigned assuming a boat transition state.
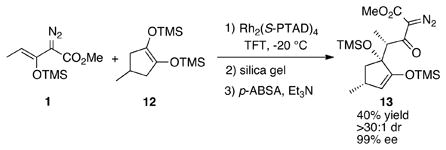 |
(4) |
In conclusion, the synthetic utility of the CHCR reaction has been greatly expanded by the design of substrates that will react through a boat transition state instead of a chair transition state. This has lead to the formation of the reversed diastereomeric series of products in a highly stereoselective manner. This study demonstrates the value of computational studies, not only to rationalize a new synthetic process, but also, to identify opportunities to develop new chemistry. The results showcase the synthetic potential of using carbenoid chemistry to achieve highly enantioselective C—H functionalization reactions.
Experimental Section
Typical procedure for the C—H functionalization: To an oven-dried 25 mL flask containing Rh2(S-PTAD)4 (16.5 mg, 0.01 equiv) and substrate (1.0 mmol, 1.0 equiv) in 6 mL dried trifluorotoluene under argon atmosphere was added a solution of (Z)-methyl 2-diazo-3-((trimethylsilyl)oxy)pent-3-enoate (1) (365 mg, 1.6 mmol, 1.6 equiv) in 6 mL dried trifluorotoluene by syringe pump over 3 h at −20 °C. The solution was warmed up to room temperature over 2 h. The mixture was concentrated under reduced pressure and then stirred with 5 g silica gel in 15 mL hexane for 30 mins. The mixture was filtrated and washed with several portions of Et2O. The organic solution was concentrated under vacuum and the residue was purified by flash chromatography on silica gel to provide a colorless oil, which was dissolved in 5 mL dried CH3CN containing p-ABSA (240 mg, 1.0 mmol, 1.0 equiv.), Et3N (0.30 ml, 2.0 mmol, 2.0 equiv.) The mixture was stirred for additional 3 h and then concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel to provide β-keto diazoacetates.
Supplementary Material
Footnotes
This material is based on work supported by the National Science Foundation under the Center for Chemical Innovation in Stereoselective C–H Functionalization (CHE-0943980) and by the National Institutes of Health (GM080337).
Supporting information for this article is available on the WWW under http://www.angewandte.org
References
- 1.a) Davies HML, Manning JR. Nature. 2008;451:417. doi: 10.1038/nature06485. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Colby DA, Bergman RG, Ellman JA. Chem Rev. 2010;110:624. doi: 10.1021/cr900005n. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Doyle MP, Duffy R, Ratnikov M, Zhou L. Chem Rev. 2010;110:704. doi: 10.1021/cr900239n. [DOI] [PubMed] [Google Scholar]; d) Giri R, Shi BF, Engle KM, Maugel N, Yu JQ. Chem Soc Rev. 2009;38:3242. doi: 10.1039/b816707a. [DOI] [PubMed] [Google Scholar]; e) Zalatan DN, Du Bois J. In: C-H Activation. Yu J-Q, Shi Z, editors. Vol. 292. WILEY-VCH Verlag; Heidelberg: 2010. pp. 347–379. [Google Scholar]; f) Davies HML, Beckwith REJ. Chem Rev. 2003;103:2861. doi: 10.1021/cr0200217. [DOI] [PubMed] [Google Scholar]
- 2.For recent examples of stereoselective C–H functionalization, see: Bigi MA, Reed SA, White MC. Nature Chem. 2011;3:218. doi: 10.1038/nchem.967.Tsai AS, Bergman RG, Ellman JA. J Am Chem Soc. 2008;130:6316. doi: 10.1021/ja8012159.Shi BF, Zhang YH, Lam JK, Wang DH, Yu JQ. J Am Chem Soc. 2010;132:460. doi: 10.1021/ja909571z.Tran DN, Cramer N. Angew Chem. 2010;122:8357. doi: 10.1002/anie.201004179.Angew Chem Int Ed. 2010;49:8181.Kang YK, Kim SM, Kim DY. J Am Chem Soc. 2010;132:11847. doi: 10.1021/ja103786c.Li Q, Yu Z. J Am Chem Soc. 2010;132:4542. doi: 10.1021/ja100409b.Suematsu H, Katsuki T. J Am Chem Soc. 2009;131:14218. doi: 10.1021/ja9065267.Coulter MM, Dornan PK, Dong VM. J Am Chem Soc. 2009;131:6932. doi: 10.1021/ja901915u.Horino Y, Yamamoto T, Ueda K, Kuroda S, Toste FD. J Am Chem Soc. 2009;131:2809. doi: 10.1021/ja808780r.Thu H, Tong GS, Huang J, Chan SL, Deng Q, Che C. Angew Chem. 2008;120:9893. doi: 10.1002/anie.200803157.Angew Chem Int Ed. 2008;47:9747.McQuaid KM, Sames D. J Am Chem Soc. 2009;131:402. doi: 10.1021/ja806068h.Chen K, Richter JM, Baran PS. J Am Chem Soc. 2008;130:7247. doi: 10.1021/ja802491q.Liang C, Collet F, Robert-Peillard F, Müller P, Dodd RH, Dauban P. J Am Chem Soc. 2008;130:343. doi: 10.1021/ja076519d.Trost BM, Malhotra S, Olson DE, Maruniak A, Du Bois J. J Am Chem Soc. 2009;131:4190. doi: 10.1021/ja809697p.
- 3.a) Dai X, Wan Z, Kerr R, Davies HML. J Org Chem. 2007;72:1895. doi: 10.1021/jo0618167. [DOI] [PubMed] [Google Scholar]; b) Davies HML, Denton JR. Chem Soc Rev. 2009;38:3061. doi: 10.1039/b901170f. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Davies HML, Dai X, Long MS. J Am Chem Soc. 2006;128:2485. doi: 10.1021/ja056877l. [DOI] [PubMed] [Google Scholar]; d) Davies HML, Walji AM. Angew Chem. 2005;117:1761. [Google Scholar]; Angew Chem Int Ed. 2005;44:1733. [Google Scholar]; e) Davies HML, Jin Q. Proc Natl Acad Sci U S A. 2004;101:5472. doi: 10.1073/pnas.0307556101. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Davies HML, Jin Q. J Am Chem Soc. 2004;126:10862. doi: 10.1021/ja047185k. [DOI] [PubMed] [Google Scholar]; g) Davies HML, Dai X. Tetrahedron. 2006;62:10477. [Google Scholar]; h) Davies HML, Manning JR. J Am Chem Soc. 2006;128:1060. doi: 10.1021/ja057768+. [DOI] [PubMed] [Google Scholar]; i) Davies HML, Stafford DG, Hansen T. Org Lett. 1999;1:233. doi: 10.1021/ol9905699. [DOI] [PubMed] [Google Scholar]
- 4.Hansen JH, Davies HML. Coord Chem Rev. 2008;252:545. doi: 10.1016/j.ccr.2007.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hansen JH, Gregg TM, Ovalles SR, Lian Y, Autschbach J, Davies HML. J Am Chem Soc. 2011;133:5076. doi: 10.1021/ja111408v. [DOI] [PubMed] [Google Scholar]
- 6.a) Lian Y, Davies HML. J Am Chem Soc. 2010;132:440. doi: 10.1021/ja9078094. [DOI] [PubMed] [Google Scholar]; b) Lian Y, Davies HML. Org Lett. 2010;12:924. doi: 10.1021/ol9028385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Nadeau E, Ventura DL, Brekan JA, Davies HML. J Org Chem. 2010;75:1927. doi: 10.1021/jo902644f. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lian Y, Miller LC, Born S, Sarpong R, Davies HML. J Am Chem Soc. 2010;132:12422. doi: 10.1021/ja103916t. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Schwartz BD, Denton JR, Lian Y, Davies HML, Williams CM. J Am Chem Soc. 2009;131:8329. doi: 10.1021/ja9019484. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Reddy RP, Davies HML. J Am Chem Soc. 2007;129:10312. doi: 10.1021/ja072936e. [DOI] [PubMed] [Google Scholar]
- 8.p-ABSA: p-Acetamidobenzenesulfonyl Azide (CAS 2158-14-7).
- 9.CCDC 815184 (ent-3b from Rh2(R-PTAD)4-catalyzed reaction), CCDC 815183 (7), CCDC 815179 (9aderived from 9), and CCDC 815180 (10bderived from 10) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.