

Abstract
NOV-002 is a novel glutathione disulfide mimetic that when administered in combination with standard chemotherapeutic regimens has resulted in increased efficacy (survival, tumor response) and improved tolerance to chemotherapy (e.g., hematologic recovery) in advanced non–small cell lung cancer patients. We show that NOV-002, which is not cytotoxic as a single agent, generated time- and concentration-dependent oxidative signals at the cell surface (reduction in protein thiols) and intracellularly [altered oxidized glutathione (GSSG) and reduced glutathione levels and ratio; increased reactive oxygen species] in the premyeloid HL-60 cell line and that this was associated with an increase in S-glutathionylation of cell proteins, particularly actin. Commensurate with these effects, NOV-002 activated p38, c-Jun-NH2-kinase, and extracellular signal-regulated kinase and caused a dose-dependent increase in phosphorylation of three proteins that have previously been linked with hematopoiesis, AKT, JAK2, and STAT5. The effect of NOV-002 on enzymes involved in glutathione metabolism was evaluated. Relative to oxidized glutathione, NOV-002 was an equivalent substrate for glutathione reductase and was an inhibitor of protein disulfide isomerase, one of the components of the redox-sensitive unfolded protein response pathway. These redox-stimulated cell signaling actions occurred in the context of increased HL-60 cell proliferation after treatment with NOV-002. Overall, the pleiotropic pharmacologic effects of NOV-002 can be attributed to the GSSG component of the drug, and modulation of cellular redox balance is a feature central to the mechanism of action of NOV-002. Such modulation may underlie its clinical actions, including hematologic recovery and immunostimulation in the face of chemosuppression.
Introduction
The structure of NOV-002 is shown in Fig. 1. It is oxidized glutathione formulated with cisplatin at an approximately 1,000:1 ratio. Initial interest in the drug stemmed from clinical data generated in the Russian Federation where it is approved and marketed. In particular, in chemotherapy-naïve advanced non–small cell lung cancer (NSCLC) patients receiving NOV-002 + standard chemotherapy displayed 63% 1-year survival compared with 17% in the chemotherapy alone group (1). NOV-002 treatment also increased tolerance to chemotherapy (patients were able to receive more and more frequent cycles of chemotherapy), improved hematologic and immune parameters and normalized kidney/liver toxicity markers, and improved quality of life (Karnofsky performance score). Clinical activity of NOV-002 was confirmed in a U.S. multicenter, open-label, randomized phase 1/2 trial in the same patient population. In this trial, treatment with NOV-002 + carboplatin/paclitaxel resulted in a significantly greater objective tumor response (70%) compared with carboplatin/paclitaxel alone (30%; ref. 1). Also, as in Russian studies, NOV-002–treated patients were able to receive more cycles of chemotherapy. Safety results confirmed Russian findings in that NOV-002 was well tolerated and did not add to chemotherapy-related toxicities.
Figure 1.

Effects of NOV-002 on growth and differentiation in HL-60 cells. A, NOV-002 is composed of the disodium salt of glutathione disulfide in a 1,000:1 ratio with cisplatin. B, the growth rate of untreated HL60 cells (▲); HL60 cells + 300 μmol/L NOV-002, “acute” (●); or HL60 cells + 300 μmol/L NOV-002 every 24 h, “chronic” (◆), was measured using a cell Coulter counter every 6 to 12 h. HL60 cells were treated with increasing concentrations of PABA/NO in the presence or absence of 300 μmol/L NOV-002. Flow cytometry was used to measure cell surface markers for differentiation: cd11 (C) and cd14b (D). Columns, mean from triplicate experiments; bars, SE.
Given this unique clinical profile of increased efficacy and improved tolerance when combined with standard chemotherapeutic drugs, it was of interest to further examine the pharmacologic activities of NOV-002. Nonclinical studies conducted in the Russian Federation suggested that the drug might enhance hematologic and immune recovery in the face of chemosuppression by increasing the production of regulatory cytokines and growth factors.4 Other clinical results have implicated glutathione in regulation of immune response. In particular, manipulation of blood glutathione (GSH) levels has been shown to influence survival and quality of life in HIV patients (2, 3). It would be of some significance to elucidate a plausible general mechanism linking glutathione with immunoregulation or hematopoiesis and NOV-002 could provide a platform to achieve this.
There are precedents for pharmacologic manipulation of glutathione pathways. For example, modulation of GSH levels and glutathione S-transferase (GST) activity has been attempted as a means to improve response to cancer drugs. Use of buthionine sulfoximine and ethacrynic acid, although effective in their experimental effects on each system, were not successful enough in the clinic to merit continued development (4, 5). One consequence of these approaches was the conceptual design of a peptidomimetic inhibitor of GSTp, TLK199 [γ-glutamyl-S-(benzyl)-cysteinyl-R(−)phenyl glycine diethyl ester]. Preclinical and mechanism of action studies with this agent revealed an unexpected effect in animals, namely that the drug possessed myeloproliferative activity (6, 7). This agent, now named Telintra, is in phase 2 clinical trial; for review, see ref. (8). There are indications that NOV-002 has a similar effect on myeloproliferation. However, to date, no mechanism of action data have been published.
Sulfur has the essential chemical properties to exist in a biologically reduced sulfhydryl state where the pKa of the thiol group is ~9.65, accounting for the nucleophilicity of reduced glutathione. GSH homeostasis is maintained in cells by a complex series of balanced pathways. De novo synthesis can occur through the γ-glutamyl cycle, where the three constituent amino acids (glu-cys-gly) are combined with rate-limiting catalysis through γ-glutamylcysteine synthetase and glutathione synthetase. Salvage of GSH can occur through the cleavage activity of the membrane-associated γ-glutamyl transpeptidase (GGT) that can recycle constituents of the molecule. Whereas intracellular concentrations of GSH may vary considerably, 0.1 to 10 mmol/L are not uncommonly found in mammalian cells (10–30 μmol/L in plasma). Glutathione can occur in reduced (GSH), oxidized (GSSG), or in mixed disulfide forms and its ubiquitous abundance is testament to its biological importance. More recently, S-glutathionylation of proteins has been recognized as an important posttranslational modification. S-glutathionylation can influence conformation of structural proteins such as actin (9) or enzyme function such as protein kinase C-ζ (10) or protein phosphatases (11). Significant evidence has accumulated that protein S-glutathionylation regulates protein function, including elements of cell signaling pathways (12–14). Because GSSG can be involved as the proximal donor in the S-glutathionylation reaction, the implications are that NOV-002 may also provide donor substrate. We provide evidence here that this is indeed the case and that, as a consequence of this and other effects on cellular redox balance, NOV-002 influences multiple cell processes and functions.
Materials and Methods
Chemicals
GSH, GSSG, 5,5′-dithiobis-2-nitrobenzoic acid (DTNB), 2-vinylpyridine, NADPH, EDTA, and the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide reagent were purchased from Sigma. Amplex Red was purchased from Molecular Probes. NOV-002 was provided by Novelos Therapeutics, Inc. Antibodies were purchased from the following sources: anti-glutathionylation (Virogen); anti–phospho-c-Jun NH2-terminal kinase (JNK) and anti-p38 (Promega); phosphospecific extracellular signal-regulated kinase (ERK; Santa Cruz Biotechnology); anti-JNK2 (BD Biosciences PharMingen); and anti-p38 and anti-ERK2 (Santa Cruz Biotechnology).
Cell lines and cell culture
Mouse embryo fibroblasts (MEF) were generated from wild-type and GSTπ deficient mice as previously described (6, 15). MEF and HL60 cells were maintained in DMEM containing 10% FCS, 100 μg/mL streptomycin, 100 units/mL penicillin, and 2 mmol/L l-glutamine at 5% CO2 and 37°C.
Fluorometric detection of reactive oxygen species
HL60 cells were treated with 300 μmol/L NOV-002 for 15 min. The Amplex Red fluorescent dye [1 (μmol/L)/L] and horseradish peroxidase (5 units/2 mL) were added and the release of H2O2 in the medium was detected at 37°C in a fluorescence spectrophotometer. The excitation wavelength was 550 nm and the fluorescence emission was measured at 585 nm. The signal was generated from a calibration using known amounts of H2O2..
Flow cytometry
HL60 cells from control and treated groups were stained with anti-CD11b or CD14 (PharMingen), Annexin, or Alexa fluor C5 Maleimide (Invitrogen). Fluorescence-activated cell sorting analysis was performed in the Flow Cytometry Facility at the Medical University of South Carolina using a FACSCalibur (Becton Dickinson).
Measurement of GSH and GSH reductase
Total glutathione (GSH plus GSSG) was determined by photometric determination of 5-thio-2-nitrobenzoate, which was produced from DTNB in a kinetic assay (16) in the Drug Metabolism and Clinical Pharmacology Core Facility at the Hollings Cancer Center. After experimental treatments, cells were harvested and washed with ice-cold PBS. Cells were then suspended in 0.1 mol/L sodium phosphate-5 mmol/L EDTA (pH 8.0) and sonicated to obtain the cell homogenate. Two microliters of whole-cell lysate were removed for protein quantification. An equal volume of 2 mol/L HClO and 4 mmol/L EDTA was added to the cell extract, and the precipitated proteins were sedimented by centrifugation at 8,000 × g for 10 min at 4°C. The supernatant was neutralized with 6 μL triethanolamine. For the spectrophotometric determination, 10 μL of the cell extract supernatant or of the standard glutathione solution, in the same phosphate-EDTA buffer, were mixed with 70 μL of 0.3 mmol/L NADPH in 0.5% (w/v) NaHCO3, 10 μL of 50 units/mL glutathione reductase in phosphate-EDTA buffer, and 10 μL of 6 mmol/L DTNB in 0.5% NaHCO3. The increase in absorbance was measured at 412 nm. For GSSG determination, 2-vinylpyridine was added to a final concentration of 0.1% (v/v), and then incubated for 1 h at room temperature. At this concentration, 2-vinylpyridine is able to react with all GSH without interfering with the GSSG determination. The GSH/GSSG content was expressed as nmol/mg protein. Values were represented as the mean of three independent experiments ± SD.
Quantitative real-time PCR
mRNA expression of BiP and CHOP, two endoplasmic reticulum stress response genes, was quantitated by real-time PCR using the TaqMan PCR core reagent kit according to the manufacturer’s directions. RNA was extracted from control and treated cells using the RNAeasy kit (Sigma). One microgram RNA was used to prepare complementary DNA (cDNA) in a 50 μL reaction containing 1 μmol/L oligo-dT15 primer and Omniscript reverse transcriptase (Qiagen). Real-time PCR was performed using 5 μL cDNA in a reaction mixture of 25 μL containing brilliant SYBR green master mix (Stratagene). All reactions were performed in triplicate and data were analyzed using the 2−ΔΔCT method (17). Thermal cycler conditions were as follows: 50°C for 2 min, 95°C for 10 min, and 45 cycles of 95°C for 0.3 min and 60°C for 1 min. Glyceraldehyde-3-phosphate dehydrogenase was used as a gene control to assess differences in transcript expression between groups.
GST activity assay
The effects of NOV-002 on enzyme activity were evaluated with the 1-chloro-2,4-dinitrobenzene (CDNB) substrate as previously described (18). The specific activity of GST was assayed following incubation of 0.5 μg of recombinant GSTp at 37°C for 30 min in the presence of 300 μmol/L NOV-002 or vehicle. The reaction mixture was carried out in 100 mmol/L potassium phosphate buffer with 1.0 mmol/L EDTA (pH 6.5), containing 1 mmol/L GSH and CDNB. The absorbance at 340 nm was monitored every 30 s for 2 min in a Bio-Rad Benchmark Plus Microplate Spectrophotometer (Bio-Rad). The rate of spontaneous conjugation of GSH to CDNB was subtracted from the rates of GST-catalyzed reactions. The extinction coefficient [5.3 (mmol/L)−1] for the CDNB conjugate at 340 nm in 96-well plates was used to calculate the specific activity. The specific activity was normalized to protein and expressed as the mean [(μmol/L)/mL/min] ± SD of three independent assays.
Protein disulfide isomerase activity assay
Recombinant human protein disulfide isomerase (PDI) in the expression vector pET-28a was provided (Dr. Lana Lee, Department of Chemistry and Biochemistry, University of Windsor, Ontario, Canada) and expressed using the Escherichia coli strain BL21 (DE3) as previously described (19). PDI was incubated with various concentrations of NOV-002 in 0.1 mol/L sodium phosphate, 2 mmol/L EDTA buffer for 30 min at 37°C. The activity of PDI was monitored using a fluorescence-based assay (20) whereby reduction of the substrate (FITC-peptide)ox was measured at 25°C in a photon-counting fluorimeter following excitation at 494 nm. Maximal fluorescence was measured by addition of DTT to fully reduce the peptide substrate.
Protein preparation
Cells (5 × 105 per treatment group) were harvested and washed with 1× PBS. Cell pellets were suspended in lysis buffer [20 mmol/L Tris-HCl (pH 7.5), 15 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1% Triton X-100, 2.5 mmol/L sodium PPi, and 1 mmol/L β-glycerophosphate with freshly added phosphatase inhibitors, 5 mmol/L NaF and 1 mmol/L Na3VO4] and incubated for 30 min on ice. Lysates were sonicated for 10 s and centrifuged for 30 min at 10,000 × g at 4°C. Protein concentrations in the supernatant were assayed with the Bradford reagent (Bio-Rad Laboratories) using IgG as a standard.
Western blot analysis
Equal amounts of protein were separated on 10% SDS-polyacrylamide gels and transferred overnight onto nitrocellulose membranes (Bio-Rad). Nonspecific binding was reduced by incubating the membrane in 10% blocking buffer for 1 h containing 20 mmol/L Tris-HCl (pH 7.5), 150 mmol/L NaCl, 10% bovine serum albumin, 0.1% Tween 20, and 1× protease inhibitors. Protein expression was determined by incubating membranes with specific primary antibody in 5% blocking buffer according to the manufacturer’s recommendation. Briefly, the membranes were washed thrice and incubated with the appropriate secondary antibodies, conjugated with horseradish peroxidase, in 5% blocking buffer for 1 h. The membranes were washed thrice and developed with enhanced chemiluminescence detection reagents (Amersham Biosciences). The relative abundance of proteins was evaluated using Quantity One software (ver.4.5.2; Bio-Rad) and plotted as arbitrary units in relation to actin.
Statistical analysis
Mean values and SE were computed for each group. These data were analyzed for statistically significant differences between groups with Student’s t test. Differences were considered statistically significant if the P value was <0.05.
Results
NOV-002 stimulates proliferation of the premyeloid cell line HL-60
Patients treated with NOV-002 plus chemotherapy have elevated levels of circulating leukocytes compared with those treated with chemotherapy alone.5 To analyze whether NOV-002 affects growth or differentiation, we measured the growth rate and doubling time of HL60 cells treated with a single dose of 300 μmol/L NOV-002 (acute) or cells exposed to daily treatments of 300 μmol/L NOV-002 for 5 days (chronic). NOV-002 treatment increased the proliferative index and decreased the doubling time in HL60 cells (Fig. 1). The average doubling time for untreated, control cells was 15.8 hours, whereas NOV-002 cells had a doubling time of 12.3 hours, a 22% decrease (P < 0.05). Using flow cytometry, cell surface markers indicative of differentiation (CD14 and CD11b, which are, respectively, decreased and increased in differentiated cells) were assessed. NOV-002 treatment did not promote cell differentiation.
NOV-002 oxidizes cell surface protein thiols in vitro
Cell surface protein thiols are believed to act as sensors of extracellular redox status, and their modification has been linked to regulation of cell signaling and other functions in a variety of cell types (21). Intact GSSG is plasma membrane impermeable. However, HL60 cells express GGT and thereby have the potential to metabolize NOV-002 into its constituent amino acids that can cross cell membranes. To assess the effect of NOV-002 on cell surface protein thiols, we preincubated HL60 cells with 300 μmol/L NOV-002 or vehicle for 1 hour at 37°C (Fig. 2). To prevent internalization of the probe, the cells were placed on ice and extracellular thiol content was fluorescently labeled using Alexa-NEM. NEM has been shown to bind to cell surface thiols present in large molecules, presumably proteins, on human lymphocytes (22). Also, using maleimide-based fluorescent dyes, GSSG has been reported to decrease cell surface thiol content in human peripheral blood mononuclear cells (23). Figure 2A shows that exposure of HL60 cells to NOV-002 resulted in a 15% decrease in total cell surface thiol content (P < 0.05). It is important to keep in mind that protein thiol oxidation (i.e., S-glutathionylation) by NOV-002 would only occur at that fraction of total protein thiols, which (a) are sterically accessible and (b) display a relatively low pKa. Thus, the proportion of this subset of cell surface protein thiols modified by NOV-002 may be considerably higher than 15%.
Figure 2.

Effects of NOV-002 on cell surface and intracellular protein S-glutathionylation in HL60 cells. A, HL60 cells were treated with 300 μmol/L NOV-002 or vehicle for 1 h, placed on ice in the presence of fluorescently labeled NEM. Cell surface cysteine modifications were semiquantitated by flow cytometry. B, H2O2 production was measured in medium following treatment with 300 μmol/L NOV-002. Concentration-dependent (C) and time-dependent (D) effects of NOV-002 on intracellular protein S-glutathionylation in HL60 cells were evaluated by dot-blot. Cells (1.5 × 106) were treated with varying concentrations of NOV-002, GSSG, PABA/NO, or cisplatin (CDDP). Following treatment for 1 h, 1 μL was dotted onto a nitrocellulose membrane and analyzed for S-glutathionylation by immunoblot. Columns, mean from triplicate experiments; bars, SE.
NOV-002 generates multiple intracellular oxidative signals in vitro
Maintenance of intracellular homeostatic redox balance is a function of multiple intersecting pathways. Nevertheless, intracellular GSH/GSSG levels and ratios can provide a meaningful measure of the oxidative status of the cell. Table 1 illustrates that by 1 hour after treatment of HL60 cells with 300 μmol/L of NOV-002 intracellular levels of both GSH and GSSG were elevated and returned to control levels by 24 hours (P < 0.05). Although absolute levels of GSH and GSSG increased transiently following treatment, the ratio of GSH/GSSG was decreased by ~36% after NOV-002 treatment, indicating the generation of a mild oxidative signal within the cell.
Table 1.
Intracellular thiol levels in HL60 cells treated with 300 μmol/L NOV-002
| Time, h | GSH (μmol/L)/mg | GSSG, (μmol/L)/mg | Total, (μmol/L)/mg | GSH/GSSG |
|---|---|---|---|---|
| Control | 24.2 ± 7.7 | 0.78 ± 0.06 | 25.0 ± 7.8 | 31 ± 9.0 |
| 1 | 50.5 ± 3.2* | 2.52 ± 0.54* | 53.1 ± 3.8* | 20 ± 3.8* |
| 4 | 38.8 ± 6.2* | 1.50 ± 0.14* | 40.7 ± 6.6* | 25 ± 1.4 |
| 24 | 22.0 ± 1.5 | 0.89 ± 0.09 | 23.4 ± 1.4 | 25 ± 2.4 |
NOTE: The results are expressed as the mean ± SE from triplicate experiments.
P < 0.05.
We explored whether NOV-002 treatment leads to the formation of reactive oxygen species (Fig. 2B). Fluorescent Amplex red was used to measure the H2O2 production in the medium of cells treated with NOV-002, H2O2, or PABA/NO. NOV-002–treated HL60 cells have an ~2.5-fold increase in H2O2 production compared with untreated cells (P < 0.05). PABA/NO, a nitrosative stress-inducing agent, caused no statistically significant difference in Amplex red signal. These data suggest that NOV-002 induces mild oxidative stress that does not lead to cell death.
S-glutathionylation of intracellular proteins
Protein S-glutathionylation is increasingly recognized as an important mechanism underlying redox regulation of signaling pathways and other cell functions (review refs. 12, 24). Figure 2 shows dose response (C) and time course (D) results for total protein S-glutathionylation in HL-60 cells exposed to NOV-002 and/or PABA/NO (a positive control, nitrosating agent that has been shown previously to cause high levels of S-glutathionylation; ref. 14). NOV-002 treatment resulted in a dose-dependent increase in total intracellular protein S-glutathionylation. Quantitatively similar results were obtained after treatment with GSSG; however, cisplatin (present in trace amount in NOV-002) did not cause S-glutathionylation, even at a highly cytotoxic dose (100 μmol/L). Although in some instances the S-glutathionylation antibody can interfere with the specific protein antibody, our overall experience is that NOV-002 treatment does not alter actin expression levels (for an example, see Fig. 3A).
Figure 3.

Concentration- and time-dependent effects of NOV-002 on actin S-glutathionylation in HL60 cells. A, 1.5 × 106 cells were treated with varying concentrations of NOV-002 for 1 h. B, 1.5 × 106 cells were treated with 50 μmol/L NOV-002 for 0 to 240 min. Following treatment, 20 μg of lysate were separated by SDS-PAGE under nonreducing conditions and analyzed for S-glutathionylation by immunoblot (A). Actin modification was observed by placing control and treated cells on polylysine-coated coverslips and staining for phalloidin (B).
Gel electrophoretic separation of the total protein pool following treatment of HL60 cells with NOV-002 revealed that the major S-glutathionylated protein was actin (Fig. 3A). Although NOV-002 treatment did not alter the expression levels of actin, S-glutathionylation modification of the protein increased in a dose- and time-dependent manner. S-glutathionylation of actin alters the ratio of F (filamentous) and G (globular) actin, leading to marked changes in the overall cytoskeletal architecture and concomitant changes in cellular trafficking (25). Using confocal microscopy, we evaluated the cytoskeletal architecture and stress fibers with phalloidin staining (which binds specifically to actin; Fig. 3B). NOV-002 treatment resulted in a reduction of focal adhesions compared with untreated controls indicative of significant cytoskeletal alterations, which could have important functional implications as well.
Phosphorylation of kinase pathway proteins
Cell signaling pathways regulated by the mitogen activated protein kinases (MAPK) are one example of linkage between redox-mediated signaling and phosphorylative cascades (26). Given the alterations in redox balance at the cell surface and intracellularly resulting from exposure to NOV-002, the effect of treatment of HL60 cells with NOV-002 on kinase expression and phosphorylation patterns were examined. As shown in Fig. 4, under conditions where NOV-002 resulted in protein S-glutathionylation, phosphorylation of several MAPK pathway kinases (JNK, ERK, and p38) were all also increased in a dose-dependent and a time-dependent (data not shown) manner. At the same time, the actual protein levels of each of the three kinases were not influenced by NOV-002 treatment. In addition, Fig. 5 shows that NOV-002 treatment in a dose-dependent manner caused an increase in the phosphorylation of AKT, JAK2, and STAT5, three proteins the modification of which has been associated with signal transduction (27) and myeloproliferation (7).
Figure 4.

Effects of NOV-002 on the MAPK pathway. Concentration-dependent effects of NOV-002 on stress kinases and their phosphorylated products are consistent with actin glutathionylation patterns. HL60 cells were treated for 1 h with 0 to 500 μmol/L NOV-002 in complete medium. Fifty micrograms of protein lysate were separated by SDS-PAGE and analyzed by immunoblot for phosphorylated and total levels of (A) JNK, (B) p38, and (C) ERK. The corresponding relative abundance of phosphorylated proteins was plotted as the relative ratio to actin.
Figure 5.

Effects of NOV-002 on the JAK-STAT kinase pathway. Concentration-dependent effects of NOV-002 on stress kinases and their phosphorylated products are consistent with actin glutathionylation patterns. HL60 cells were treated for 1 h with 0 to 500 μmol/L NOV-002 in complete medium. Fifty micrograms protein lysate were separated by SDS-PAGE and analyzed by immunoblot for phosphorylated and total levels of (A) AKT, (B) STAT-5, and (C) JAK-2. The corresponding relative abundance of phosphorylated proteins was plotted as the relative ratio to actin.
NOV-002 effects on glutathione pathway-related enzymes
The proposed active component of NOV-002 is oxidized glutathione. We evaluated the effects of NOV-002 treatment on glutathione pathway–related enzymes. Inhibition of GSTp activity, independent of changes in intracellular redox status, has been shown to lead to increased proliferation in myeloid cells (7). In a cell-free system, NOV-002 did not alter GSTp activity [49 ± 2 (μmol/L)/mL/min versus control 53 ± 2 (μmol/L)/mL/min], suggesting that the proliferative effect of NOV-002 on HL60 cells was independent of GSTp activity. Oxidized glutathione is the endogenous substrate for glutathione reductase and NOV-002 was shown to be equivalent substrate to GSSG for glutathione reductase (Fig. 6), a factor that may be relevant to the observation that NOV-002 treatment leads to a transient increase in intracellular GSH as well as GSSG levels. PDI is an endoplasmic reticulum resident chaperone and also found at the surface of cells, including myeloid cells. PDI cycles between an oxidized and reduced state and is influenced by redox balance. Incubation with NOV-002 led to extensive inhibition of PDI activity in a cell-free system (Fig. 6B).
Figure 6.
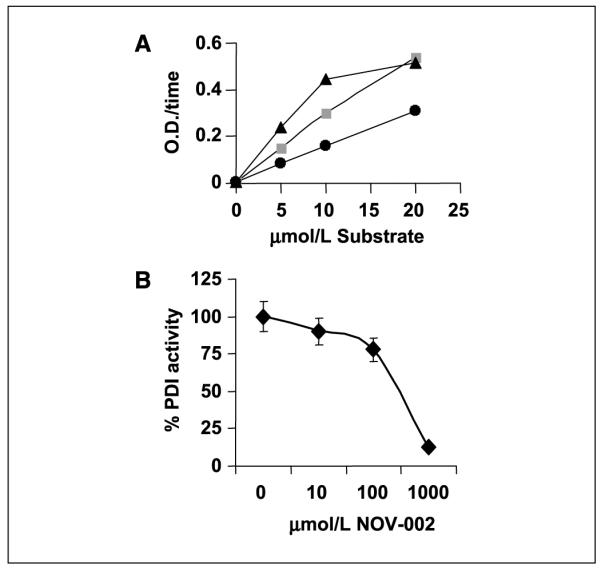
A, the ability of NOV-002 (▲), GSSG (■), or GSH (●) to serve as a substrate for glutathione reductase was assessed. B, the isomerase activity of PDI was measured following incubation of PDI with various concentrations of NOV-002 for 30 min at 37°C. Points, mean from triplicate experiments; bars, SE.
Discussion
NOV-002 is a novel therapeutic agent in late-stage development for oncology indications (1). It is composed of GSSG formulated with cisplatin at a ~1,000:1 molar ratio. The active pharmaceutical ingredient is GSSG, with the cisplatin appearing to increase exposure to GSSG after systemic administration without contributing to the therapeutic action of NOV-002.6 A typical therapeutic dose of NOV-002, 60 mg, administered daily over 6 months would result in a cumulative total dose of cisplatin equivalent to <2% of a typical single dose of cisplatin when used to treat cancer patients. NOV-002 is not cytotoxic alone even at high doses, and whereas NOV-002 causes protein S-glutathionylation, the platinum component does not. Instead, findings reported here collectively suggest that the capacity for modulation of redox conditions at the cell surface and/or intracellularly may underlie the pharmacologic properties of NOV-002. Clinical studies have shown that NOV-002 stimulates myeloproliferation, with increases in circulating lymphocyte, monocyte, T-cell, and natural killer cell counts.7 In this context, other clinical reports have implicated GSH in regulation of immune response. Manipulation of blood GSH levels has been shown to influence survival and quality of life in HIV-infected patients (2, 3) and administration of N-acetyl cysteine (a precursor of GSH) has been advanced as a therapy to enhance immune response (28). Additional approaches to manipulate glutathione pathways and thiol balance have also been shown to effect proliferation of myeloid progenitor cells, and it now seems reasonable to suggest a linkage between the two (8). In this regard, elucidating mechanism(s) of action of NOV-002 could contribute to a general understanding of how glutathione pathways are linked with hematologic and immunoregulation and, ultimately, with aspects of the clinical profile of NOV-002.
Our data suggest that the NOV-002 has pleiotropic effects on model cell systems, each of which is consistent with altered redox conditions at the cell surface or intracellularly, and with the conclusion that NOV-002 exerts pharmacologic effects through its GSSG component. It is generally accepted that GSSG will not passively cross the cell membrane (29). Thus, the effects of NOV-002 on cells may be mediated via direct effects on cell surface targets. Alternatively, the interaction of NOV-002 with a membrane-associated enzyme such as GGT (for which GSSG is a substrate) could result in hydrolysis into constituent amino acids, whereupon extra availability of cysteine could stimulate GSH metabolism. In addition, there has been a report that GGT, through Fenton chemistry activity, can result in the liberation of H2O2, which is cell permanent and capable of transmitting an oxidative signal into the cell (30). The increased bioavailability of GSSG through NOV-002 administration could increase the flux of H2O2 as a consequence of stimulating the GGT activity.
With respect to activity at the cell surface, NOV-002 treatment of HL-60 cells reduced cell surface thiol content, suggesting oxidative modification of cell surface proteins. A growing body of evidence suggests that redox-based modulation of surface proteins is capable of regulating cellular function (31). One potential target of such modification is cell surface PDI, which has been shown to regulate, for example, viral entry into cells (e.g., HIV-1 due to redox modulation of CD4 on lymphocytes and the HIV-1 envelope glycoprotein gp120; ref. 32), cell-mediated adhesion by integrins (33), and tumor cell invasiveness (34). Because NOV-002 was also found to inhibit PDI activity, this enzyme may represent a cell surface target for this drug candidate, a possibility further supported by the finding that GSSG can act as a proximal donor in the S-glutathionylation of PDI,8 thus regulating its function.
Intracellularly, NOV-002 treatment of HL-60 cells resulted in multiple changes indicative of alterations in redox balance against the backdrop of stimulating the rate of cell proliferation with no effect on markers of cell differentiation. Redox conditions can regulate a number of signaling pathways and directly control cell division and differentiation responses. In neuronal progenitor cells, a fine tuning of the redox balance has led some investigators to suggest that as little as a 15% increase/decrease in cellular redox can turn on pathways that channel cells toward either proliferation and cell division, or differentiation (35, 36). In the results described here, NOV-002 altered intracellular levels of GSH and GSSG by >35% as well as caused a time- and concentration-dependent increase in the phosphorylation of three kinases that play direct roles in cell proliferation (JNK, p38, and ERK) and in AKT, a kinase that acts in concert with JAK2 and STAT5 to regulate marrow proliferation. The JAK-STAT signaling pathway is intimately involved in governing the response of cells to cytokines and growth factors (27). Activation of the pathway by NOV-002 indicates that the drug is effecting those pathways that lead to hematopoiesis/myeloproliferation. These results are consistent with earlier work with TLK199, another small-molecule myeloproliferative agent (7). In each case, these stimulations were temporally coincident with the induction of S-glutathionylation of actin and could suggest a cause-effect relationship between signaling and cytoskeleton morphology. S-glutathionylation occurs to certain target proteins when a cell is exposed to oxidative or nitrosative stress (37). Filomeni and colleagues have used a number of model systems to show that under certain conditions, GSSG can act as a pro-oxidant activator of the p38 MAPK death pathway (26, 38). Indeed, their studies generally reflect the important role that GSSG has in mediating early response pathways through redox changes that transduce to a kinase signaling cascade that affects cell survival pathways. As such, the concomitant increases in intracellular GSSG, actin S-glutathionylation, kinase phosphorylation, and cell proliferation in HL-60 cells treated with NOV-002 may be causally interrelated.
In summary, NOV-002 was found to exert pleiotropic effects in HL-60 cells that are consistent with the conclusion that its active pharmaceutical ingredient is GSSG and that its central mode of action may involve modulation of redox balance at the cell surface and intracellularly. Although identification of the specific molecular targets of NOV-002 requires further study, redox modulation regulates downstream kinase events that may be responsible for its stimulation of proliferation in this premyeloid cell line, an effect that could be related to the clinical profile of NOV-002 in oncology patients, which includes myeloproliferation and improved tolerance of cytotoxic chemotherapy.
Acknowledgments
Grant support: National Cancer Institute grants CA08660 and CA117259.
We thank the Drug Metabolism and Pharmacokinetics, Flow Cytometry, and Proteomics Core Facilities of the Hollings Cancer Center.
Footnotes
Novelos, unpublished data.
D.M. Townsend et al., submitted.
Novelos, unpublished data.
Novelos, unpublished data.
D.M. Townsend et al., submitted for publication.
References
- 1.Pazoles CJ, Gernstein H. NOV-002, a chemoprotectant/immunomodulator, added to first line carboplatin/paclitaxel in advanced non-small cell lung cancer (NSCLC): a randomized phase 1/2, open label, controlled study [abstract 17021] American Society of Oncology. 2006 [Google Scholar]
- 2.Herzenberg LA, De Rosa SC, Dubs JG, et al. Glutathione deficiency is associated with impaired survival in HIV disease. Proc Natl Acad Sci U S A. 1997;94:1967–72. doi: 10.1073/pnas.94.5.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peterson JD, Herzenberg LA, Vasquez K, Waltenbaugh C. Glutathione levels in antigen-presenting cells modulate Th1 versus Th2 response patterns. Proc Natl Acad Sci U S A. 1998;95:3071–6. doi: 10.1073/pnas.95.6.3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bailey HH, Ripple G, Tutsch KD, et al. Phase I study of continuous-infusion L-S R-buthionine sulfoximine with intravenous melphalan. J Natl Cancer Inst. 1997;89:1789–96. doi: 10.1093/jnci/89.23.1789. [DOI] [PubMed] [Google Scholar]
- 5.O’Dwyer PJ, LaCreta F, Nash S, et al. Phase I study of thiotepa in combination with the glutathione transferase inhibitor ethacrynic acid. Cancer Res. 1991;51:6059–65. [PubMed] [Google Scholar]
- 6.Ruscoe JE, Rosario LA, Wang T, et al. Pharmacologic or genetic manipulation of glutathione S-transferase P1-1 (GSTpi) influences cell proliferation pathways. J Pharmacol Exp Ther. 2001;298:339–45. [PubMed] [Google Scholar]
- 7.Gate L, Majumdar RS, Lunk A, Tew KD. Increased myeloproliferation in glutathione S-transferase pi-deficient mice is associated with a deregulation of JNK and Janus kinase/STAT pathways. J Biol Chem. 2004;279:8608–16. doi: 10.1074/jbc.M308613200. [DOI] [PubMed] [Google Scholar]
- 8.Townsend D, Tew K. Cancer drugs, genetic variation and the glutathione-S-transferase gene family. Am J Pharmacogenomics. 2003;3:157–72. doi: 10.2165/00129785-200303030-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang J, Boja ES, Tan W, et al. Reversible glutathionylation regulates actin polymerization in A431 cells. J Biol Chem. 2001;276:47763–6. doi: 10.1074/jbc.C100415200. [DOI] [PubMed] [Google Scholar]
- 10.Ward NE, Chu F, O’Brian CA. Regulation of protein kinase C isozyme activity by S-glutathiolation. Methods Enzymol. 2002;353:89–100. doi: 10.1016/s0076-6879(02)53039-1. [DOI] [PubMed] [Google Scholar]
- 11.Rao RK, Clayton LW. Regulation of protein phosphatase 2A by hydrogen peroxide and glutathionylation. Biochem Biophys Res Commun. 2002;293:610–6. doi: 10.1016/S0006-291X(02)00268-1. [DOI] [PubMed] [Google Scholar]
- 12.Ghezzi P. Regulation of protein function by glutathionylation. Free Radic Res. 2005;39:573–80. doi: 10.1080/10715760500072172. [DOI] [PubMed] [Google Scholar]
- 13.Biswas S, Chida AS, Rahman I. Redox modifications of protein-thiols: emerging roles in cell signaling. Biochem Pharmacol. 2006;71:551–64. doi: 10.1016/j.bcp.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 14.Townsend DM, Findlay VJ, Fazilev F, et al. A glutathione S-transferase pi-activated prodrug causes kinase activation concurrent with S-glutathionylation of proteins. Mol Pharmacol. 2006;69:501–8. doi: 10.1124/mol.105.018523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosario LA, O’Brien ML, Henderson CJ, Wolf CR, Tew KD. Cellular response to a glutathione S-transferase P1-1 activated prodrug. Mol Pharmacol. 2000;58:167–74. doi: 10.1124/mol.58.1.167. [DOI] [PubMed] [Google Scholar]
- 16.Griffith OW. Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal Biochem. 1980;106:207–12. doi: 10.1016/0003-2697(80)90139-6. [DOI] [PubMed] [Google Scholar]
- 17.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 18.Boyland E, Chasseaud LF. Glutathione S-aralkyl-transferase. Biochem J. 1969;115:985–91. doi: 10.1042/bj1150985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raturi A, Vacratsis PO, Seslija D, Lee L, Mutus B. A direct, continuous, sensitive assay for protein disulphide-isomerase based on fluorescence self-quenching. Biochem J. 2005;391:351–7. doi: 10.1042/BJ20050770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tomazzolli R, Serra MD, Bellisola G, Colombatti M, Guella G. A fluorescence-based assay for the reductase activity of protein disulfide isomerase. Anal Biochem. 2006;350:105–12. doi: 10.1016/j.ab.2005.11.037. [DOI] [PubMed] [Google Scholar]
- 21.Dominici S, Valentini M, Maellaro E, et al. Redox modulation of cell surface protein thiols in U937 lymphoma cells: the role of γ-glutamyl transpeptidase-dependent H2O2 production and S-thiolation. Free Radic Biol Med. 1999;27:623–35. doi: 10.1016/s0891-5849(99)00111-2. [DOI] [PubMed] [Google Scholar]
- 22.Sahaf B, Heydari K, Herzenberg LA, Herzenberg LA. Lymphocyte surface thiol levels. Proc Natl Acad Sci U S A. 2003;100:4001–5. doi: 10.1073/pnas.2628032100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sahaf B, Heydari K, Herzenberg LA, Herzenberg LA. The extracellular microenvironment plays a key role in regulating the redox status of cell surface proteins in HIV-infected subjects. Arch Biochem Biophys. 2005;434:26–32. doi: 10.1016/j.abb.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 24.Townsend DM, Findlay VL, Tew KD. Glutathione S-transferases as regulators of kinase pathways and anticancer drug targets. Methods Enzymol. 2005;401:287–307. doi: 10.1016/S0076-6879(05)01019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Findlay VJ, Townsend DM, Saavedra JE, et al. Tumor cell responses to a novel glutathione S-transferase-activated nitric oxide-releasing prodrug. Mol Pharmacol. 2004;65:1070–9. doi: 10.1124/mol.65.5.1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Filomeni G, Aquilano K, Civitareale P, Rotilio G, Ciriolo MR. Activation of c-Jun-N-terminal kinase is required for apoptosis triggered by glutathione disulfide in neuro-blastoma cells. Free Radic Biol Med. 2005;39:345–54. doi: 10.1016/j.freeradbiomed.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 27.O’Shea JJ, Gadina M, Schreiber RD. Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell. 2002;109(Suppl):S121–31. doi: 10.1016/s0092-8674(02)00701-8. [DOI] [PubMed] [Google Scholar]
- 28.Atkuri KR, Mantovani JJ, Herzenberg LA, Herzenberg LA. N-Acetylcysteine—a safe antidote for cysteine/glutathione deficiency. Curr Opin Pharmacol. 2007;7:355–9. doi: 10.1016/j.coph.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brennan JP, Miller JI, Fuller W, et al. The utility of N,N-biotinyl glutathione disulfide in the study of protein S-glutathiolation. Mol Cell Proteomics. 2006;5:215–25. doi: 10.1074/mcp.M500212-MCP200. [DOI] [PubMed] [Google Scholar]
- 30.Pompella A, Corti A, Paolicchi A, Giommarelli C, Zunino F. γ-Glutamyltransferase, redox regulation and cancer drug resistance. Curr Opin Pharmacol. 2007;7:360–6. doi: 10.1016/j.coph.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 31.Jordan PA, Gibbins JM. Extracellular disulfide exchange and the regulation of cellular function. Antioxid Redox Signal. 2006;8:312–24. doi: 10.1089/ars.2006.8.312. [DOI] [PubMed] [Google Scholar]
- 32.Matthias LJ, Hogg PJ. Redox control on the cell surface: implications for HIV-1 entry. Antioxid Redox Signal. 2003;5:133–8. doi: 10.1089/152308603321223621. [DOI] [PubMed] [Google Scholar]
- 33.Lahav J, Wijnen EM, Hess O, et al. Enzymatically catalyzed disulfide exchange is required for platelet adhesion to collagen via integrin α2β1. Blood. 2003;102:2085–92. doi: 10.1182/blood-2002-06-1646. [DOI] [PubMed] [Google Scholar]
- 34.Goplen D, Wang J, Enger PO, et al. Protein disulfide isomerase expression is related to the invasive properties of malignant glioma. Cancer Res. 2006;66:9895–902. doi: 10.1158/0008-5472.CAN-05-4589. [DOI] [PubMed] [Google Scholar]
- 35.Smith J, Ladi E, Mayer-Proschel M, Noble M. Redox state is a central modulator of the balance between self-renewal and differentiation in a dividing glial precursor cell. Proc Natl Acad Sci U S A. 2000;97:10032–7. doi: 10.1073/pnas.170209797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Noble M, Smith J, Power J, Mayer-Proschel M. Redox state as a central modulator of precursor cell function. Ann N Y Acad Sci. 2003;991:251–71. doi: 10.1111/j.1749-6632.2003.tb07481.x. [DOI] [PubMed] [Google Scholar]
- 37.Tew KD. Redox in redux: Emergent roles for glutathione S-transferase P (GSTP) in regulation of cell signaling and S-glutathionylation. Biochem Pharmacol. 2007;73:1257–69. doi: 10.1016/j.bcp.2006.09.027. [DOI] [PubMed] [Google Scholar]
- 38.Filomeni G, Rotilio G, Ciriolo MR. Glutathione disulfide induces apoptosis in U937 cells by a redox-mediated p38 MAP kinase pathway. FASEB J. 2003;17:64–6. doi: 10.1096/fj.02-0105fje. [DOI] [PubMed] [Google Scholar]