

Abstract
Staphylococcus aureus causes significant illnesses throughout the world, including toxic shock syndrome (TSS), pneumonia, and infective endocarditis. Major contributors to S. aureus illnesses are secreted virulence factors it produces, including superantigens and cytolysins. This study investigates the use of superantigens and cytolysins as staphylococcal vaccine candidates. Importantly, 20% of humans and 50% of rabbits in our TSS model cannot generate antibody responses to native superantigens. We generated three TSST-1 mutants; G31S/S32P, H135A, and Q136A. All rabbits administered these TSST-1 toxoids generated strong antibody responses (titers>10,000) that neutralized native TSST-1 in TSS models, both in vitro and in vivo. These TSST-1 mutants lacked detectable residual toxicity. Additionally, the TSST-1 mutants exhibited intrinsic adjuvant activity, increasing antibody responses to a second staphylococcal antigen (β-toxin). This effect may be due to TSST-1 mutants binding to the immune co-stimulatory molecule CD40. The superantigens TSST-1 and SEC and the cytolysin α-toxin are known to contribute to staphylococcal pneumonia. Immunization of rabbits against these secreted toxins provided complete protection from highly lethal challenge with a USA200 S. aureus strain producing all three exotoxins; USA200 strains are common causes of staphylococcal infections. The same three exotoxins plus the cytolysins β-toxin and γ-toxin contribute to infective endocarditis and sepsis caused by USA200 strains. Immunization against these five exotoxins protected rabbits from infective endocarditis and lethal sepsis. These data suggest that immunization against toxoid proteins of S. aureus exotoxins protects from serious illnesses, and concurrently superantigen toxoid mutants provide endogenous adjuvant activity.
Keywords: Staphylococcus aureus, Superantigen, Cytolysin, Vaccine, Rabbit Model
INTRODUCTION
Staphylococcus aureus is a major pathogen worldwide, responsible for significant illnesses, many of which are life threatening such as toxic shock syndrome (TSS), infective endocarditis, sepsis, and pneumonia [1, 2]. S. aureus has the ability to cause a wide variety of infections by production of numerous virulence factors, both cell-surface and secreted exoproteins [1, 2]. Treatment of S. aureus infections can be challenging and expensive, especially with the high occurrence of antibiotic resistant infections, such as caused by methicillin-resistant S. aureus (MRSA) [3].
Infective endocarditis is a life threatening infection of the heart endothelium caused by many organisms [4, 5]. In the past decade, S. aureus has emerged as a primary cause of infective endocarditis throughout the world, largely in elderly patients and intravenous drug users [4-8]. The illness is characterized by formation of large “cauliflower-like” vegetations on the endothelium of the heart. These vegetations are composed of host factors (tissue factor, fibronectin, and fibrinogen) and host cells, as well as microbial colonies. Infective endocarditis is difficult to treat, and there are many risks associated with the illness, including cardiac failure, embolisms, renal dysfunction, and mycotic aneurysms [4, 5]. Treatment of S. aureus infective endocarditis typically requires extensive antibiotic regimens, often lasting ≥6 weeks, and many times surgery is required [4, 5, 7, 8].
Although cell-surface virulence factors are critical for S. aureus attachment and vegetation initiation, recent research has also implicated secreted virulence factors as major contributors to infective endocarditis progression with S. aureus. Pragman et al. showed that the superantigen TSS toxin-1 (TSST-1) is highly important for infective endocarditis vegetation formation caused by strains that produce the superantigen [9]. In a rabbit model, the researchers showed that strains producing native TSST-1 had significantly larger vegetation sizes and increases of nearly 7 Logs colony-forming units (CFUs)/vegetations compared to isogenic strains lacking TSST-1. We have further noted that strains lacking superantigens do not induce infective endocarditis in rabbits [9]. A research group recently published a study examining the genotype of strains isolated from infective endocarditis patients with persistent bacteremia and observed that the majority of them are pulsed-field gel electrophoresis type USA200 and carried the tstH gene that encodes TSST-1 [10]; there is a one:one correlation between the presence of tstH and TSST-1 protein production. Additionally, it has been published that 90% of infective endocarditis cases are associated with USA200 strains and production of TSST-1 [11]. These studies collectively suggest that TSST-1 is highly important for S. aureus in its ability to cause infective endocarditis. Recent studies from Mattis et al. showed that another superantigen, staphylococcal enterotoxin (SE) C, is highly important for infective endocarditis caused by strains that produce that superantigen (Mattis, D.M., A.R. Spaulding, O.N. Chuang-Smith, E.J. Sundberg, P.M. Schlievert, and D.M. Kranz. Enterotoxin C Contributes to USA400 Methicillin-Resistant Staphylococcus aureus Infective Endocarditis in Rabbits Submitted Infect. Immun.). When these investigators treated rabbits with a specific SEC inhibitor after challenge with a strain known to cause infective endocarditis at a high level in the rabbit model, the microbes were significantly reduced in ability to cause disease. Studies have also shown that secreted cytolysins contribute to infective endocarditis. Huseby et al. recently published that the cytolysin β-toxin facilitates infective endocarditis progression [12]. Cheung et al. showed that a S. aureus mutant that no longer produced α-toxin, β-toxin, γ-toxin, and δ-toxin was drastically reduced in its ability to cause infective endocarditis [13], although because these studies used a regulatory mutant for their studies, numerous other factor may also have contributed to reduced ability to cause illness.
In the rabbit model of infective endocarditis, we also gain important information on the role of exoproteins in lethal sepsis, since S. aureus is administered intravenously in high concentrations. Our prior studies strongly suggest that superantigens are important in lethal sepsis [14].
We and others have shown that superantigens and cytolysins are critical determinants of staphylococcal pneumonia [15-18]. Rabbits actively immunized against TSST-1 and SEC, and animals passively protected from SEB are protected from highly lethal intra-pulmonary S. aureus challenge [17]. In addition, mice immunized against α-toxin are protected from lethal pneumonia [16].
These data led us to consider the possibility of a vaccine against serious S. aureus infections using the major secreted virulence factors (cytolysins and superantigens) as immunizing agents. Here we report our studies related to TSST-1, SEC, α-toxin, β-toxin, and γ-toxin, alone and in combination for protection against staphylococcal pneumonia, infective endocarditis, and sepsis. Our studies show that vaccines containing these important secreted virulence factors lead to immunity that protects against illness and increases survival. Additionally, TSST-1 mutant toxoids have endogenous adjuvant activities, dependent on interaction with the immune co-stimulatory molecule CD40 that amplifies immune responses to other antigens.
MATERIAL AND METHODS
Bacterial strains and growth
S. aureus strain RN4220 containing plasmids encoding TSST-1, TSST-1 mutants, or SEC were used as sources of TSST-1, TSST-1 mutants, or SEC [19-21]. Strain RN4220 does not produce detectable endogenous superantigens. RN4220 was also used as the source of β-toxin [22]. S. aureus strain MNPE was the source of native α-toxin [23]. Escherichia coli clones were the sources of mutant α-toxin (H35L), as provided by Dr. Juliane Bubeck-Wardenburg, University of Chicago, and γ-toxin as expressed from a pET vector [16]. S. aureus strain MNPE was used in microbial challenge studies; this organism caused a fatal case of post-influenza TSS in Minnesota [24]. This organism is USA200; these organisms cause the majority of TSS cases [25]. MNPE has the following secreted virulence factor phenotype of importance for our studies: TSST-1high+, SEChigh+, α-toxinhigh+, β-toxinhigh+, and γ-toxin+ [23]. For use in pneumonia and infective endocarditis/sepsis studies, the organism was grown overnight in 25 ml of Todd-Hewitt (Difco Laboratories, Detroit, MI) broth at 37 °C with shaking at 200 revolutions per min under standard air conditions [26]. The organism was washed one time with phosphate-buffered saline (PBS; 0.005M sodium phosphate, pH 7.2; 0.15 M NaCl) through centrifugation at 14,000 × g, 5 min, and then resuspended in Todd Hewitt medium at 2 × 109/0.2 ml volume for high-dose injection in pneumonia studies [17], and in PBS at 1 × 108/ml, with 2 ml being injected intravenously for infective endocarditis/sepsis studies [27].
Secreted virulence factor purification
All reagents used in preparation of superantigens were maintained pyrogen-free. For production of TSST-1, TSST-1 toxoids, SEC, native α-toxin, and native β-toxin, the organisms were grown overnight in dialyzed beef-heart media [28]. TSST-1, TSST-1 toxoids, SEC and β-toxin were precipitated from culture fluids with 4 volumes of absolute ethanol for two days (80% final concentration), resolubilized in distilled water, and then purified by thin-layer isoelectric focusing. Isoelectric focusing pH gradients were pH 3.5-10 for initial separation, followed by gradients of pH 6-8 for TSST-1, TSST-1 toxoids, and α-toxin and 7-9 for SEC and β-toxin [28]. Native α-toxin was produced comparably from S. aureus MNPE, except the toxin was precipitated from culture fluids with 80% final saturation of ammonium sulfate, followed by solubilization in distilled water and three days dialysis, and then followed by isoelectric focusing. The biologically inactive mutant of α-toxin (H35L) and an enriched preparation of γ-toxin were produced from Escherichia coli clones in pET vectors and purified on nickel columns [16]. TSST-1, TSST-1 mutants, and SEC were homogeneous when tested by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and reversed-phase high-performance liquid chromatography [28]. Additionally, these proteins were negative for contaminating lipopolysaccharide (LPS), peptidoglycan, cytolysins, lipase, and proteases. Native α-toxin was further purified by reversed-phase high-performance liquid chromatography and was homogeneous [23]. The α-toxin mutant H35L and γ-toxin, as produced in E. coli contained minor E. coli contaminates that did not affect experimentation. Purified toxins were quantified using the BioRad protein assay [17].
Production of TSST-1 mutants
Three site-specific mutants of TSST-1 were prepared through use of the Quikchange method (Stratagene, La Jolla, CA). The initial plasmid was native tstH, on a shuttle plasmid pCE104, cloned into E. coli [20]. After performing mutagenesis, the resultant plasmids were cloned first into E. coli, verified to have the correct TSST-1 mutations by sequencing the entire structural genes, and then cloned into S. aureus RN4220 for production and purification. The mutants were TSST-1 (G31S/S32P) that fails to interact with major histocompatibility complex (MHC) class II molecules [20, 29, 30], H135A that fails to bind the variable part of the β chain of the T cell receptor (Vβ-TCR) [20, 29, 30], and Q136A that also fails to bind to Vβ-TCR [20, 29, 30].
Rabbits
All rabbits were used in compliance with regulations established by the University of Minnesota Institutional Animal Care and Use Committee (IACUC) through approved protocols. Dutch-belted rabbits, male and female, weighing 1-2 Kg were used for pneumonia studies. New Zealand white rabbits, male and female, weighing 2-3 Kg were used for endocarditis/sepsis studies. All rabbits were purchased from Bakkom Rabbitry, Red Wing, MN.
Immunizations
Dutch-belted and New Zealand white rabbits were hyperimmunized against biologically inactivated proteins (TSST-1 and α-toxin mutants) by emulsifying 25 μg of each alone or in combination in PBS with an equal volume of Freund’s incomplete adjuvant. Immunizations were in multiple subcutaneous sites in the nape of the necks. Native toxins (TSST-1, SEC, α-toxin, β-toxin, and γ-toxin) were used at a dose of approximately 10 μg/ml for immunization following the same protocol. Immunizations for all experiments were every-other-week (days 0, 14, and 28) for three injections. One week after the last immunization of animals, blood was drawn from the marginal ear veins, sera collected, and antibody titers determined by ELISA. In one experiment, following the initial three injections, animals were immunized monthly for up to six months. One week after each immunization, blood was drawn from the marginal ear veins, sera collected, and antibody titers determined by ELISA.
ELISA antibody quantification
Enzyme-linked immunosorbent assays (ELISAs) were used to determine antibody titers of immunized animals as described previously [31]. Briefly, flat-bottomed 96-well plates (NUNC Maxisorp, Portsmouth, NH) were coated with 1.0 μg/well of purified native homologous superantigen or cytolysin and then washed. Rabbit serum samples were serially diluted 2-fold beginning with a 1:10 dilution; plates were incubated for a minimum of 1.5 hr at room temperature, and then washed. Horseradish peroxidase-conjugated anti-rabbit IgG antibodies (Sigma-Aldrich, St. Louis, MO) were added to the wells. The plates were again incubated for a minimum of 1.5 hr, and the wells were washed. The relative levels of IgG were determined by 100 μl/well addition of an o-phenylenediamine and H2O2 substrate. Colorimetric reactions were halted by the addition of 50 μl of a 12.5% sulfuric acid solution. Plates were scanned for absorbance at 490 nm wavelength using a spectrophotometer.
TSST-1 mutant toxicity studies
Three assays were used to assess residual toxicity of TSST-1 mutants that are proposed for use as toxoids. Mutants tested included TSST-1 (G31S/S32P), TSST-1 (H135A), and TSST-1 (Q136A). Three assays were used.
TSST-1 amplifies the lethal effects of LPS by as much as 106 fold [32]. Our prior studies in rabbits indicate the LD50 of LPS alone in Dutch-belted rabbits is 500 ug/kg given intravenously [32]. Additionally, bolus administration of as much as 2 mg/kg of TSST-1 alone intravenously to rabbits is not lethal; TSST-1 is more lethal when continuous exposure occurs over several days [33, 34]. The relationship between TSST-1 and LPS in this enhancement phenomenon is log:log, such that for each 10-fold increase in TSST-1 pre-treatment, the amount of LPS administered 4 hr later to kill the animals is reduced by 10-fold. Thus, we used 500 μg/kg native TSST-1 or the three TSST-1 mutants for pre-treatment of 5 Dutch-belted rabbits/group intravenously, followed by 100 μg/kg of LPS from Salmonella enteritidis serovar typhimurium, as prepared in our laboratory by the hot-phenol method [35], at the 4 hr time-point. Fevers were recorded at the 4 hr time-point, just prior to administration of LPS compared to pre-injection of TSST-1 or mutants, and deaths were recorded over a 48 hr time-period. If mutants lacked lethality, this would indicate they were >500,000-fold inactivated [32].
TSST-1 alone is lethal to rabbits when administered in subcutaneously implanted miniosmotic pumps [33, 34]; a lethal dose in this model is 75 μg/animal (11 ug/day). Native TSST-1 and each TSST-1 mutant (1000 μg/animal; 143 μg/day or 10× lethal dose) were administered in miniosmotic pumps (Alza Corporation, Vacaville, CA) to 5 rabbits/group [33, 34]. Pumps were implanted while animals were anesthetized with ketamine (25 mg/kg) and xylazine (25 mg/kg) (Phoenix Pharmaceuticals, Burlingame, CA). Rabbits were monitored for 7 days for the development of TSS symptoms (fever, diarrhea, reddening of conjunctivae, and evidence of hypotension) and lethal illness, defined as the point 100% predictive of impending death, including simultaneous failure of the animals to remain upright and failure to exhibit flight responses. Animals were euthanized with intravenous injection of 1 ml/kg of Beuthanasia-D (Shering-Plough. Westlake, TX). Surviving rabbits were euthanized at the end of 7 days.
The most sensitive measure of TSST-1 toxicity in vitro is tests of superantigenicity in a 4-day assay [36]. Native TSST-1 is superantigenic across the toxin range from 10 μg/well down to 10−6 μg/well. Native TSST-1 and all three TSST-1 mutants were tested in this assay for superantigenicity with use of rabbit splenocytes in dose ranges of 10 μg/well to 10−8 μg/well. Proliferation was measured by incorporation of 3H-thymidine into DNA [36].
Immunization against TSST-1 mutant proteins protection of rabbits from native TSST-1
We examined the ability of TSST-1 (G31S/S32P), TSST-1 (H135A), and TSST-1 (Q136A) to elicit protective antibodies against the native toxin. For these studies, 10 rabbits per group were immunized three times, every-other-week, with the individual TSST-1 mutants, and then these rabbits and control, non-immunized animals, (5/group) were challenged with otherwise lethal doses of native TSST-1, either (10 μg/kg) plus LPS (10 μg/kg) intravenously (5000 × LD50) [32] or alone (500 μg/kg) in miniosmotic pumps (71 μg/day; 5.5 × LD50) [33, 34].
In vitro test of antibodies against TSST-1 mutants to neutralize native TSST-1
Pre- and post-immunization sera from 10 rabbits per group immunized three times with TSST-1 (G31S/S32P), TSST-1 (H135A), and TSST-1 (Q136A) were pooled and tested for capacity to neutralize superantigenicity of native TSST-1 (1 μg/well) with use of rabbit splenocytes in a standard 4 day assay [36].
Rabbit pulmonary illness model
Dutch-belted rabbits were administered MNPE (2 × 109 colony-forming units [CFUs] in 0.2 ml volumes) via intra-tracheal inoculation as described previously [31]. Briefly, rabbits were anesthetized with subcutaneous injections of ketamine (25 mg/kg) and xylazine (25 mg/kg) (Phoenix Pharmaceuticals, Burlingame, CA). Their necks were shaved, and small incisions were made to expose the tracheas. Small (3 mm) incisions were made into the tracheas before inserting 1 mm diameter polyethylene catheters (Becton, Dickinson, and Co, Sparks, MD) and threading them into the left bronchi. MNPE was administered through the catheters, and then catheters removed and incision sites closed. Rabbits were monitored for 7 days for the development of TSS symptoms (fever, diarrhea, reddening of conjunctivae, and evidence of hypotension) and lethal illness, defined as the point 100% predictive of impending death, including simultaneous failure of the animals to remain upright and failure to exhibit flight responses. Animals were euthanized with intravenous injection of 1 ml/kg of Beuthanasia-D (Shering-Plough. Westlake, TX). Surviving rabbits were euthanized at the end of 7 days.
Rabbit infective endocarditis and sepsis model
New Zealand white rabbits were used for the rabbit model of infective endocarditis and sepsis, as previously described [27]. Briefly, the rabbits were anesthetized with ketamine (25 mg/kg) and xylazine (25 mg/kg). Incisions were made on the left side of the necks to expose the left common carotid arteries. Catheters were inserted into the left carotid arteries and threaded until against the aortic valves, where they remained in place for 2 hr to induce damage to the endothelia. After 2 hr, the catheters were removed and the surgical sites closed. Doses of 2 × 108 CFUs of S. aureus MNPE were injected into the marginal ear veins. Because the animals were injected intravenously, we were able to monitor progression to lethal sepsis as well as infective endocarditis. Rabbits were monitored for 4 days for signs of illness and lethality, as described above. At the time of impending death or after 4 days, rabbits were euthanized. Hearts were removed and examined for vegetations. If vegetations were observed, they were excised, weighed, homogenized and serially diluted to determine CFUs. If vegetations were not present, scrapings of the aortic valves were taken, serially diluted, and plated.
CD40 and antibodies
Purified CD40 and CD40 ligand (CD154) were purchased from R and D Systems, Minneapolis, MN. Monoclonal antibodies that neutralize CD40 interaction with CD40 ligand (CD154) and CD40 ligand were also purchased from R and D Systems.
Human Vaginal Epithelial Cells (HVECs)
HVECs from a pre-menopausal woman were described previously [37]. A second HVEC line was purchased from ATCC. HVECs were cultured in keratinocyte serum-free medium (KSFM) with antibiotics until 24 hr before use. At that time, the cells were changed to KSFM without antibiotics. Experiments were performed in KSFM medium without antibiotics. These cells were determined by flow cytometry to lack MHC II molecules on their surfaces.
TSST-1 binding to CD40 by Western immunoblotting
CD40 (2 μg/lane) and control protein (ovalbumin; 2 μg) were electrophoresed in non-denaturing PAGE and then transblotted onto PVDF membranes [38]. Membranes were blocked by addition of 1% bovine serum albumin and 1% human serum for 30 min. Subsequently, 0.033 μg/ml to 33 μg/ml of TSST-1, TSST-1 (Q136A), or TSST-1 (G31S/S32P) were incubated with the membranes for 24 hr at room temperature. The membranes were then washed and incubated successively with rabbit antibodies against TSST-1, alkaline phosphatase-conjugated antibodies against rabbit IgG, and finally substrate, with washing between steps.
Kd Determination for TSST-1 binding to CD40
Various concentrations of TSST-1, ranging from 0.033 μg/ml to 33 μg/ml, were incubated individually with 2 μg CD40 on PVDF membranes overnight to ensure equilibrium in binding. Subsequently, the membranes were washed and incubated successively with rabbit antibodies against TSST-1, alkaline phosphatase-conjugated antibodies against rabbit IgG, and finally substrate. The density of protein bands was compared to standard amounts of purified TSST-1 treated similarly, with concentrations compared by NIH program ImageJ (http://rsbweb.nih.gov/ij/). For Kd determination, Scatchard analysis was performed.
CD40 pull-down assay
Magnetic beads (Dynabeads, Invitrogen Life Sciences, Grand Island, NY) coated with protein A were treated with goat IgG antibodies against TSST-1, then TSST-1, and finally CD40 (2 ug), with washing between steps and after incubation with CD40. The resultant preparations were treated with sodium dodecyl sulfate (SDS) PAGE sample buffer, electrophoresed by SDS-PAGE [39], and then tested by Western immunoblotting for CD40.
Monoclonal antibodies that neutralize CD40 ligand binding to CD40 competition with TSST-1 for CD40 binding on HVECs
Monoclonal antibodies against CD40 alone (xCD40; 20 μl undiluted), TSST-1 alone (100 μg/ml), isotype-matched monoclonal antibodies against streptococcal pyrogenic exotoxin A (xSPEA) and monoclonal antibodies against CD40 + TSST-1, and monoclonal antibodies against streptococcal pyrogenic exotoxin + TSST-1 were incubated with HVECs for 6 hr. Subsequently IL-8 production was measured by ELISA. As an important control, we showed the same monoclonal antibodies against CD40 block CD40 ligand stimulation of cytokine production from HVECs.
Statistics
Unpaired Student’s t test was used to compare fever responses between groups. Log-rank test was used to compare differences in animal survival curves among groups. Bonferroni method was used to adjust for multiple pairwise comparisons between groups. In assays of TSST-1 interaction with CD40 on HVECs, values presented are standard errors of the mean; mean differences were determined by Student t test analysis.
RESULTS
Rabbit antibody responsiveness
We determined the ability of rabbits to develop protective antibody responses to native TSST-1 as an in vivo model to understand the mechanism for the lack of protective antibody responses in humans. Rabbits, as opposed to mice, are highly susceptible to superantigens and make an excellent model for studying factors important for the development of TSS [33, 34]. Immunization of 20 Dutch-belted rabbits with 25 μg/dose of native TSST-1 emulsified in incomplete adjuvant every-other-week for three injections resulted in only 10/20 rabbits developing antibody titers against TSST-1 and those were >10,000 as tested by ELISA, where titer refers to the reciprocal of the last well dilution to give a positive color change above background. For comparison, humans who are susceptible to TSS have antibody titers of ≤40 against TSST-1, and humans who do not develop TSS have titers of ≥80 [33, 34]. Thus, the 10 rabbits that developed antibodies may be considered hyperimmune to TSST-1.
In contrast, the 10 remaining animals had antibody titers of <10, the lower limit of our detection. These 10 non-responsive animals were next continuously immunized monthly for up to 6 months or for as long as they survived. The rabbits were also monitored for development of antibodies to TSST-1 by ELISA monthly. All 10 animals succumbed to the vaccination attempts, with 7 dying after 6 months. At all tested time-points, all of these 10 rabbits had antibody titers of ≤10. Thus, the rabbit model appears to duplicate the human situation in that a significant percentage of both humans and rabbits appear unable to develop antibody responses to TSST-1. We have observed the same phenomenon for rabbit antibody responses to the superantigens SEB and SEC (data not shown).
Immunization against mutant TSST-1 proteins
We next performed studies to evaluate whether or not rabbits could develop antibodies to TSST-1 mutants that were inactivated in ability to bind to MHC II or Vβ-TCR. For these studies, three site-specific mutants were constructed in tstH, leading to production of TSST-1 proteins G31S/S32P that fails to interact with MHC II and H135A and Q136A that fail to bind Vβ-TCR [30]. These three proteins were used separately to immunize 10 Dutch-belted rabbits each, with 25 μg/dose for three every-other-week injections. Upon drawing blood one week after the third injection, all 10 animals in each group (30 total) had antibody titers of >10,000 against native TSST-1 as tested by ELISA. These data indicate the prior failure of 50% of rabbits to develop antibody responses resulted from TSST-1 induced dysregulation of immune responses, rather than genetic inability to recognize TSST-1 as a foreign protein.
Residual toxicity of TSST-1 mutants
We used the three potential TSST-1 toxoids (G31S/S32P, H135A, and Q136A) as proof of principle for ability to produce effective superantigen toxoids. None of the 30 immunized rabbits above exhibited signs of TSS as a result of vaccination, suggesting the three mutant proteins were biologically inactivated. We tested the extent of inactivation of the mutant proteins through three assays, maximizing the chances to observe residual toxicity.
We used 500 μg/kg TSST-1 or TSST-1 mutants for pre-treatment of 5 Dutch-belted rabbits per group intravenously, followed by 100 μg/kg of LPS at the 4 hr time-point. Native TSST-1 as expected caused high fevers, whereas all 3 mutants were non-pyrogenic (p<0.001 for comparison of TSST-1 to any mutant) (Figure 1A). Additionally, all 5 rabbits receiving native TSST-1 followed by LPS, succumbed within 1 hr, but none of the 5 rabbits receiving mutant TSST-1 proteins followed by LPS succumbed by 48 hr (p<0.008 for TSST-1 compared to any mutant) (Figure 1A). These data suggest that all 3 mutant proteins were ≥500,000-fold inactivated, and thus could be considered as toxoids.
Figure 1.
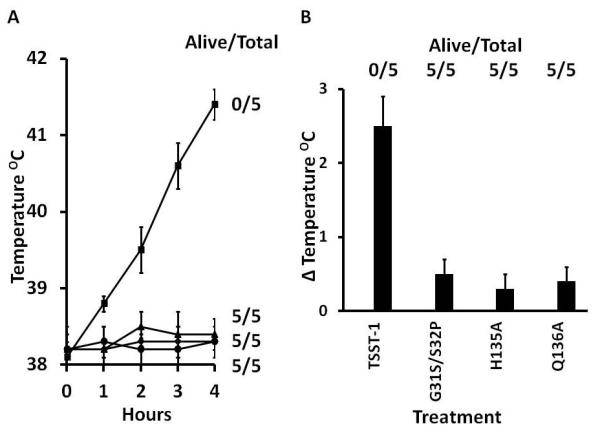
A. Pyrogenicity ± standard deviation over a 4 hr period and enhancement of lethal LPS shock (Alive/Total) by 500 μg/kg TSST-1 (■), G31S/S32P (◆), H135A (▲), and Q136A (●). TSST-1, G31S/S32P, H135A, and Q136A were administered intravenously at 0 hr; LPS (100μg/kg) was administered intravenously at 4 hr, just after taking the 4 hr temperatures. Alive/Total refers to the number of animals that survived as measured 48 hr post LPS injection.
B. Pyrogenicity on day 2 ± standard deviation and lethality over a 7 day period due to TSST-1, G31S/S32P, H135A, and Q136A administered subcutaneously as 1000 μg/miniosmotic pump.
Administration of the 3 TSST-1 mutants (1000 μg/animal; 143 μg/day or 10× LD50) in miniosmotic pumps to 5 rabbits/group did not induce fevers, as measured on day 2 post-implantation (p<0.001 for TSST-1 compared to any mutant), did not cause any TSS symptoms, and did not cause deaths in any animals (Figure 1B). In contrast, native TSST-1 was pyrogenic, induced TSS symptoms, and caused the deaths of all 5 animals by 48 hr (p<0.008 for TSST-1 compared to any mutant).
Native TSST-1 was superantigenic across the toxin range from 10 μg/well down to 10−6 μg/well (Figure 2). None of the 3 TSST-1 mutants exhibited superantigenic activity, even at the 10 μg/well dose. These studies indicate the superantigencity of the mutants was reduced by >107-fold.
Figure 2. Superantigenicity ± standard deviation of TSST-1 (■), G31S/S32P (◆), H135A (▲), and Q136A (●) for rabbit splenocytes in a 4-day assay.
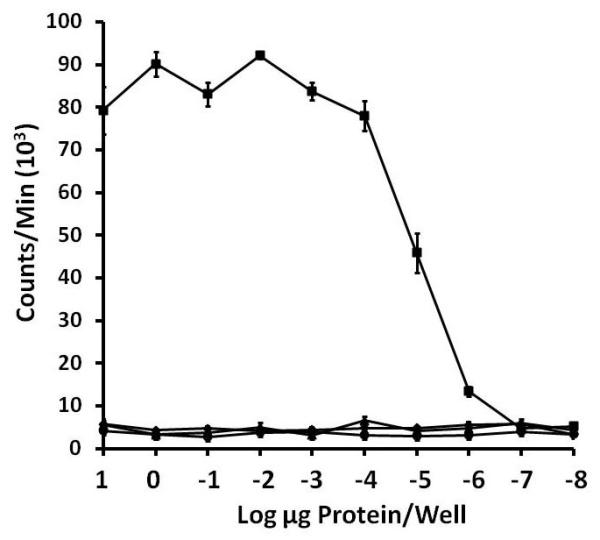
Rabbit splenocytes (2 × 105/well) were incubated with TSST-1 and mutants for 3 days, and then 1 μCi 3H-thymidine per well added for 24 hr. DNA was harvested, and counts per minute determined as a measure of T cell proliferation.
Immunization against TSST-1 mutants protects rabbits from native TSST-1 lethality
With evidence that the three mutant TSST-1 proteins (G31S/S32P, H135A, and Q136A) were converted into biologically inactive proteins that stimulate antibodies reactive against TSST-1 as tested by ELISA, we examined the ability of these proteins to elicit protective antibodies against the native toxin. Ten rabbits per group were immunized three times with the individual TSST-1 mutants, their antibody titers were determined to be >10,000, and then these rabbits and control, non-immunized animals, (5/group), were challenged one week after the last immunization with otherwise lethal doses of native TSST-1, either (10 μg/kg) plus LPS (10 μg/kg) intravenously (5000 × LD50) or alone (500 μg/kg) in miniosmotic pumps (71 μg/day; 5.5 × LD50). None of the 5 rabbits per group developed fevers when challenged with TSST-1 in the LPS enhancement model, and none of the 5 animals/group succumbed after being given LPS at the 4 hr time-point. In contrast, all 5 control, non-immunized animals developed TSST-1 induced fevers, and all succumbed in <6 hr post-administration of LPS. In the miniosmotic pump model, none of the 5 animals/group developed fevers, as measured on day 2 post-implantation, none showed TSS symptoms, and none succumbed. In contrast, all 5 control, non-immunized animals showed fevers, and all succumbed by 2 days post-implantation.
TSST-1 neutralization by antibodies
Prior to their challenge with native TSST-1, serum collected from the 10 rabbits above that were immunized three times every-other-week with the mutant proteins were pooled. These pooled sera and pooled sera from pre-vaccinated animals were tested in vitro for ability to neutralize TSST-1 superantigenicity, as tested with rabbit splenocytes and 1 μg/well of native TSST-1 (Figure 3). In these assays, undiluted and 1/10 and 1/100 diluted sera from immune animals completely neutralized TSST-1 superantigenicity; even 1/1000 diluted pooled sera from immune animals partially neutralized native TSST-1 superantigenicity. In contrast, 20 μl of undiluted pre-immune pooled serum failed to neutralize superantigenicity. The data suggest the mechanism of immunizing against TSST-1 lethality is neutralization of superantigenicity.
Figure 3. Comparison of pooled rabbit sera from non-immune animals versus animals hyperimmune to TSST-1 mutants G31S/S32P, H135A, and Q136A to inhibit superantigenicity of TSST-1 (1 μg/well), as tested in a 4 day assay with rabbit splenocytes.
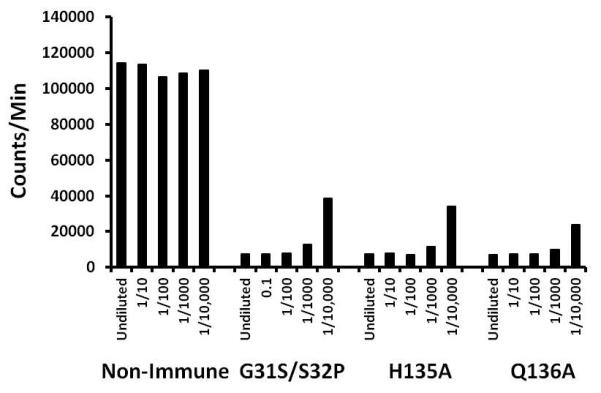
Splenocytes were incubated with designated dilutions of sera + TSST-1 for 3 days, and then 1 μCi 3H-thymidine added for 24 hr. DNA was harvested and counts/min determined as a measure of lymphocyte proliferation. Counts/min splenocytes + TSST-1 = 110,801 ± 8647. Counts/min splenocytes alone = 7248 ± 1164.
TSST-1 (Q136A) and TSST-1 (G31S/S32P) function as adjuvants to stimulate antibody responses to other antigens
In a previous study with streptococcal superantigens, it was suggested that non-toxic mutant superantigens may have intrinsic adjuvant activity [40]. This observation was formally tested in immunization studies using a second staphylococcal antigenic toxin (β-toxin) with and without two TSST-1 mutants (G31S/S32P and Q136A). Rabbits were immunized three times and then assayed one week after immunization. Rabbits immunized with β-toxin alone developed immune response antibody titers that increased from 100 after the first immunization to 600 after the third immunization (Figure 4). In contrast, co-immunization with β-toxin and either TSST-1 (G31S/S32P) or TSST-1 (Q136A) resulted in antibody titers to β-toxins increasing from 200-300 after the first immunization to nearly 106 after the third immunization. These data indicate that the MHC II mutant TSST-1 (G31S/S32P) and the TCR mutant TSST-1 (Q136A) function as effective adjuvants.
Figure 4. Antibody titer to β-toxin of rabbit (5/group) immunized against staphylococcal β-toxin alone and β-toxin mixed with TSST-1 mutants G31S/S32P and Q136A.
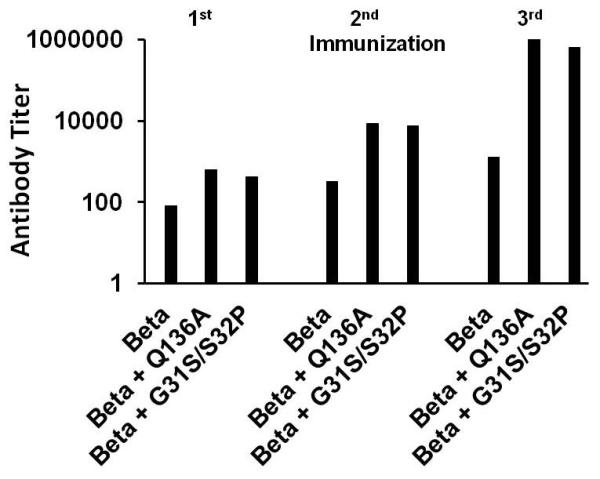
Titers represent the reciprocal of the serum dilution to give a positive color change by ELISA when tested against β-toxin.
Possible mechanism of adjuvanticity through the immune co-stimulatory molecule CD40
Our vaccination studies were performed in rabbits because these animals, like humans, are highly susceptible to the toxic effects of superantigens. However, studies in rabbits restrict our ability to determine the mechanism of adjuvanticity. Because of this problem, we performed in vitro studies with use of human vaginal epithelial cells (HVECs) to determine possible mechanisms of intrinsic adjuvanticity, based on our hypothesis that adjuvanticity must occur through TSST-1 and immune cell receptor interactions independent of superantigen interaction with MHC II and Vβ-TCR molecules. Thus, our studies focused on additional TSST-1 receptors that could explain amplified antibody responses.
Our prior studies examined changes in HVEC gene expression following exposures to TSST-1 by microarray analysis [37]. In addition to increasing the expression of cytokines and chemokines, these studies indicated that CD40 RNA transcription was up-regulated when ATCC HVECs were incubated with TSST-1 (unpublished data). CD40 is an important immune co-stimulatory molecule required for optimal production of antibodies by B cells [41]. Additionally, our studies determined that HVECs lack MHC II molecules on their surfaces (data not shown). Thus, these cells provided an important cell line that we could use to determine if TSST-1 interacts with CD40 as the potential receptor needed for adjuvanticity.
Through use of non-denaturing PAGE, we demonstrated that TSST-1, TSST-1 (Q136A), and TSST-1 (G31S/S32P) bound to CD40 in Western immunoblots (Figure 5), but did not bind to electrophoresed ovalbumin as a negative control (not shown). The binding of all three TSST-1 proteins appeared comparable. The comparable binding of all three proteins indicates that regions of TSST-1 that interact with Vβ-TCR (Q136) and α-chain MHC II (G31/S32) do not interact with CD40.
Figure 5. Wild-type TSST-1, TSST-1 (Q136A), and TSST-1 (G31S/S32P) bind to non-denatured CD40 in Western immunoblots.
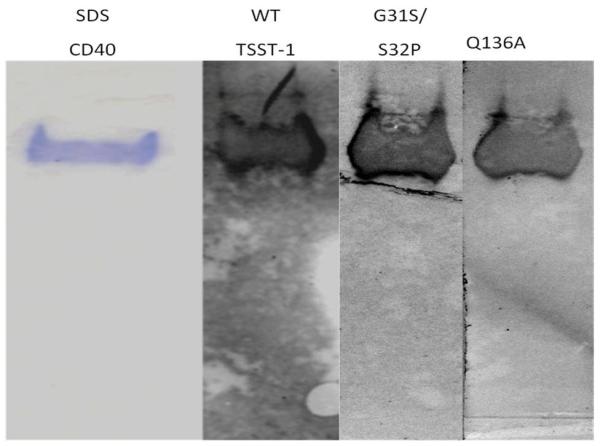
CD40 (2 ug/lane) was subject to non-denaturing PAGE and either stained with Coomassie blue (lane 1) or tranblotted to PVDF membranes. Blots were incubated consecutively with TSST-1 proteins, rabbit antibodies against TSST-1, alkaline phosphatase-conjugated antibodies against rabbit IgG, and substrate.
Through use of a fixed concentration of CD40 in Western immunoblots (2 μg), combined with incubation with dilutions of TSST-1 ranging from 0.033 μg/ml to 33 μg/ml, and comparison to standard amounts of TSST-1, the Kd of the interaction of CD40 with TSST-1, as determined by NIH program ImageJ and Skatchard analysis, was approximately 2.7 × 10−6 M.
In order to have an independent method to assess CD40 interaction with TSST-1, we used pull-down assays to confirm binding. In this assay, magnetic beads coated with protein A were treated with goat IgG antibodies against TSST-1, then TSST-1, and finally CD40 (2 μg), with washing between steps and after incubation with CD40. The resultant preparations were treated with sodium dodecyl sulfate (SDS) PAGE sample buffer, electrophored by SDS-PAGE, and then tested by Western immunoblotting for CD40 (Figure 6). Controls consisted of treating the beads without TSST-1 but with CD40. In the presence of TSST-1 on the beads, more CD40 was pulled down than in the absence of TSST-1, confirming that TSST-1 bound to CD40.
Figure 6. Wild-type TSST-1 immobilized on beads pulls-down CD40.
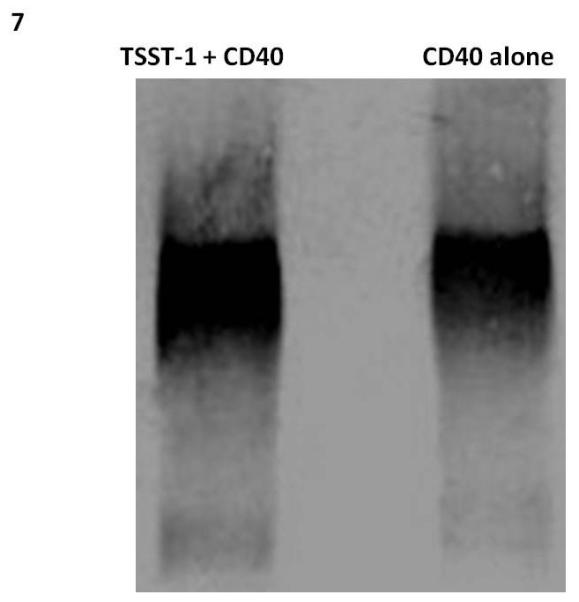
TSST-1 was immobilized on magnetic Dynabeads and then used to bind purified CD40. Control beads lacked TSST-1 and were treated comparably with CD40. After washing, beads were treated with SDS-PAGE buffer and samples electrophoresed in SDS-PAGE gels. Subsequently, proteins were transferred to PVDF membranes and probed with monoclonal antibodies to CD40, alkaline phosphatase-conjugated antibodies to mouse immunoglobulin, and then substrate.
We hypothesized that co-incubation of TSST-1 and monoclonal antibodies that neutralize CD40 interaction with CD40 ligand on T cells with HVECs would result in interference with IL-8 chemokine production. Unexpectedly, we observed a nearly 3-fold synergy in IL-8 chemokine production when both TSST-1 and monoclonal antibodies against CD40 were incubated with the HVECs compared to TSST-1 alone (Figure 7); the monoclonal antibodies to CD40 did not induce cytokine production. Additionally, an irrelevant monoclonal antibody (monoclonal antibodies against streptococcal pyrogenic exotoxin A (SPEA) did not synergize with TSST-1 to cause amplified IL-8 production. Finally, the same monoclonal antibodies against CD40 block CD40 ligand stimulation of chemokine production from HVECs (data not shown).
Figure 7. HVECs treated with TSST-1 and monoclonal antibodies against CD40, that neutralize interaction with T cell CD40 ligand, synergize to cause chemokine production.
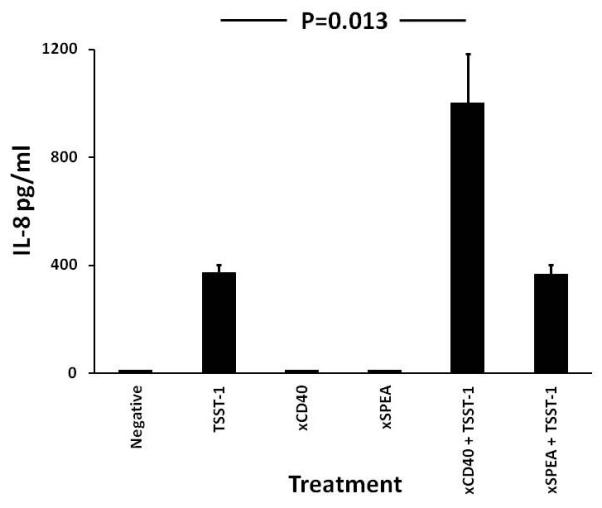
Monoclonal antibodies against CD40 alone (xCD40; 20 μl undiluted), TSST-1 alone (100 μg/ml), isotype-matched monoclonal antibodies against streptococcal pyrogenic exotoxin A (xSPEA) and monoclonal antibodies against CD40 + TSST-1, and monoclonal antibodies against streptococcal pyrogenic exotoxin + TSST-1 were incubated with confluent HVECs in 96 well microtiter plates in triplicate for 6 hr. Subsequently IL-8 production was measured by ELISA. Bars represent standard error of the means.
Collectively, these data suggest that TSST-1 binds to the immune co-stimulatory molecule CD40, which is required for optimal stimulation of B cell to produce neutralizing antibodies. This may account for the TSST-1 mutant toxoid adjuvanticity. It is likely that native TSST-1 interacts more prominently with MHC II and Vβ-TCR to mask the adjuvant effect.
Tri-valent vaccine prevents S. aureus pneumonia
We have shown previously that immunity to TSST-1 and SEC protects rabbits from intra-pulmonary challenge with TSST-1, SEC or USA200 S. aureus producing TSST-1 and SEC [17]. Other investigators have shown that immunity to staphylococcal α-toxin protects mice from lethal pneumonia [16]. We thus evaluated the ability of a trivalent vaccine, composed of TSST-1 (G31S/S32P), a low dose of native SEC, and combined with a non-toxic dose of α-toxin (H35L) (5 rabbits) or wild-type α-toxin (6 rabbits) to protect from lethal pneumonia with a high dose challenge with USA200 S. aureus MNPE (2 × 109 CFUs) that produces high levels of TSST-1, SEC, and α-toxin. We also evaluated immunization against the non-toxic mutant of α-toxin (H35L) (5 animals) or native α-toxin (5 animals) alone to protect rabbits from similar challenge with S. aureus MNPE. Since we observed no differences in antibody responses or protection from pneumonia in rabbits that had been immunization with α-toxin (H35L) versus wild-type α-toxin (low dose immunization), we combined the groups in data presented. All animals were immunized every-other-week for 3 injections in incomplete adjuvant, shown to have high antibody titers (>10,000) against all three native toxins by ELISA, and were challenged intra-pulmonary one week after the last immunization, along with non-immune controls, with 2 × 109 MNPE. There were significant differences in survivals among the groups (p<0.001). For rabbits immunized against the trivalent vaccine containing TSST-1 G31S/S32P + SEC + α-toxin (H35L or native), all were protected from lethal pneumonia (Figure 8a). In contrast, all 11 non-immunized animals succumbed to the lethal challenge (p<0.001). Rabbits immunized with the α-toxin H35L alone or native α-toxin alone showed delayed deaths due to challenge with MNPE, but ultimately, 9/11 succumbed (p=0.001, compared to non-immunized controls). Rabbits immunized against the trivalent vaccine had better survival than rabbits immunized against α-toxin (H35L or native) alone (p<0.001).
Figure 8.
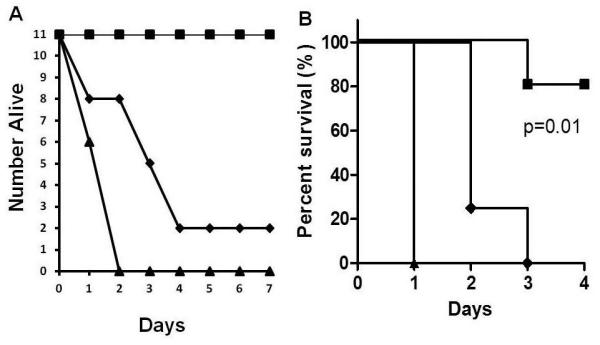
A. Protection of rabbits from lethal S. aureus USA200 pneumonia by vaccination against TSST-1 (G31S/S32P) + SEC + α-toxin (H35L or wild-type) and partial protection by vaccination against α-toxin (H35L). Rabbits (11/group) were immunized three times with antigens, TSST-1 (G31S/S32P) + SEC + α-toxin (H35L or wild-type) as a cocktail (■) or α-toxin (H35L) alone (◆), or remained non-immunized (▲). Antigens were emulsified in incomplete adjuvant and immune animals plus non-immune control animals challenged intrapulmonary with 2 × 109 S. aureus MNPE. Rabbits immunized against TSST-1 (G31S/S32P) + SEC + α-toxin (H35L or wild-type) were significantly protected from lethality compared to non-vaccinated animals or animals vaccinated against α-toxin (H35L or wild-type) alone (p<0.001). Animals vaccinated against α-toxin (H35L or wild-type) were significantly delayed in lethality compared to non-vaccinated controls (p=0.001). B. Protection of rabbits from lethal sepsis by immunization against five S. aureus exotoxins. Rabbits were immunized three times with TSST-1 (G31S/S32P), SEC, α-toxin (H35L), β-toxin, and γ-toxin (■) or α-toxin (H35L) alone (◆), or remained non-vaccinated (▲). Challenge organism was intravenous USA200 S. aureus MNPE (2 × 108/2 ml volume in PBS).
Pentavalent vaccine prevents infective endocarditis and sepsis
We have previously shown that TSST-1, SEC, and staphylococcal β-toxin contribute to infective endocarditis [9, 12]. These toxins are produced by USA200 S. aureus strains, the strains most commonly associated with persistent bacteremia and infective endocarditis [10]. We hypothesized that prior vaccination against the three major cytolysins of S. aureus (α-toxin [H35L], β-toxin, and γ-toxin), combined with vaccination against TSST-1 (G31S/S32P) and SEC, may protect rabbits from infective endocarditis and sepsis due to USA200 MNPE.
Rabbits (4-5/group) were immunized against these 5 proteins or α-toxin H35L alone every-other-week for 3 injections. Control animals remained non-immunized. After immunization, all animals were highly immune to each toxin by ELISA, and then all immune plus non-immunized animals were challenged one week after the last immunization with MNPE in our previously established model of infective endocarditis and sepsis. Rabbits previously immunized with the pentavalent vaccine were significantly protected from lethal sepsis (Figure 8b). Vegetation sizes for MNPE are typically up to 100 mg (data not shown). One rabbit from the pentavalent immunized group died late on day 2 and had a vegetation of 6 mg with 1×108 CFU. The largest vegetation seen in the immunized rabbits was 14 mg while the smallest was 1 mg, vastly smaller than the typical size associated with MNPE. The data suggest that prior immunization against these 5 secreted toxins provided immune protection against otherwise lethal challenge and significantly reduced vegetation size.
For rabbits previously immunized against α-toxin H35L alone, three of four developed small vegetations (2-3 mg), and all succumbed from lethal sepsis (Figure 8b), though lethality was delayed compared to the non-immunized control group (day 2 or 3 for H35L immunized rabbits versus day 1 for control rabbits). There were significant differences in overall survival among the groups (p=0.01). The survivals of animals immunized against α-toxin alone and pentavalent vaccine were both significantly better than non-immunized animals (p=0.002 and 0.001, respectively). Furthermore, rabbits immunized against pentavalent vaccine were also significantly protected from lethal sepsis than rabbits immunized against α-toxin alone (p=0.004).
All control, non-immunized animals in this study succumbed to lethal sepsis by 24 hr post injection of MNPE (Figure 8b). None of the animals had significant vegetations, presumably due to the rapidity with which the animals succumbed, though there was visual confirmation of small vegetations forming on their aortic valves.
DISCUSSION
We have shown that immunization with secreted virulence factors produced by S. aureus protects against pneumonia, infective endocarditis, and lethal sepsis caused by high-dose challenge with a highly-pathogenic USA 200 strain. The challenge organism came from a patient who succumbed to post-influenza TSS [24]. Immunization against TSST-1, SEC, and α-toxin protects rabbits against pneumonia caused by the USA200 strain producing these three exotoxins, and immunization with a pentavalent vaccine against TSST-1, SEC, α-toxin, β-toxin, and γ-toxin protects rabbits against infective endocarditis and sepsis due to the same organism. Control, non-immune rabbits succumbed to the infections, dying before the end of the experiments.
Superantigens and cytolysins function critically in a number of illnesses caused by S. aureus including necrotizing pneumonia, sepsis, and infective endocarditis [9, 13, 15, 16]. We sought to create a vaccine based on the most common secreted virulence factors known to participate in these diseases. The superantigens TSST-1, SEC, as well as the cytolysins α-toxin, β-toxin, and γ-toxin were prime candidates; however we believe that a more effective vaccine will additionally include SEB and SE-like X toxoids. Both of these exotoxins have been shown recently also to participate in serious S. aureus illnesses [17, 42]. Thus, the studies in the present research are proof of principle that a polyvalent toxoid vaccine can be developed to protect against serious staphylococcal infections.
Prior studies reported the inability of people to produce antibodies to superantigens [43-45]. There were two possible reasons for this: persons have genetic inabilities to recognize and respond to the superantigens; or these individuals have hyperimmune responses to the superantigens, resulting in immune dysregulation and lack of antibody responses. This latter effect has been observed in patients with streptococcal TSS [46]. We sought to understand the underlying mechanism, as genetic inability to respond would hinder using superantigen toxoid vaccines. Typically, only small antigens with one or two epitopes would be expected result in inability to recognize an antigen as foreign. Superantigens range in size from 19,000 to 30,000 molecular weight, and in comparison to other antigens, would be expected to have multiple epitopes for antibody recognition [47, 48]. Although formally possible, this makes it less likely that genetic inability to respond accounts for the lack of antibody responses in 20% of adults. We showed in a rabbit model that 50% of animals appear unable to develop antibody responses to native TSST-1, suggesting the same mechanism causes their unresponsiveness as humans. However, 100% of rabbits are able to make antibody responses when superantigenicity is removed through mutation. This suggests that the inability to produce antibodies to superantigens results from hyper-responsiveness to superantigens, leading to immune dysfunction through an unknown mechanism, rather than genetic inability to respond.
We also performed extensive studies to show that the TSST-1 mutant proteins used in our studies possess three important properties of toxoids. First, the mutant proteins contain no detectable residual toxicities, appearing to be >107-fold inactivated by the most sensitive test, superantigenicity, as tested in vitro. Additionally, with use of the most sensitive assays in rabbit models of TSS, we demonstrated that the proteins are >500,000-fold inactivated. Other studies with use of streptococcal superantigens to develop toxoid vaccines have also shown that mutation of immune cell contact residues greatly reduces residual activities [40, 49]. From a safety perspective double mutants such as TSST-1 (G31S/S32P) would be the most useful in vaccines since reversion to toxicity is unlikely. Like TSST-1 mutants, our studies and prior studies [16] show that the H35L mutation of α-toxin inactivates the cytolysin activity. This is particularly important since rabbits are killed by 0.1 μg intravenously of native α-toxin in 1 hr [23]. Second, prospective toxoid proteins must be immunogenic, preferably with few vaccinations. Our studies showed that exceptionally high-titer antibodies are formed by three injections. Importantly, prior studies suggested but did not prove that superantigens have intrinsic adjuvanticities [40]. The present studies demonstrate this activity clearly, showing that non-toxic superantigen mutants amplify immune responses to a second antigen, staphylococcal β-toxin, by 10 to 100-fold. We have also shown this adjuvanticity with multiple other antigens, including HIV proteins and sheep erythrocytes (data not shown). Third, toxoids must elicit antibodies that protect against native toxin. By multiple assays, we demonstrated that antibodies against TSST-1 toxoids are capable of neutralizing superantigenicity and capable of protecting rabbits from lethal challenge by native TSST-1. The rabbits have antibody titers >10,000. From prior studies, we and others have shown that healthy humans who do not develop TSS most often have antibody titers against TSST-1 between 80 and 320 [44, 45, 50].
We have explored the possible mechanism of adjuvanticity of TSST-1 mutants. This effect is not seen with use of wild-type TSST-1 which is more likely to result in antibody immunosuppression than in adjuvanticity. Rabbits like humans are highly susceptible to superantigens, but insufficient reagents are available to determine the mechanism of adjuvanticity. Thus, we used data obtained from prior studies with human epithelial cells exposed to TSST-1 to study this possible effect, with the hypothesis that TSST-1 mutant toxoids that function as adjuvants must interact with a host cell receptor other than the two known receptors that cause TSS, namely MHC II and Vβ-TCR. Our studies suggest that TSST-1 binds to the immune co-stimulatory molecule CD40 that is required for optimal production of antibodies by B cells [41]. Our studies showed that TSST-1 directly binds to purified CD40 in Western immunoblots and in CD40 pull-down assays. Additionally, our previous studies show TSST-1 interaction with HVECs leads to increased chemokine production despite HVECs lacking T cell receptors and MHC II molecules [37]. Interestingly, monoclonal antibodies that mimic T cell CD40 ligand binding to CD40 do not block cytokine production, but in fact lead to synergy in chemokine production. We hypothesize that this effect accounts for the adjuvant effect that leads to amplified antibody production in the presence of TSST-1 mutant toxoids. Additional studies are needed to verify this hypothesis.
As proof of principle for protective ability of a cocktail vaccine against secreted virulence factors, rabbits were immunized with a trivalent vaccine of TSST-1 (G31S/S32P), native SEC, and α-toxin (H35L or wild-type) a protein previously used as to protect mice from lethal staphylococcal pneumonia. The animals were challenged intra-pulmonary with a highly lethal dose of a USA200 strain producing all three of these secreted virulence factors. All of the vaccinated animals survived and exhibited immunity to the organisms. These data suggest that these secreted virulence factors are critical for establishing pulmonary infections, in addition to their causing lethality. Similarly, a pentavalent vaccine composed of 5 secreted virulence factors significantly protected rabbits against lethal sepsis and reduced the severity of infective endocarditis due to challenge with the same organism. In some studies, we immunized rabbits against α-toxin (H35L) or native α-toxin only and challenged with the highly lethal dose of MNPE. Partial protection was observed. It is possible that greater protection would have been observed if the challenge dose was not as high. However, the survival studies with high dose challenge clearly emphasize the advantage of having multiple secreted factors in the vaccine.
In our studies, we did not have available toxoids for all of the superantigens and cytolysins we wished to assess. However, our studies demonstrate that an appropriate vaccine with multiple toxoids may be protective. In our prior studies, we have already identified the critical amino acids necessary for activity of SEB, and SEC, just as we have used previous studies to identify the critical residues in TSST-1 [19]. We have also shown that these toxoids are highly inactivated [19]. Additionally, others have identified key residues in α-toxin [16], and we have completely inactivated β-toxin by mutation of two active site residues [51]. There is no compelling reason to mutagenize γ-toxin since these heptamer pore-forming toxins require two different chains for activity [52]. We suggest that immunization can be accomplished with use of the shared B chain. Our current efforts are being directed towards identification of important residues for mutation in SE-like X.
Collectively, these results suggest that a vaccine composed of common secreted virulence factors produced by Staphylococcus aureus is capable of protecting rabbits from lethal staphylococcal disease. Future studies in our laboratory seek to explore the possibility of adding in other secreted virulence factors, including SEB and the newly discovered SE-like X.
CONCLUSIONS
Patients with staphylococcal TSS do not develop neutralizing antibody responses to the superantigen TSST-1, and thus they remain susceptible to TSS recurrences. This effect results from immune dysfunction due to TSST-1, rather than genetic inability to recognize the superantigen as foreign. TSST-1 may be >107 inactivated by genetic modification of amino acid residues in the MHC II and Vβ-TCR sites; resultant mutants stimulate protective immunity against native TSST-1 and function as adjuvants to amplify antibody responses to secondary antigens. Immunization against cocktails of secreted superantigens and cytolysins protects rabbits from lethal pneumonia, infective endocarditis, and sepsis.
Highlights.
Vaccination against Staphylococcus aureus is achieved with toxoids.
Toxoids are >107 inactivated and stimulate immunity to S. aureus.
Superantigen toxoids exhibit adjuvant activity dependent on Immun Co-stimulatory molecule CD40 binding activity.
Protective neutralizing antibodies are produced in rabbits to toxic shock syndrome toxin-1.
ACKNOWLEDGEMENTS
This work was supported by National Institutes of Health Grants R01 AI074283, R01 AI73366, and U54 AI57153. P.M.S. is a member of the Great Lakes Regional Center of Excellence in Biodefense and Emerging Infectious Diseases. Dr. Juliane Bubeck-Wardenburg is gratefully acknowledged for providing α-toxin (H35L) used in these studies.
List of Abbreviations
- CFUs
Colony-forming units
- LPS
Lipopolysaccharide
- MHC
Major Histocompatibility complex
- MRSA
Methicillin-resistant S. aureus
- PBS
Phosphate-buffered saline
- SDS-PAGE
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SE
Staphylococcal enterotoxin
- TSS
Toxic shock syndrome
- TSST-1
TSS toxin-1
- Vβ-TCR
Variable part of the β-chain of the T cell receptor
- HVECs
Human vaginal epithelial cells
- KSFM
Keratinocyte serum-free medium
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest:
None of the authors have conflict of interests to declare.
Submission declaration and verification:
This research has not been published elsewhere, is not under review elsewhere, is published with approval by all authors, and if accepted will not be published elsewhere in the same form, in English or in any other language, including electronically without the written permission of the copy-right holder.
Contributors:
ARS performed direct experimentation and data analyses, wrote, and edited the manuscript. JAM, Y-CL, AJB, MLP, and PMS performed direct experimentation and data analyses, and edited the manuscript. All have approved the final version for submission.
REFERENCES
- [1].Lowy FD. Staphylococcus aureus infections. N Engl J Med. 1998;339:520–32. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- [2].McCormick JK, Yarwood JM, Schlievert PM. Toxic shock syndrome and bacterial superantigens: an update. Annu Rev Microbiol. 2001;55:77–104. doi: 10.1146/annurev.micro.55.1.77. [DOI] [PubMed] [Google Scholar]
- [3].Mulligan ME, Murray-Leisure KA, Ribner BS, Standiford HC, John JF, Korvick JA, et al. Methicillin-resistant Staphylococcus aureus: a consensus review of the microbiology, pathogenesis, and epidemiology with implications for prevention and management. Am J Med. 1993;94:313–28. doi: 10.1016/0002-9343(93)90063-u. [DOI] [PubMed] [Google Scholar]
- [4].Mylonakis E, Calderwood SB. Infective endocarditis in adults. N Engl J Med. 2001;345:1318–30. doi: 10.1056/NEJMra010082. [DOI] [PubMed] [Google Scholar]
- [5].Beynon RP, Bahl VK, Prendergast BD. Infective endocarditis. BMJ. 2006;333:334–9. doi: 10.1136/bmj.333.7563.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bayer AS, Ramos MD, Menzies BE, Yeaman MR, Shen AJ, Cheung AL. Hyperproduction of alpha-toxin by Staphylococcus aureus results in paradoxically reduced virulence in experimental endocarditis: a host defense role for platelet microbicidal proteins. Infect Immun. 1997;65:4652–60. doi: 10.1128/iai.65.11.4652-4660.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fowler VG, Jr., Miro JM, Hoen B, Cabell CH, Abrutyn E, Rubinstein E, et al. Staphylococcus aureus endocarditis: a consequence of medical progress. Jama. 2005;293:3012–21. doi: 10.1001/jama.293.24.3012. [DOI] [PubMed] [Google Scholar]
- [8].Miro JM, Anguera I, Cabell CH, Chen AY, Stafford JA, Corey GR, et al. Staphylococcus aureus native valve infective endocarditis: report of 566 episodes from the International Collaboration on Endocarditis Merged Database. Clin Infect Dis. 2005;41:507–14. doi: 10.1086/431979. [DOI] [PubMed] [Google Scholar]
- [9].Pragman AA, Yarwood JM, Tripp TJ, Schlievert PM. Characterization of virulence factor regulation by SrrAB, a two-component system in Staphylococcus aureus. J Bacteriol. 2004;186:2430–8. doi: 10.1128/JB.186.8.2430-2438.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Xiong YQ, Willard J, Yeaman MR, Cheung AL, Bayer AS. Regulation of Staphylococcus aureus alpha-toxin gene (hla) expression by agr, sarA, and sae in vitro and in experimental infective endocarditis. J Infect Dis. 2006;194:1267–75. doi: 10.1086/508210. [DOI] [PubMed] [Google Scholar]
- [11].Nienaber JJ, Sharma Kuinkel BK, Clarke-Pearson M, Lamlertthon S, Park L, Rude TH, et al. Methicillin-susceptible Staphylococcus aureus endocarditis isolates are associated with clonal complex 30 genotype and a distinct repertoire of enterotoxins and adhesins. J Infect Dis. 204:704–13. doi: 10.1093/infdis/jir389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Huseby MJ, Kruse AC, Digre J, Kohler PL, Vocke JA, Mann EE, et al. Beta toxin catalyzes formation of nucleoprotein matrix in staphylococcal biofilms. Proc Natl Acad Sci U S A. 2010;107:14407–12. doi: 10.1073/pnas.0911032107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cheung AL, Eberhardt KJ, Chung E, Yeaman MR, Sullam PM, Ramos M, et al. Diminished virulence of a sar-/agr-mutant of Staphylococcus aureus in the rabbit model of endocarditis. J Clin Invest. 1994;94:1815–22. doi: 10.1172/JCI117530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Spaulding AR, Satterwhie EA, Lin Y-C, Chuang-Smith ON, Frank KL, Merriman JA, Schaefers MM, Yarwood JM, peterson ML, Schlievert PM. Comparison of Staphylococcus aureus strains for ability to cause infective endocarditis and lethal sepsis in rabbits. Front Cell Infect Microbiol. 2012;2:1–9. doi: 10.3389/fcimb.2012.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bubeck Wardenburg J, Bae T, Otto M, Deleo FR, Schneewind O. Poring over pores: alpha-hemolysin and Panton-Valentine leukocidin in Staphylococcus aureus pneumonia. Nat Med. 2007;13:1405–6. doi: 10.1038/nm1207-1405. [DOI] [PubMed] [Google Scholar]
- [16].Bubeck Wardenburg J, Schneewind O. Vaccine protection against Staphylococcus aureus pneumonia. J Exp Med. 2008;205:287–94. doi: 10.1084/jem.20072208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Strandberg KL, Rotschafer JH, Vetter SM, Buonpane RA, Kranz DM, Schlievert PM. Staphylococcal superantigens cause lethal pulmonary disease in rabbits. J Infect Dis. 2010;202:1690–7. doi: 10.1086/657156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Voyich JM, Otto M, Mathema B, Braughton KR, Whitney AR, Welty D, et al. Is Panton-Valentine leukocidin the major virulence determinant in community-associated methicillin-resistant Staphylococcus aureus disease? J Infect Dis. 2006;194:1761–70. doi: 10.1086/509506. [DOI] [PubMed] [Google Scholar]
- [19].Leder L, Llera A, Lavoie PM, Lebedeva MI, Li H, Sekaly RP, et al. A mutational analysis of the binding of staphylococcal enterotoxins B and C3 to the T cell receptor beta chain and major histocompatibility complex class II. J Exp Med. 1998;187:823–33. doi: 10.1084/jem.187.6.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Murray DL, Earhart CA, Mitchell DT, Ohlendorf DH, Novick RP, Schlievert PM. Localization of biologically important regions on toxic shock syndrome toxin 1. Infect Immun. 1996;64:371–4. doi: 10.1128/iai.64.1.371-374.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Murray DL, Prasad GS, Earhart CA, Leonard BA, Kreiswirth BN, Novick RP, et al. Immunobiologic and biochemical properties of mutants of toxic shock syndrome toxin-1. J Immunol. 1994;152:87–95. [PubMed] [Google Scholar]
- [22].Gaskin DK, Bohach GA, Schlievert PM, Hovde CJ. Purification of Staphylococcus aureus beta-toxin: comparison of three isoelectric focusing methods. Protein Expr Purif. 1997;9:76–82. doi: 10.1006/prep.1996.0664. [DOI] [PubMed] [Google Scholar]
- [23].Lin YC, Anderson MJ, Kohler PL, Strandberg KL, Olson ME, Horswill AR, et al. Proinflammatory exoprotein characterization of toxic shock syndrome Staphylococcus aureus. Biochemistry. 2011;50:7157–67. doi: 10.1021/bi200435n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].MacDonald KL, Osterholm MT, Hedberg CW, Schrock CG, Peterson GF, Jentzen JM, et al. Toxic shock syndrome. A newly recognized complication of influenza and influenzalike illness. Jama. 1987;257:1053–8. doi: 10.1001/jama.257.8.1053. [DOI] [PubMed] [Google Scholar]
- [25].Schlievert PM, Strandberg KL, Lin YC, Peterson ML, Leung DY. Secreted virulence factor comparison between methicillin-resistant and methicillin-sensitive Staphylococcus aureus, and its relevance to atopic dermatitis. J Allergy Clin Immunol. 2010;125:39–49. doi: 10.1016/j.jaci.2009.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Schlievert PM, Blomster DA. Production of staphylococcal pyrogenic exotoxin type C: influence of physical and chemical factors. J Infect Dis. 1983;147:236–42. doi: 10.1093/infdis/147.2.236. [DOI] [PubMed] [Google Scholar]
- [27].Schlievert PM, Gahr PJ, Assimacopoulos AP, Dinges MM, Stoehr JA, Harmala JW, et al. Aggregation and binding substances enhance pathogenicity in rabbit models of Enterococcus faecalis endocarditis. Infect Immun. 1998;66:218–23. doi: 10.1128/iai.66.1.218-223.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Blomster-Hautamaa DA, Schlievert PM. Preparation of toxic shock syndrome toxin-1. Methods Enzymol. 1988;165:37–43. doi: 10.1016/s0076-6879(88)65009-9. [DOI] [PubMed] [Google Scholar]
- [29].Jardetzky TS, Brown JH, Gorga JC, Stern LJ, Urban RG, Chi YI, et al. Three-dimensional structure of a human class II histocompatibility molecule complexed with superantigen. Nature. 1994;368:711–8. doi: 10.1038/368711a0. [DOI] [PubMed] [Google Scholar]
- [30].McCormick JK, Tripp TJ, Llera AS, Sundberg EJ, Dinges MM, Mariuzza RA, et al. Functional analysis of the TCR binding domain of toxic shock syndrome toxin-1 predicts further diversity in MHC class II/superantigen/TCR ternary complexes. J Immunol. 2003;171:1385–92. doi: 10.4049/jimmunol.171.3.1385. [DOI] [PubMed] [Google Scholar]
- [31].Strandberg KL, Rotschafer JH, Vetter SM, Buonpane RA, Kranz DM, Schlievert PM. Staphylococcal superantigens cause lethal pulmonary disease in rabbits. J Infect Dis. 2010;202:1690–7. doi: 10.1086/657156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Schlievert PM. Enhancement of host susceptibility to lethal endotoxin shock by staphylococcal pyrogenic exotoxin type C. Infect Immun. 1982;36:123–8. doi: 10.1128/iai.36.1.123-128.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lee PK, Deringer JR, Kreiswirth BN, Novick RP, Schlievert PM. Fluid replacement protection of rabbits challenged subcutaneous with toxic shock syndrome toxins. Infect Immun. 1991;59:879–84. doi: 10.1128/iai.59.3.879-884.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Parsonnet J, Gillis ZA, Richter AG, Pier GB. A rabbit model of toxic shock syndrome that uses a constant, subcutaneous infusion of toxic shock syndrome toxin 1. Infect Immun. 1987;55:1070–6. doi: 10.1128/iai.55.5.1070-1076.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Westphal O, Luderitz O, Bister F. Uber die extraktion von vacterium mit phenol/wasser. Z Naturforsch. 1952;7b:148–55. [Google Scholar]
- [36].Schlievert PM, Shands KN, Dan BB, Schmid GP, Nishimura RD. Identification and characterization of an exotoxin from Staphylococcus aureus associated with toxic-shock syndrome. J Infect Dis. 1981;143:509–16. doi: 10.1093/infdis/143.4.509. [DOI] [PubMed] [Google Scholar]
- [37].Peterson M, Ault K, Kremer MJ, Klingelhutz AJ, Davis CC, Squier CA, Schlievert PM. Innate immune system is activated by stimulation of vaginal epithelial cells with Staphylococcus aureus and toxic shock syndrome toxin-1. Infect Immun. 2005;73:2164–74. doi: 10.1128/IAI.73.4.2164-2174.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Schlievert PM, Case LC, Nemeth KA, Davis CC, Sun Y, Qin W, et al. Alpha and beta chains of hemoglobin inhibit production of Staphylococcus aureus exotoxins. Biochemistry. 2007;46:14349–58. doi: 10.1021/bi701202w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- [40].Roggiani M, Stoehr JA, Olmsted SB, Matsuka YV, Pillai S, Ohlendorf DH, et al. Toxoids of streptococcal pyrogenic exotoxin A are protective in rabbit models of streptococcal toxic shock syndrome. Infect Immun. 2000;68:5011–7. doi: 10.1128/iai.68.9.5011-5017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev. 2009;229:152–72. doi: 10.1111/j.1600-065X.2009.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wilson GJ, Seo KS, Cartwright RA, Connelley T, Chuang-Smith ON, Merriman JA, et al. A novel core genome-encoded superantigen contributes to lethality of community-associated MRSA necrotizing pneumonia. PLoS Pathog. 2011;7:e1002271. doi: 10.1371/journal.ppat.1002271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Osterholm MT, Davis JP, Gibson RW, Mandel JS, Wintermeyer LA, Helms CM, et al. Tri-state toxic-state syndrome study. I. Epidemiologic findings. J Infect Dis. 1982;145:431–40. doi: 10.1093/infdis/145.4.431. [DOI] [PubMed] [Google Scholar]
- [44].Parsonnet J, Hansmann MA, Delaney ML, Modern PA, Dubois AM, Wieland-Alter W, et al. Prevalence of toxic shock syndrome toxin 1-producing Staphylococcus aureus and the presence of antibodies to this superantigen in menstruating women. J Clin Microbiol. 2005;43:4628–34. doi: 10.1128/JCM.43.9.4628-4634.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Vergeront JM, Stolz SJ, Crass BA, Nelson DB, Davis JP, Bergdoll MS. Prevalence of serum antibody to staphylococcal enterotoxin F among Wisconsin residents: implications for toxic-shock syndrome. J Infect Dis. 1983;148:692–8. doi: 10.1093/infdis/148.4.692. [DOI] [PubMed] [Google Scholar]
- [46].Kotb M, Norrby-Teglund A, McGeer A, El-Sherbini H, Dorak MT, Khurshid A, et al. An immunogenetic and molecular basis for differences in outcomes of invasive group A streptococcal infections. Nat Med. 2002;8:1398–404. doi: 10.1038/nm1202-800. [DOI] [PubMed] [Google Scholar]
- [47].Blomster-Hautamaa DA, Novick RP, Schlievert PM. Localization of biologic functions of toxic shock syndrome toxin-1 by use of monoclonal antibodies and cyanogen bromide-generated toxin fragments. J Immunol. 1986;137:3572–6. [PubMed] [Google Scholar]
- [48].Murphy BG, Kreiswirth BN, Novick RP, Schlievert PM. Localization of a biologically important epitope on toxic-shock-syndrome toxin-1. J Infect Dis. 1988;158:549–55. doi: 10.1093/infdis/158.3.549. [DOI] [PubMed] [Google Scholar]
- [49].McCormick JK, Tripp TJ, Olmsted SB, Matsuka YV, Gahr PJ, Ohlendorf DH, et al. Development of streptococcal pyrogenic exotoxin C vaccine toxoids that are protective in the rabbit model of toxic shock syndrome. J Immunol. 2000;165:2306–12. doi: 10.4049/jimmunol.165.4.2306. [DOI] [PubMed] [Google Scholar]
- [50].Bergdoll MS, Crass BA, Reiser RF, Robbins RN, Davis JP. A new staphylococcal enterotoxin, enterotoxin F, associated with toxic-shock-syndrome Staphylococcus aureus isolates. Lancet. 1981;1:1017–21. doi: 10.1016/s0140-6736(81)92186-3. [DOI] [PubMed] [Google Scholar]
- [51].Huseby M, Shi K, Brown CK, Digre J, Mengistu F, Seo KS, et al. Structure and biological activities of beta toxin from Staphylococcus aureus. J Bacteriol. 2007;189:8719–26. doi: 10.1128/JB.00741-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sugawara-Tomita N, Tomita T, Kamio Y. Stochastic assembly of two-component staphylococcal gamma-hemolysin into heteroheptameric transmembrane pores with alternate subunit arrangements in ratios of 3:4 and 4:3. J Bacteriol. 2002;184:4747–56. doi: 10.1128/JB.184.17.4747-4756.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]