

Abstract
Cancer nanotheranostics aims to combine imaging and therapy of cancer through use of nanotechnology. The ability to engineer nanomaterials to interact with cancer cells at the molecular level can significantly improve the effectiveness and specificity of therapy to cancers that are currently difficult to treat. In particular, metastatic cancers, drug-resistant cancers, and cancer stem cells impose the greatest therapeutic challenge that requires targeted therapy to treat effectively. Targeted therapy can be achieved with appropriate designed drug delivery vehicles such as nanoparticles, adult stem cells, or T cells in immunotherapy. In this article, we first review the different types of materials commonly used to synthesize nanotheranostic particles and their use in imaging. We then discuss biological barriers that these nanoparticles encounter and must bypass to reach the target cancer cells, including the blood, liver, kidneys, spleen, and particularly the blood-brain barrier. We then review how nanotheranostics can be used to improve targeted delivery and treatment of cancer cells using nanoparticles, adult stem cells, and T cells in immunotherapy. Finally, we discuss development of nanoparticles to overcome current limitations in cancer therapy.
Keywords: nanomedicine, immunotherapy, cancer stem cells, metastasis, multidrug resistance
1. Introduction
Cancer remains the second leading cause of death in the Americas and Europe after heart disease, and the third leading cause of death in the world after heart and infectious diseases.[1] Years of intense research and billions of dollars in spending have dramatically increased our knowledge of the causes and biology of cancer, leading to the development of many improved treatment strategies. Yet, an estimated 7.5 million deaths in 2008 alone were caused by cancer,[1] signaling the pressing need for newer, even more effective therapies. Current cancer therapies are largely limited by 1) inability to bypass biological barriers, 2) non-specific delivery and poor biodistribution of drugs, 3) ineffectiveness against metastatic disease, 4) drug resistance of cancers, and 5) lack of effective modality for treatment monitoring.[2-5]
The application of nanotechnology to cancer therapy has the potential to overcome these challenges by enabling the engineered nanomedicines to navigate the body in very specific ways. In the past 20 years a number of nanomedicines have been approved for clinical use.[6, 7] Some have even become the standard of care for specific cancer types.[8] Drawing on these previous successes is the field of cancer nanotheranostics (therapeutics and diagnostics in nanomedicine) which utilizes nanotechnology for the combined imaging and treatment of cancer using a single nanomedicine.[9] Theranostic nanomedicines, mostly nanoparticles (NPs) carrying therapeutics, are designed to improve current cancer therapies by addressing the specific existing limitations.
The ability to monitor treatment in real-time will allow physicians to adjust the type and dosing of drug for each patient to prevent overtreatment that would result in harmful side-effects, or undertreatment that would lead to incomplete cancer remission. The ability to see when a drug reaches a maximum tolerable concentration in off-target organs and a sufficient concentration in the tumor would be a significant advantage over current treatments, or separate treatment and monitoring systems. NPs can enable treatment monitoring by either attaching different imaging moieties or taking advantage of the intrinsic properties of some NP materials (e.g., superparamagnetism for MRI). While these theranostic NP formulations are more complex to develop, their potential clinical utility substantiates the investment required at the front-end.
Bypass of biological barriers such as the immune system, liver, kidneys, spleen, and blood-brain barrier can increase the amount of therapy that can reach target cancer cells. These barriers are highly efficient at removing foreign materials from the body and preventing access to tumors. NPs can be engineered to bypass these barriers for proper trafficking throughout the body and accumulate in target cells or tissues. Further, systemic distribution or off-target accumulation of therapeutics can be detrimental to patient health and targeted biodistribution of NPs can help to diminish these side-effects. NPs can be targeted to cancer cells in various ways to improve the specificity of treatment. The improved specificity can also help improve therapy of metastatic disease which involves the cancer spread throughout the body, and eradicate cancer stem cells which are thought to drive primary and metastatic tumor growth. Finally, the ability to overcome drug resistance could greatly improve cancer therapy. NPs can be specifically designed to be insensitive to the resistance mechanisms acquired by these drug-resistant cancer cells.
In this review, we first outline the different types of NPs currently being developed and their mechanisms for imaging. We then provide an overview of the different barriers that may be encountered by nanomedicines in the body and discuss the strategies to bypass these barriers. A significant focus is given to the blood-brain barrier as this is a major hurdle in the treatment of brain tumors. We then present various strategies for targeted delivery of cancer therapeutics, including NP or adult stem cell mediated delivery and cancer immunotherapy. We further discuss the development of NPs to overcome the drug resistance and treat cancer stem cells, the major challenges in current cancer therapies. Finally, we review the nanomedicines which are found most promising for clinical translation.
2. Nanomaterials for Cancer Imaging
Many different types of nanomaterials have been developed to provide contrast in medical imaging.[10] Some of these materials incorporate an imaging moiety into their design, while others provide contrast as a result of their intrinsic material properties. Multiple imaging modalities can also be implemented into a single nanotheranostic design by incorporating multiple moieties to provide a more complete picture of the disease. Molecular imaging can identify tumor cell location within the body, and aims to provide information such as metabolism, expression profile, and stage of the disease.[11] Furthermore, molecular imaging can reveal early tumor response to therapy that will aid in improving treatment regimens.[5] An overview of the different types of NPs and examples of the images obtained with these NPs is provided in Figure 1. Here NPs are broadly classified by the materials they are made of, which includes liposomes and micelles, polymers and dendrimers, noble metals, semiconductors, carbon nanotubes and fullerenes, transition metal oxides, metal-organic frameworks, and lanthanide series.
Figure 1.

Typical nanomaterial formulations for imaging and therapy of cancers, their mechanism for imaging, and associated representative images. Example images reproduced with permission for liposomes and micelles (SPECT image overlaid with CT),[15] polymers and dendrimers (PET image overlaid with CT),[275] noble metals (near-IR optical imaging),[276] semiconductors (fluorescence imaging),[32] carbon nanotubes and fullerenes (photoacoustic imaging),[35] transition metal oxides (MRI),[81] metal-organic frameworks (MRI),[51] and lanthanide series (X-ray radioluminescence imaging).[57]
2.1. Liposomes, Micelles, Polymers, and Dendrimers
Liposomes[12] and micelles[13] are by far the most widely used and studied nanomaterials for cancer therapy. These lipid-based nanoparticles (LNPs) are synthesized from lipids containing a hydrophilic head group and lipophilic tail that spontaneously form spheres at critical concentrations. Imaging of LNPs is achieved through incorporation of moieties that can be detected through various imaging modalities. For example, liposomes can encapsulate fluorescent dyes for optical detection or radionuclides for positron emission tomography (PET) imaging. The radionuclide copper-64 (64Cu) loaded into liposomes can be monitored using PET imaging after injection into human colon adenocarcinoma xenograft mice.[14] Liposomes loaded with rhenium-188 (188Re) can be imaged in mice using single-photon emission computed tomography (SPECT).[15] Furthermore, magnetic NPs (discussed below), which are detectible in magnetic resonance imaging (MRI), can be loaded into LNPs for imaging purposes.[16] LipoCEST agents composed of lanthanide(III)-based complexes loaded into LNPs provide fairly sensitive MRI detection.[17] Similar to LNPs, polymer- and dendrimer-based NPs can be imaged through attachment of these imaging moieties.[18] Also, nanoemulsions of perfluorocarbon (PFC) polymers can be used for targeted ultrasound and MR imaging.[19] Similarly, liposomes and microbubbles loaded with stabilized gas bubbles are used in ultrasound imaging owing to the high acoustic reflectivity of the gas bubbles.[20]
2.2. Noble Metal NPs
Noble metal NPs, such as gold and silver, are optically active due to their unique properties that arise at the nanoscale, known as surface plasmon resonance which can be used for imaging applications.[21] Surface plasmon resonance occurs in nanosized noble metal NPs through excitation and relaxation of surface plasmons at the interface of the NP surface and surrounding solution. The optical properties of these NPs can be tuned by changing their size, shape, and surface properties. Their optical activity in the visible spectrum allows for their detection intraoperatively; however, their low quantum yields make detection difficult.[22, 23] Engineering these NPs to have more sharp edges (such as in nanocubes) can improve the quantum yield of gold NPs to allow for their detection in biological tissues.[23, 24] Furthermore, dark-field imaging of light scattering from noble metal NPs can detect single NPs highlighting the sensitivity of this method.[25] Nevertheless, their use in cancer detection is limited to superficial sites due to the limited penetration depth of light, even in the near infrared range where tissue absorbance is minimal.[26] However, these NPs can also provide contrast in CT imaging due to their high densities as compared to human soft tissue which enables non-invasive, real-time imaging of the vast majority of solid tumors.[27] The high density of the NPs attenuates the X-rays resulting in high contrast regions where NPs are present. These NPs provide a significant advantage for molecular imaging over the commonly used CT contrast agents such as iodine owing to their higher X-ray absorption coefficient, long circulation time in blood, and high surface area for easy attachment of targeting and therapeutic agents.[28] Furthermore, gold NPs can be used in photoacoustic imaging where absorbed light causes the NP to emit ultrasonic waves through thermo-elastic expansion that can be detected by an ultrasound detector.[29] However, this method is still limited to an imaging depth that is penetrable by the photons used to excite the NPs.
2.3. Semiconductor materials
Semiconductor materials have been widely studied for synthesizing NP cores because of their unique optical properties that arise from the quantum confinement of an exciton at the nanoscale.[30] The absorption and emission spectra of a semiconductor NP (also known as a quantum dot or QD) are size-dependent, and thus the optical spectrum of a QD can be fine-tuned by altering the size of the NP core. Optical properties of QDs can also be engineered through controlling their shape and surface properties. These QDs show very bright fluorescence that does not photobleach as organic chromophores do, which allows for long-term, repeated imaging. Furthermore, the radiative emission from the QD can be tuned to the visual spectrum to allow for intraoperative imaging. However, the heavy metals commonly used to synthesize these semiconductor NPs, most commonly cadmium, are highly toxic so their use in humans may be limited. Strategies have been developed to synthesize cadmium-free quantum dots to improve their clinical translation.[31] In addition, use of these QDs for detection or diagnosis of cancer is limited to superficial sites such as skin and esophageal cancers due to the limited penetration depth of visible light. Near-infrared-emitting NPs have been developed for deeper tissue penetration, but imaging is still limited to about 3 cm.[32]
2.4. Carbon Nanotubes and Fullerenes
Carbon nanotubes and fullerenes (CNTs) have been investigated for cancer imaging applications.[33] Both single-walled and multi-walled CNTs have a high surface area and internal volume for loading of drugs and imaging agents, but alone CNTs are not soluble in most organic or aqueous solutions. Therefore, surface modification of CNTs is critical for their use in theranostic applications.[34] Polyhydroxy fullerene can be detected using photoacoustic imaging and used for photothermal ablation therapy after intratumoral injection.[35] Furthermore, multi-walled CNTs can be used for photothermal ablation therapy owing to their release of vibrational energy upon near-infrared light exposure.[36] However, potential toxicity associated with CNTs must be addressed before clinical translation.[37]
2.5. Metal Oxide NPs
Magnetic metal oxide NPs have been very widely studied for use as contrast agents in magnetic resonance imaging (MRI).[38, 39] MRI is a powerful tool for medical imaging owing to its virtually unlimited tissue penetration depth and thus NPs can be detected anywhere in the body.[40] Iron oxide NPs, in particular, have received significant attention owing to their proven biocompatibility and biodegradability. Iron from degraded NPs is used in the body’s natural iron stores such as hemoglobin in red blood cells.[41, 42] These NPs develop superparamagnetism at the nanoscale as each particle becomes a single magnetic domain which is free to rotate at room temperature. In MRI, the superparamagnetic NPs generate local inhomogeneities in the magnetic field decreasing the signal. Therefore, regions in the body that have iron oxide NPs appear darker in MR images as a result of the negative contrast. The relaxivity of iron oxide NPs, or their ability to provide contrast in MRI, can be improved by tuning the size, shape, and defect type of the NP core.[43] However, detection of these negative contrast NPs is difficult in low signal intensity tissues such as the lungs and blood clots. Positive contrast can also be achieved with magnetic NPs, which can improve detection in low signal body regions.[44] For example, manganese oxide[45] and gadolinium oxide[46] NPs provide positive contrast in MRI. Furthermore, iron oxide NPs with core sizes less than 10 nm can provide positive contrast in MRI.[47] Hyperthermia can be achieved with iron oxide NPs using a rapidly changing magnetic field.[48] High frequency alternating magnetic fields cause the magnetic moment of the superparamagnetic NPs to quickly shift through Néel fluctuations, which creates very high local temperatures. This mechanism can be used for tumor cell destruction after iron oxide NPs are internalized by the target cells.
2.6. Metal-Organic Frameworks
Metal-organic frameworks (MOFs) are nanosized structures comprising metal cations and electron donors such as carboxylates or amines that form coordination bonds and are self-assembled into highly porous materials.[49] They contain organic molecules that impart synthetic flexibility so that the crystalline structure, size, and porosity can be engineered depending on the combination of organic linker and metal cation used in synthesis. Furthermore, the metal cation chosen can impart magnetic properties for detection in MRI. For example, gadolinium (Gd) and manganese (Mn) based MOFs have been synthesized for MR and potential multi-modal imaging.[50] Iron (Fe) based MOFs are detectible in MRI after intravenous injection into rats, indicating their utility in vivo.[51] Their ease in synthesis makes MOFs a promising theranostic agent, but scale-up for mass production and reduction of synthesis times have been difficult.[49, 52]
2.7. Upconverting Nanophosphors
Upconverting nanophosphors (UCNPs) are generally prepared through lanthanide-doping of NPs.[53] The optical properties of UCNPs are vastly different than those of conventional fluorophores or QDs. Instead of absorbing a single photon to excite an electron from the ground state to an excited state of higher energy, UCNPs utilize the accumulation of multiple low-energy exciting photons to emit a higher energy photon upon relaxation of the electron back to the ground state. This can provide high sensitivity detection for cancer cell imaging with lower autofluorescence and long fluorescence lifetimes.[53] Tumor targeted polyethylenimine-coated hexagonal-phase sodium yttrium fluoride:ytterbium, erbium/cerium (NaYF4:Yb,Er/Ce) NPs are able to target cancer cells in vitro and in vivo for tumor visualization.[54] Furthermore, folic acid activated UCNPs have been developed for targeting and imaging of HeLa cells both in vitro and in vivo.[55] However, imaging depth is still limited by the ability of light to penetrate tissue which is on the order of 3 mm for near infrared. X-ray-excitable NPs are another class of lanthanide series NPs, and can be used in a new dual molecular/anatomical imaging modality, x-ray luminescence computed tomography (XLCT).[56, 57] These NPs are excited by high energy radiation such as x-rays rather than by the much lower energy photons of the optical spectrum. This allows for detection of NPs in tissues or cancers deep in the body. However, the use of high energy x-rays limits the imaging time that can be performed in a single patient, especially in pregnant women and children.
2.8. Multimodal imaging
Each of the imaging modalities discussed above have their own advantages and disadvantages in sensitivity, resolution, and imaging depth. Combining multiple imaging modalities in a single NP design can exploit the advantages while improving disadvantages of the individual techniques.[58] For example, PET imaging is one of the most sensitive imaging techniques, but provides no anatomical information. Therefore, PET imaging has been combined with X-ray computed tomography (CT) which provides the needed anatomical information for accurate staging and localization of the disease. In fact, PET/CT imaging platforms have been commercially available for many years and are routinely used for early detection of cancer recurrence and localization.[59] Therefore, the attachment of a radionuclide on a high density NP core could provide both the high sensitivity of PET along with the anatomical localization with CT in a single theranostic agent.[60] Iron oxide NPs labeled with a fluorescent dye can potentially be used for pre-surgical planning and diagnosis using MRI, and intraoperative assistance in distinguishing tumor from healthy tissue using fluorescence imaging.[61]
Tri-modal imaging with MRI, CT, and fluorescence combines the strengths of these individual imaging modalities, including spatial and temporal resolution and sensitivity. Gold/silica NPs that have a lipid and polyethylene glycol (PEG) coating containing fluorescent molecules and paramagnets have been developed for this application.[62] In this design, the gold NP core provides contrast in CT, the paramagnetic lipid provides contrast in MRI, and a Cy5.5 fluorophore provides contrast in optical imaging (Figure 2).
Figure 2.

Concentration–signal curves of the lipid-coated gold/silica particles for (a) FI, (b) MRI and (c) CT. Note that both nanoparticle concentrations and the corresponding Gd and Au concentrations are given. The molar longitudinal relaxivity r1 of the gadolinium in the lipids was found to be 14.0 mM−1 s−1 and the slope in the CT curve was 23 HU per g gold l−1 solution. The inset in each panel shows the corresponding image of nanoparticle dilution series (concentrations indicated) associated with each imaging modality, revealing the high sensitivities of all three imaging techniques. Reproduced with permission.[62]
3. Bypassing Biological Barriers
The body has evolved many strategies to attack and remove foreign materials (e.g., bacteria, viruses, medical implants, and drugs) that have been introduced into the body. This imposes a great difficulty for nanotechnologists aiming to develop cancer nanotheranostic devices since these devices will be eliminated from the body before they have a chance to reach the target disease site. Therefore, the understanding of the barriers imposed by a biological system is critical to the design of nanomedicines. The barriers imposed by the body can be broadly classified as physiological barriers and cellular barriers.[63] Cellular barriers include the cell membrane, endosome/lysosome, and intracellular trafficking. Physiological barriers include the blood, liver, spleen, kidneys, immune system, and the barriers that prevent extravasation of foreign substances from the blood (Figure 3a-d). The extravasation from the blood to reach brain tumors is particularly difficult due to the blood-brain barrier (BBB) (Figure 3e).
Figure 3.

Physiological barriers encountered by NPs. a) Upon injection into the blood, NPs circulate throughout the body reaching the capillaries of the liver, kidneys, tumor, and brain. b) Passive accumulation in the tumor occurs with NPs with hydrodynamic diameters between 30–200 nm. i) endothelial cell, ii) tumor cell. c) The Kupffer cells of the liver readily recognize materials with hydrodynamic diameters larger than 100 nm and removes them from circulation. i) endothelial cell, ii) Kupffer cell, iii) hepatocyte. d) The pores of the glomerulus in the kidneys are around 10 nm and thus materials with hydrodynamic diameters larger than this will avoid renal filtration. i) endothelial cell, ii) glomerular basement membrane. e) The blood-brain barrier (BBB) prevents passive accumulation of materials in the brain due to tight junctions between endothelial cells, and thus active or disruptive mechanisms must be used to bypass the BBB.
3.1. Extracellular Barriers
Blood is a highly complex fluid composed of salts, sugars, proteins, enzymes, and amino acids that can destabilize NPs causing aggregation and embolism. Furthermore, blood contains immune cells such as monocytes that can recognize and remove foreign materials from circulation. NPs must be highly stable and avoid the recognition by the immune system to prolong the blood half-life and increase access to the tumor. This is commonly achieved through the passivation of the NP surface with biocompatible polymers.[64] For example, iron oxide NPs coated with triethoxysilylpropyl succinic anhydride and polyethylene glycol show good stability in biological media for five months.[65] These polymers confer a brush border on the surface of the NPs, which helps prevent NP aggregation and recognition by the immune system. Furthermore, these polymers can help reduce the zeta potential (a measure of the surface charge) of the NP towards neutral by providing a physical barrier between the blood components and charged NP surface. Highly cationic NPs readily bind anionic plasma proteins (opsonization), which can destabilize the NP and promote recognition by the immune system, therefore neutral NPs are desirable for their stability in blood. Enzymes present in the blood can degrade the NP and its therapeutic payload. Passivation of the NP surface with biocompatible polymers can also protect the NP from enzymatic degradation. Furthermore, encapsulating the therapeutic payload in the interior of the NP can help prevent enzymatic degradation.
The liver, spleen, and kidneys confer hydrodynamic size restraints on NPs to be between 10–100 nm in diameter (Figure 3c and d). Macrophage cells of the liver and spleen, such as Kupffer cells that line the hepatic sinusoids in the liver, readily eliminate and metabolize materials larger than approximately 100 nm from the blood. Therefore, NPs smaller than 100 nm show reduced liver and spleen uptake.[7, 66] The kidneys filter metabolites and toxins from the blood by filtration through the basal lamina which has pores of approximately 10 nm. NPs with hydrodynamic diameters larger than 10 nm show reduced renal filtration.[67]
Finally, the NPs must extravasate from the blood at the diseased site to enable designated functions (Figure 3b). For many tumors, NP accumulation in tumors occurs by the enhanced permeability and retention (EPR) effect wherein leaky vasculature combined with minimal lymph drainage at the tumor site promotes the accumulation of materials between 30–200 nm in size. Leaky vasculature in the tumor is a result of highly metabolizing cancer cells that stimulate rapid and poorly organized neovascularization. The new blood vessels lack structured fenestrations, which enables efficient extravasation of nanosized materials of up to several hundred nanometers into the tumor.[68] These materials are then retained in the tumor site due to the lack of lymphatic drainage. PEGylation of NPs promotes NP accumulation in the tumor through the EPR effect, generally through prolonged blood half-life which increases the probability of the NP reaching the tumor.[69] However, the case with brain tumors is much more complex due to the presence of the BBB.
3.2. The BBB
Of the physiological barriers, the BBB is one of the most difficult to overcome in order to deliver nanomedicines into the brain. The BBB is composed of a dense layer of endothelial cells connected by tight junctions that prevent passive accumulation of many molecules into the brain. This is a significant challenge in brain cancer therapy as many potentially effective therapies are unable to reach target brain cancer cells. Pathways across the BBB include both passive and active mechanisms (Figure 3e).[70] Passive pathways include aqueous paracellular in which small (< 200 Da) water soluble molecules can diffuse from the blood between tightly packed endothelial cells into the brain. Also, lipid soluble agents such as ethanol and barbiturates are able to passively accumulate in the brain through the lipophilic pathway where these lipid soluble agents can diffuse through the cell membrane. Active transport mechanisms include adsorptive transport and receptor-mediated transcytosis. Adsorptive transport occurs with charged plasma proteins that interact electrostatically with endothelial cells of the BBB. Receptor-mediated transcytosis occurs naturally for the transport of insulin and transferrin into the brain. Furthermore, there are transport proteins that bind and actively traffic small molecules such as glucose and amino acids into the brain. Both active and passive transport mechanisms can be utilized by properly-designed NPs to gain access to the brain.
Even in tumors where the BBB is disrupted, such as with metastases to the brain, uptake of drug into the tumor site is still extremely low due to the blood-tumor barrier (BTB).[71] The endothelial cells that are recruited to the brain-residing tumor during angiogenesis likely arise from parent endothelial cells that form the BBB, so they still form tight junctions and highly express efflux pumps that remove substances from the tumor site. The resulting BTB prevents chemotherapies from reaching brain metastases, resulting in little therapeutic effect.[72]
3.2.1. Disrupting BBB integrity
One way to gain access across the BBB is to physically disrupt the BBB so that delivered NPs can penetrate into the brain reaching brain cancers through the paracellular aqueous pathway. This has been commonly achieved through injection of vasodilators such as bradykinin and histamine which widen blood vessels causing the gaps between endothelial cells in the BBB to increase in size, or with hyperosmotic solutions of mannitol that cause the endothelial cells to shrink.[73] This strategy can increase accumulation of dextran coated magnetic NPs in rat glial brain tumors.[74] Another method to physically disrupt the BBB integrity is through use of low-energy burst tone, focused ultrasound in the presence of microbubbles.[75]
Magnetic targeting can be combined with the focused ultrasound BBB disruption method to further improve magnetic NP access across the BBB.[76] The ultrasound disruption provides an EPR effect for NPs to passively accumulate in the brain while the magnetic field actively pushes the NPs into the desired region of the brain. However, the clinical utility of these BBB disruption strategies are hindered by the danger associated with opening the BBB, which also allows the access of unwanted foreign substances. Furthermore, observed pre-clinical efficacy has not translated well in clinical trials,[70, 77] perhaps due to the presence of the BTB even when the BBB is disrupted.
An alternative strategy is to knockdown the expression of genes involved in the formation of the tight junction in the BBB. This could provide transient access to the brain since expression of these genes would return. Delivery of siRNA against claudin-5, a transmembrane protein present in tight junctions, to endothelial cells provides size-selective opening of the BBB.[78] This method has a significant advantage over the other disruptive strategies since the BBB could be opened just enough to allow nanomedicines to enter while maintaining the natural defense against larger bacteria and viruses.
3.2.2. Convection enhanced delivery
Convection enhanced delivery (CED) is a method for delivery of macromolecules throughout the brain by circumventing the BBB.[79] CED involves injection of solution into the interstitial space in the brain at a rate that is high enough to induce fluid convection throughout the brain by a pressure gradient, but not too high so that the fluid would leak back up the cannula tract and out of the brain. This strategy has been used for delivery of NPs throughout brain tumors in animal models.[80] For example, epidermal growth factor receptor variant III (EGFRvIII) antibody conjugated iron oxide NPs can be delivered to brain tumors through CED in a mouse model of glioblastoma.[81] The EGFRvIII antibody acts as a glioblastoma targeting agent that also provides therapy. These NPs show good distribution in the brain and accumulation in the brain tumor resulting in an increased survival rate. However, the translation of CED to widespread clinical use has been hindered by low efficacy.[82] A prospective phase I/II clinical study using CED to deliver NPs in brain cancer patients found that the therapeutic effect was restricted to a small area around the infusion site.[83] This is likely due to the size dependence of CED where smaller molecules better penetrate and distribute throughout the brain tumor.[79, 84]
A similar, but opposite approach to CED is retro-CED which removes fluid from the interstitial space in the brain to increase the pressure gradient from the vascular compartment into the brain.[85] This is achieved by placing a catheter, through which a hyperosmotic fluid is pumped, in the brain, which drives fluid flow from the interstitial space in the brain into the catheter. This can be problematic, however, if the delivered therapy can permeate through the membrane of the catheter since it will be removed from the brain.[86]
3.2.3. Active transport
Active transport across the endothelial cells can be exploited to gain access across the BBB by attaching BBB-penetrating ligands to the surface of NPs. This is advantageous over other disruptive or invasive strategies since the body’s natural defenses remain intact and is not invasive. Magnetic NPs activated with myristoylated polyarginine peptides are able to penetrate endothelial cells to gain access to stereotactically implanted brain tumors.[87] Furthermore, these cell penetrating peptides help increase uptake into tumor cells for improved retention for MRI monitoring.
Transferrin acts to move free iron in the blood into cells through active transport mediated by the transferrin receptor. This receptor is expressed by many tissues including endothelial cells of the BBB. However, the plasma concentration of transferrin is approximately 25 μM causing the transferrin receptor to be saturated, limiting the in vivo utility of transferrin as a BBB penetration molecule.[88] In fact, in a study comparing four other targeting ligands (RI7217, COG133, angiopep-2, and CRM197) attached to liposomes, transferrin did not mediate BBB penetration.[89] Of these five targeting ligands, only RI7217 is able to significantly enhance permeation across the BBB in vivo in mice. Despite this, some success with transferrin activated NPs has been achieved for brain tumor targeting, but most of these studies have been performed using in vitro models of the BBB without the transferrin receptor saturation.
As an alternative, the transferrin antibody OX26 has been used extensively for delivery of various drugs and biologics across the BBB.[90] It was developed to couple to PEGylated liposomes for drug delivery into the brain.[91] OX26 is able to gain access to the brain across the BBB through transcytosis after interacting with the transferrin receptor.[92]
Lactoferrin is a receptor in the transferrin family, and another alternative to transferrin for targeting of brain tumors across the BBB. Unlike transferrin, lactoferrin has a low endogenous plasma concentration, and its receptors are highly expressed on the surface of brain tumor cells.[93] Superparamagnetic iron oxide NPs activated with lactoferrin are able to accumulate in an intracranial model of glioma, but penetration across the BBB was not investigated although it was suggested.[94] Non-viral gene delivery to the brain has also been achieved using lactoferrin activated polyamidoamine NPs.[95]
Chlorotoxin (CTX) activated iron oxide NPs are also able to cross the BBB to target medulloblastoma brain tumors,[96] likely by receptor-medicated transport through endothelial cells. Annexin A2 is expressed on the surface of neovasculature and is a target of CTX.[97] This represents a likely mechanism for transport. A transgenic mouse model of medulloblastoma provides an ideal model for delivery of NPs across the BBB into brain tumors since the disease progresses similar to the clinical progression, and it has a viable BBB.[98] NPs without CTX are not found in the brain tumor or healthy brain tissue after intravenous injection into these transgenic mice, whereas NPs conjugated with CTX are significantly taken up by brain cancer cells after crossing the BBB.[96]
Table 1 highlights the various NPs utilized for active transport across the BBB including the associated ligands used for active transport, and their physiochemical properties (size and zeta potential). These studies reveal that controlling NP physiochemical properties alone may not be enough to breach the BBB, highlighting the necessity of BBB permeating ligands. NPs of sizes between around 30–600 nm and zeta potentials between +20 mV to −20 mV are all unable to cross the BBB alone, but are able to do so when a BBB permeating ligand is attached. Despite these many successes in animal models, BBB penetrating NPs are still a fairly new class of drug and remain in pre-clinical development.
3.2.4. Long-range axonal transport across the BBB
A potential access point across the BBB can be learned from pathogens which employ long-range axonal transport from nerve endings to cell bodies residing within the brain.[99] Neuronal infections that cause rabies, tetanus, and botulism are a result of this type of transport into the brain. These pathogens utilize receptors at nerve endings at neuromuscular junctions to be taken up into the neuron, and then are actively transported across the BBB through neuronal projections that connect with the cell body of neurons. Active transport along intracellular microtubules occurs through binding cytoplasmic kinesin, dynein, and dynactin which naturally transport organelles along microtubules. To our knowledge, this pathway has not yet been exploited for NP delivery across the BBB, but we see it as an exciting avenue that should be explored.
3.3. Cellular Barriers
Once the NP has extravasated from the blood into the tumor site it must be taken up by the cancer cells to deliver the therapeutic payload. The cell membrane consists of a negatively charged phospholipid bilayer that separates the inside of the cell from the extracellular space. Entry into the cell can occur by direct permeation through the cell membrane or by various forms of endocytosis, followed by intracellular trafficking to the target subcellular organelle (Figure 4).
Figure 4.

Cellular barriers encountered by NPs. Entry into the cell across the cell membrane can occur by direct permeation, or by various types of endocytosis mechanisms. Upon endocytosis, the NP must escape the endosome before acidification degrades the payload or the NP is exocytosed with membrane recycling. After the NP gains access to the cytoplasm of the cell, intracellular trafficking will ensure that the therapeutic payload will reach the desired site of action such as the mitochondria, endoplasmic reticulum, nucleus, or cytoskeleton.
Direct permeation through the cell membrane can be achieved by small, hydrophobic molecules, but with NPs it is more difficult due to their larger size. Attachment of certain cell-penetrating peptides to NPs has shown to bypass endocytosis and enable direct permeation through the cell membrane.[100, 101] For example, polyarginine conjugated iron oxide NPs loaded with siRNA are able to permeate across the cell membrane for direct access to the cytoplasm.[100] Transmission election microscopy (TEM) imaging reveals the NPs gain entry to the cytoplasm of the cell without endocytic vesicles.
Most NPs are taken up by cells through endocytosis mechanisms which include receptor-mediated endocytosis and adsorptive endocytosis, mainly via clathrin-coated pits.[102, 103] Uptake of NPs can also occur through phagocytosis which is the main uptake mechanism into macrophage cells, caveolae-mediated endocytosis which occurs in non-clathrin-coated plasma membrane buds present on the surface of some cells, macropinocytosis which is a fluid-phase endocytosis mechanism, and other mechanisms that do not involve clathrin or caveolae.[103] The uptake mechanism of NPs can have a determinant effect on subsequent intracellular trafficking. For example, NPs taken up via clathrin-coated pits enter acidic endosomes/lysosomes where the reduction in pH activates destructive enzymes. On the other hand, NPs taken up via caveolae in lipid rafts may not be transported to endosomes or lysosomes.[104, 105] Perfluorocarbon nanoemulsions loaded with siRNA are taken up through lipid rafts and show enhanced gene knockdown efficiency in endothelial cells in vitro as compared to liposomes taken up through clathrin-coated pits.[104] Cationic NPs that interact electrostatically with the anionic cell membrane are taken up through adsorptive endocytosis. Also, anionic NPs can interact with cationic proteins embedded in the cell membrane for adsorptive endocytosis. Polystyrene NPs with zeta potentials of +59 mV or −60 mV show similar uptake in HeLa cells indicating that electrostatic adsorptive endocytosis can occur with both highly anionic and cationic NPs.[106]
Receptor-mediated endocytosis is achieved by attaching a molecule to the surface of the NP that is recognized by receptors on the surface of the cell, a strategy used for targeted NP delivery (discussed in section 4.1). Upon binding a cell surface receptor, the cell will engulf the NP by wrapping the cell membrane around the NP and pinching off the endocytic vesicle inside the cell. However, the presence of a targeting molecule alone does not ensure optimal uptake of NPs. The size of the receptor-targeted NP also has an effect on uptake. NPs with a size around 50 nm show the greatest uptake through the receptor mediated endocytosis pathway.[107, 108] This size-dependent uptake is attributed to the “wrapping time” it takes the cell to fully engulf the NP.[108, 109] NPs smaller than 50 nm lack the free energy necessary to completely wrap the NP on the surface of the cell membrane, and NPs larger than 50 nm require longer wrapping times due to slower receptor diffusion kinetics of the cell membrane around the NP.
Once the NP has been taken up by the cell, proper trafficking to the intracellular site of action can dramatically improve the therapeutic efficacy of the delivered drug. First, if the NP is taken up through the endocytosis pathway, it must escape the endosome before enzymes become active in the reduced pH of the lysosome. Next, the NP must localize to the intracellular site of action such as the cytoplasm, nucleus, mitochondria, Golgi apparatus, or cytoskeleton. Finally, the drug or therapy must become available to interact with its target while still attached to or after release from the NP.
Endosomal escape can be achieved with cationic liposomes that fuse with the endosomal membrane to release the liposomal components into the cytoplasm through a three-step process.[110] The liposome first binds to the cell surface and becomes endocytosed. The cationic lipids from the liposome reorganize the anionic phospholipids from the endosomal membrane, which destabilizes the endosome. The destabilized endosome and neutralized liposome then release the therapeutic payload into the cytoplasm. Amphiphilic fusogenic peptides are also used to escape the endosome.[111] These peptides undergo a structural change in the environment of reduced pH from inert to hydrophobic α-helices that can fuse with and disrupt the endosomal membrane to gain access into the cytoplasm. Finally, escape from the endosome can be achieved through the proton sponge effect where a polymer, generally with tertiary amines with low pKa values, buffers the influx of protons that reduce the pH of the endosome/lysosome. This influx is followed by counter ions which disrupts the osmotic balance between the endosome and cytoplasm, causing the endosome to swell and rupture releasing the endocytosed NPs into the cytoplasm.[112]
After gaining access to the cytoplasm of the cell, the NP can be directed to the intracellular site of action of the drug.[113] For example, transport to the nucleus is required for DNA and chemotherapy drugs, such as doxorubicin and cisplatin, to be effective. Transport of DNA to the nucleus has been achieved using polyethylenimine (PEI),[114] exploiting the active nuclear transport mechanism by coupling nuclear localization signal (NLS) peptides directly to NPs,[115] and using intracellular actin polymerization as a molecular motor to traffic the delivered payload to the nucleus.[116] Cytoplasmic delivery is required for anticancer drugs such as small interfering RNA (siRNA), proteins, and some chemotherapy agents. Delivery of siRNA to the perinuclear region of the cytoplasm can improve gene knockdown efficiency since this is the region where messenger RNA (mRNA, the target for siRNA) is translated into protein. Delivery to the cytoskeleton is required for drugs such as paclitaxel and Vinca alkaloids.[117] These drugs stabilize microtubules which are a key component of the cytoskeleton, and prevent their degradation during cell division, resulting in apoptosis. Transport to the mitochondria is required for the therapy using geldanamycin, a drug that binds mitochondrial heat shock protein 90 in tumor cells, resulting in collapse of mitochondria function and tumor cell death.[118] Selective transport to the mitochondria can be achieved with cyclic guanidinium moieties, which is the strategy used in the development of gamitrinibs for intracellular targeting of geldanamycin to mitochondria.[118]
4. Targeted Therapy
Targeted therapy refers to the specific treatment of cancer cells while leaving healthy cells unharmed, but has been rarely achieved. The goal is to kill off all of the cancer cells before killing off too many healthy cells. This is difficult with standard chemotherapies which are toxic to both healthy and cancerous tissues. Nanotechnology can be used to improve drug accumulation specifically to the tumor site using various mechanisms such as passive and active targeting. Furthermore, activation of the immune system against cancer cell specific surface markers can be used as a targeted therapy since the immune system is highly evolved to specifically recognize and remove target cells.
4.1. NPs as Drug Carriers
NP-based therapy can provide a significant advantage over standard chemotherapies by increasing the drug delivery specificity to the tumor site through either passive or active means. Figure 5 provides an overview of the various methods for improving drug accumulation within the tumor and at the intracellular site of action. NPs may passively accumulate in the tumor site due to the EPR effect, with PEGylation enhancing this effect. The attachment of a targeting ligand (active targeting) can promote cellular uptake and distribution of the NP throughout the tumor,[119] and proper engineering of the NP can ensure the desired intracellular trafficking. The NP design can also utilize light, pH, ultrasound, and magnetic fields for targeting and distribution throughout the tumor.
Figure 5.

Targeting strategies to improve NP delivery throughout the tumor. I) Non-PEGylated NPs accumulate in the tumor site through the EPR effect. II) PEGylated NPs show enhanced accumulation in the tumor site through the EPR effect. III) Targeted NPs show better distribution throughout the tumor and higher cellular uptake. IV) Subcellular targeting increases NP delivery to the intracellular site of action of the drug.
Passive accumulation of NPs at the tumor site for drug delivery through the EPR effect has been exploited clinically using the NP formulations Myocet and DaunoXome which are liposomal formulations of doxorubicin and daunorubicin, respectively. For maximal tumor uptake through the EPR effect, the NP must have a long circulation time in the blood, which is commonly achieved through PEGylation of the NP.[120] This was the goal in the development of Doxil, a PEGylated liposomal formulation of doxorubicin. However, Doxil shows no improvement in time of survival in metastatic breast cancer patients.[121] This is likely due to poor penetration into the tumor and thus only a small proportion of tumor cells receive treatment.[122] The addition of a tumor targeting antibody to the surface of Doxil improves its therapeutic efficacy both in vitro and in vivo by enhancing the uptake of Doxil into tumor cells.[123]
Active targeting involves the attachment of a targeting ligand on the surface of the NP that recognizes receptors overexpressed on cancer cells. These targeting ligands can include antibodies, antibody fragments, peptides, aptamers, and small molecules such as folic acid or glucose that target the metabolism of cancer cells.[39, 63] For example, glypican-3 (GPC3) is absent in normal adult tissue, but is highly expressed on the surface of 80% of cells of hepatocellular carcinoma (HCC), a common and deadly form of liver cancer.[124] This represents an ideal target receptor since no off-target specific uptake would occur. PEG coated iron oxide NPs show significantly higher uptake using anti-GPC3 antibody targeting in GPC expressing cells as compared to GPC-negative cells, and can be detected both optically with fluorophore conjugation and using MRI due to the iron oxide magnetic core.[125] Various other antibodies that target receptors overexpressed on the surface of cancer cells such as human epidermal growth factor receptor-2 (HER2/neu), EGFR, tumor necrosis factor-α (TNF-α), and vascular endothelial cell growth factor (VEGF) are attached to NPs to achieve cancer cell targeting.[126]
Peptide activated NPs can also bind cancer cell surface receptors for targeted delivery. CTX is a peptide derived from the venom of the giant Israeli scorpion that targets matrix metalloproteinase-2 overexpressed on the cell surface of cancers such as malignant glioma, medulloblastoma, prostate cancer, intestinal cancer, and sarcoma.[127] CTX activated iron oxide NPs have desirable pharmacokinetics and biodistribution,[128] and show excellent targeting of cancer cells both in vitro and in vivo.[41, 61, 96, 129]
Also, the highly active metabolism of cancer cells can be targeted using small molecules such as folic acid attached to the surface of NPs.[130, 131] Highly metabolizing cancer cells require folic acid for various biochemical pathways such as DNA biosynthesis and DNA repair. Therefore, the folate receptor is overexpressed on the surface of many types of cancer cells to sequester folic acid. Iron oxide NPs coated with a monolayer of PEG and activated with folic acid show cancer cell specific uptake in human adenocarcinoma cells, and can be monitored using MRI.[130]
Pre-targeting is an alternative approach to targeted deliver therapies. The pre-targeting strategy employs a targeting ligand conjugate to pre-label cells, followed by treatment with NPs that recognize the targeting ligand conjugate on the cell surface. This strategy provides the ability to use a single NP formulation for targeting multiple cancer cell targets. For example, biotin activated iron oxide NPs can be used to specifically bind to fusion proteins that contain an antibody fragment and an avidin.[132] Therefore, the same NP system can be used for any antibody-avidin fusion protein that is developed for specific cancer cell types. Furthermore, a cycloaddition reaction can be utilized for NP recognition of pre-labeled cells.[133, 134] Here, antibodies are modified with trans-cyclooctene (TCO) and NPs with tetrazine (Tz).[134] The bioorthogonal reaction between TCO and Tz is similar to the avidin-biotin reaction in that it is fast, chemoselective, does not require a catalyst, and can occur in serum, but unlike avidin-biotin this reaction is covalent. This strategy provides a 3-fold higher labeling efficiency of cells with NPs as compared to the avidin-biotin system.
While many studies have shown that targeting increases the accumulation of NPs at the tumor site, recent work has shown the targeting agent actually improves NP uptake into target cancer cells and distribution throughout the tumor.[135, 136, 137] For example, targeted polymeric NPs composed of heparin, folate (the targeting agent), and paclitaxel (HFT-T) show similar biodistribution and tumor accumulation as compared to non-targeted polymeric NPs composed of heparin and paclitaxel (HT-T) (Figure 6a and b).[136] However, histological and flow cytometry analyses reveal HFT-T provides a significantly higher uptake into cancer cells (Figure 6c and d), which results in improved tumor growth inhibition.
Figure 6.

In vivo distribution of HFT-T in KB-3-1 tumor-bearing mice. Near infrared dye (cy5.5)-labeled HFT-T or HT-T was injected i.v. into KB-3-1 tumor-bearing mice. (a) Imaging of mice at 1, 24, and 48 h after injection. (b) Biodistribution of HFT-T and HT-T in major organs at 48 h after injection. (c) The cellular internalization of HFT-T versus HT-T in KB-3-1 xenografts 24 h after injection (i.v.). HFT-T showed marked internalization in KB-3-1 cells identified by human EpCAM expression (green). In contrast, HT-T showed much less internalization by KB-3-1 cells and was predominantly found in the extracellular space. (d) Flow cytometry analyses of cells obtained from disaggregated KB-3-1 xenografts 24 h after i.v. injection of HFT-T or HT-T. Two-dimensional event density plots of disaggregated tumor cell suspensions from animals injected with HFT-T or HT-T. The cells were stained with anti-EpCAM Ab-FITC conjugate to identify human cancer cells. The cells in Q4-2 and Q2-2 were human tumor cells (EpCAM positive), the cells in Q1-2 and Q2-2 contained nanoparticles (bodipy 564 positive), and the cells in Q2-2 were human tumor cells containing nanoparticles (double positive). Reproduced with permission.[136] Copyright 2009, American Chemical Society.
NPs developed for targeted gene delivery to brain cancer also show improved cell uptake and distribution throughout the tumor.[137-139] Iron oxide NPs coated with a copolymer comprising chitosan, PEG, and PEI and loaded with green fluorescent protein (GFP) encoding plasmid DNA offer a means to monitor tumor uptake with MRI of the NP core and successful intracellular delivery through for optical detection of GFP expressing transfected cells.[137] The NPs loaded with DNA (NP:DNA) and activated with CTX (NP:DNA-CTX) to achieve targeted DNA delivery. Both NP:DNA and NP:DNA-CTX accumulate at the tumor site to a similar degree in mice containing xenograft tumors of brain cancer, showing the addition of CTX does not increase the localization of NP to the tumor. However, fluorescence imaging of the tumor to detect GFP expression shows significantly higher GFP expression in tumors from NP:DNA-CTX treated mice as compared to NP:DNA treated mice. Histological analysis confirms the increased GFP expression is due to the enhanced distribution of NP:DNA-CTX throughout the tumor, and thus a higher proportion of cells are exposed to and transfected by the NP. These examples highlight the importance of a targeting agent in the delivery of therapies to solid tumors.
Active targeting can also direct the NP to specific cells of the tumor stroma that promote tumor growth, as well as cancer cells. The tumor stroma includes the non-cancerous cells in the tumor microenvironment such as endothelial cells which increase blood flow to the tumor, macrophages which diminish anti-tumor immune responses and promote tumor growth, and fibroblasts which also inhibit anti-tumor immune responses and provide structural support. This tumor stroma targeting has been shown with PEGylated iron oxide NPs activated with one of two peptides: arginine-glycine-aspartic acid (RGD) to target tumor-associated endothelial cells or CTX to target brain cancer cells.[140] Mice bearing xenograft human brain tumors injected intravenously with either NP-RGD or NP-CTX show similar tumor contrast enhancement in MRI. Fluorescence imaging reveals, however, that NP-RGD accumulates in the endothelial cells of neovasculature whereas NP-CTX distributes throughout the tumor in both endothelial cells and cancer cells. This selective targeting can help localize delivered drug to desired target cells.
Active targeting of tumors can also be achieved through surface engineering of NPs to alter their tumor penetration properties. Modeling and tumor cylindroid studies reveal that cationic NPs are readily taken up by tumor cells, but do not penetrate into the core of the tumor sphere, whereas anionic NPs can readily penetrate deeply into the tumor but are poorly taken up by tumor cells.[141] In order to increase the penetration into the tumor and promote cellular uptake, NPs can be engineered to reverse charge in the acidic tumor microenvironment.[142] This charge-reversal strategy generally protects amine groups on the surface of NPs through a pH sensitive bond to render an anionic or neutral NP, ideal for proper navigation through the body. Upon entering the acidic tumor microenvironment, the amine groups become deprotected and generate a cationic NP that can be readily taken up by cells.
Deprotection of amine groups can also aid in therapy by exposing target cells to highly cationic, toxic molecules such as PEI, along with enhancing cell uptake. PEI with primary amines blocked with citraconic anhydride and attached to iron oxide NPs coated with PEG can be modified with anti-GFP siRNA to knockdown transgene expression and CTX as a targeting agent.[143] Blocking the primary amines of PEI completely suppresses the toxic effect of PEI which is be reversed at acidic pH present in the tumor microenvironment and endosome of the cell. These NPs show selective gene knockdown in and toxicity to target cells at acidic pH 6.2 as compared to normal physiological pH 7.4. This shows how the tumor microenvironmental approach can be used to improve targeted therapy to cancer.
This microenvironment targeting approach can be used along with a tumor targeting ligand to enable higher specificity to the tumor. It has been employed for targeted delivery of chemotherapeutic drug to tumors.[144] Polymeric micelles loaded with doxorubicin and activated with trans-activator of transcription (TAT) peptide provide efficient cellular internalization and tumor cell kill. Under physiological conditions the TAT peptide is hidden by the micelle surface whereas in the acidic tumor microenvironment the TAT peptide becomes exposed and interacts with the tumor cell surface for uptake. These NPs are able to reduce xenograft tumor size and slow subsequent growth after intravenous injection.
Furthermore, activatable cell penetrating peptides (ACPPs) show enhanced tumor accumulation by selectively becoming active in the tumor microenvironment of high enzymatic activity.[145] These ACPPs comprise a polycationic cell penetrating peptide (CPP) that is linked to a polyanionic peptide through a protease cleavable linker. Therefore, the CPP does not interact with cells until the polyanionic peptide is cleaved by proteases present in the tumor microenvironment. This strategy improves the delineation of tumor boundaries when attached to fluorescent NPs resulting in more thorough tumor removal during surgery in mice containing xenografts.[146]
Strategies utilizing external forces can also improve penetration of NPs into tumors. This provides another means for affecting a larger proportion of target cells without the need of a targeting ligand. Pulsed ultrasound enhances the penetration of NPs or microbubbles into tumor spheroids in vitro.[147] The ultrasound reduces the packing density of cells, which is a major barrier to the penetration of drugs deep into tumors,[148] through the cavitation of microbubbles which produce significant mechanical impacts on the cells and extracellular matrix. Similarly, magnetic NPs can be pulled into the tumor site by a driving magnetic field. Iron oxide NPs injected intravenously into mice bearing xenograft breast tumors show significantly higher accumulation and retention in tumors at the presence of a magnetic field generated by a neodymium iron boron magnet placed over the tumor for 1 hr after injection of NPs.[149] However, this magnetic field-mediated targeting is only feasible with tumors accessible to an external magnet. The strong decrease in magnetic field strength in deeper tissues such as lung, liver, and brain, limits active accumulation of magnetic NPs. Tumors in these tissues could be magnetically targeted through magnetic resonance navigation (MRN) which utilizes a modified MRI scanner to produce gradients of up to 400 mT m−1 to direct NPs to specific locations in the body.[150]
4.2. Stem Cells as Drug Carriers for Targeted Delivery
Adult stem cells such as mesenchymal stem cells and neural stem cells have recently received considerable attention for use as drug and NP carriers. The goal is to exploit the tumor homing properties of these stem cells to actively deliver the therapeutic payload or imaging agent to the tumor site. This active targeting is different than the active targeting achieved with NPs alone; stem cells are able to home to the tumor site whereas NPs with active targeting increase the chances of sticking to and being internalized by tumor cells. Two different types of adult stem cells have been studied, mesenchymal stem cells (MSCs) and neural stem cells (NSCs). Both can be loaded with NPs without affecting their normal cellular function, and can be tracked using MRI for at least 6 weeks after implantation into rats.[151] Using stem cells and NPs together can promote the active targeting of NPs to a larger proportion of cancer cells, and can help improve stem cell-mediated drug delivery through imaging.
MSCs were first discovered in the stromal compartment of bone marrow and give rise to connective tissue, skeletal muscle cells, and cells of the vascular system.[152] MSCs home to wound sites to aid in healing, and since the tumor microenvironment consists of many signaling factors also present in a site of inflammation, MSCs migrate to tumor regions as well. Signaling factors in the tumor microenvironment that aid in MSC recruitment include tumor growth factor-β (TGF-β), interleukin-6 (IL-6), cyclophilin B, hepatoma-derived growth factor (HDGF), urokinase-type plasminogen activator (uPA), monocyte chemotactic protein-1 (MCP-1), VEGF, and fibroblast growth factor-2 (FGF2).[153] While MSCs promote tumorigenesis, they can be employed to deliver drugs specifically to the tumor site.[154] MSCs are seen as a promising cell-based therapy owing to their ease in isolation (mainly from bone marrow) and expansion in vitro, in sharp contrast to NSCs which are difficult to prepare in sufficient amounts. Systemically-injected interferon-β (IFN-β) and cytokine expressing MSCs are able to reduce tumor growth through induced local immune response against tumor cells.[155] Furthermore, MSCs have been used to deliver tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) to tumor sites to induce apoptosis in cancer cells.[156] MSCs stably transfected to produce TRAIL are able to penetrate the tumor and act as a reservoir that slowly releases the therapeutic protein.
MSCs have been used to deliver many other types of drugs including conditionally replicating adenoviruses which inhibit tumor growth both in vitro and in vivo,[157] pro-toxin converting enzymes such as herpes simplex virus-thymidine kinase (HSV-tk) and cytosine deaminase which convert harmless compounds into toxic drugs,[158] and antibodies to inhibit cancer cell function.[159] MSCs can actively penetrate deep into the tumor microenvironment so that a higher proportion of cells would be exposed to the therapy. The variety of therapies MSCs have delivered to tumor sites indicates that they should be an effective vehicle for targeted delivery of theranostic NPs which may allow for both drug delivery and monitoring. In fact, a variety of NP formulations have been loaded into MSCs for targeted delivery to tumor sites.[160, 161]
Furthermore, magnetic NP and QD labeled MSCs[162] can be tracked using MRI and fluorescence imaging, respectively.[163] This strategy of MRI tracking of MSCs has been utilized to monitor MSC homing to lung metastases.[164] MSCs loaded with iron oxide NPs and injected intravenously into a mouse model of pulmonary metastases, could be observed homing to metastatic sites 1 hr after injection through MRI (Figure 7).
Figure 7.

Intravenously-delivered superparamagnetic iron oxide (SPIO) NP-loaded MSCs localize to lung metastases and can be visualized by MRI. (A) Representative coronal MRI sections (n = 4 mice) of a normal mouse lung (Normal), mouse lung with metastases 35 d after i.v. delivery of MDAMB231 cells (pre-MSC), and the same mouse lung 1 h after SPIO-loaded MSC injection (post-MSC). The metastases (circled) are visualized as focal regions of increased signal. These areas correspond to metastases on H&E histologic sections (bar, 100 μm). One hour after SPIO-loaded MSC injection, there is a decrease in signal intensity caused by the iron oxide in MSCs (+, ribcage; *, trachea; ^, diaphragm with upper abdomen below; ~, fissure separating lobes). (B) The reduction in signal intensity secondary to the NP-loaded MSCs 1 and 24 h after MSC injection was further confirmed and quantified by comparing signal-to-noise ratio (SNR) between the lung parenchyma and the deltoid muscle in three consecutive MR slices in three mice; there was a significant (P = 0.005) reduction in SNR across all four radiological areas [left upper (LU), left lower (LL), right upper (RU), and right lower (RL)]. (C) Tumor histology from mice harvested at day 35, 1 h after NP-loaded MSC injection and MRI. Prussian blue (i) and DiI staining (ii; red) on contiguous sections from mice, showing that MSCs migrate to and incorporate into lung metastases after i.v. delivery (bar, 20 μm). iii, macrophage immunohistochemistry (brown) stains different cells from NP-loaded cells (blue stain). iv, macrophage immunofluorescence (green) stains different cells from DiI-labeled (red) cells (bar, 5 μm). Reproduced with permission.[164] Copyright 2009 American Association for Cancer Research.
Neural stem cells (NSCs) can also be utilized for tumor specific NP delivery. Adult NSCs reside in the CNS and give rise to cells of the neuroectodermal lineage. NSCs show extensive tropism to experimental glioma and thus should function well as a delivery vehicle for multifunctional NP delivery to brain tumors.[165] Differently engineered NSCs have been shown to improve survival and cancer cell kill in animal models of glioma.[160] Furthermore, magnetic NPs loaded into NPCs are able to attenuate melanoma tumor growth through hyperthermia by an alternating magnetic field in a xenograft mouse model.[166] However, the technical challenges involved in the isolation of NSCs remain a major hurtle in their widespread use and development as vectors for NP delivery.
4.3. NPs in T Cell Based Immunotherapy
T cells have been the target for some NP based therapies for diabetes, arthritis, and transplant rejection.[167] In these cases the goal is to inhibit the immune response to prevent inflammation or rejection of implants or transplants. Conversely, cancer immunotherapy requires the activation of the immune system which is normally suppressed in the tumor microenvironment.[168] T cells are a type of white blood cell of the immune system and are involved in cell-mediated immunity. Adoptive T cell therapy involves removing T cells from patients and stimulating them against a tumor antigen ex vivo before injecting back into the patient to induce specific cancer cell kill.[169] This autologous T cell therapy has shown promise in some melanoma patients,[170] and, in fact, has become the standard of care in some relapsed cancer patients. The application of nanotechnology to T cell based immunotherapies can help improve the knowledge of T cell trafficking by providing a means of imaging in real time, and improve therapy as an adjuvant. Also, T cells can be used as a delivery vehicle for targeting NPs to tumors.[171]
T cell tracking is important for monitoring infiltration into the tumor site. For example, indium-111 (111In)-labeled tumor-specific T cells can be tracked in breast cancer patients using single-photon emission computed tomography (SPECT).[172] These T cells are able to target and kill metastatic breast cancer cells in the bone marrow, but are unable to penetrate solid tumor masses as determined by SPECT imaging. While radiolabeling for SPECT and PET imaging provides a strong signal for in vivo imaging, these imaging modalities do not provide the spatial resolution required for in vivo tracking of T cells. To overcome this, T cells loaded with magnetic NPs can be detected in MRI for real-time tracking and better localization.[173]
Immune cell labeling and tracking using MRI can provide information on therapeutic efficacy, and procedures have been developed for efficient labeling and tracking.[174] The high soft tissue contrast and spatial resolution of MRI as compared to PET and SPECT allows for accurate localization of T cells. Additionally, single T cells loaded with iron oxide NPs can be visualized after implanting into mice. T cells loaded with NPs of 1.3 pg iron equivalent per cell and injected into mice bearing flank xenografts of ovalbumin-expressing lymphoma cells (EG7-OVA) can be tracked using MRI.[175] Imaging reveals single labeled T cells throughout the xenograft tumor (Figure 8). This specific information can provide clues into improving T cell immunotherapy and allow physicians to adjust treatment dosing in real-time.
Figure 8.

In vivo MRI detection of labeled lymphocytes in a mouse tumor. a,b: Sequential MR images (3D-SPGR, voxel (60 μm)3) of the tumor in mice 48 h after injection of three million unlabeled lymphocytes (a) or the same number of magnetically labeled cells (b; iron load 1.3 pg/cell at the time of injection). Control tumors (a) give a homogeneous signal, whereas punctuate signal voids (white arrows) distributed throughout the tumor are observed in tumors of mice that received labeled lymphocytes (b). c,d: Zoom of the tumor image containing a signal void (d; labeled lymphocytes) or no signal void (c; control). Reproduced with permission.[175]
The surface of T cells can be labeled for applications in pseudo-autocrine stimulation and adjuvant drug-loaded NPs for tumor specific delivery.[176] T cells labeled with liposomes with hydrodynamic diameters of 200–300 nm are able to infiltrate EG7-OVA tumors in vivo while minimizing off-target sequestration by the liver and spleen.[176] This provides an advantage over non-targeted, and even targeted NPs, since T cells are able to actively penetrate the tumor and can deliver drug-loaded NPs to regions not accessible by targeted NPs alone. Furthermore, when these liposomes are loaded with T cell stimulating factors that are slowly released, the T cells are able to rapidly proliferate in vivo to maintain an immune response against the tumor and eradicate lung and bone marrow B16 melanomas.[176] Figure 9 shows how the attachment of adjuvant loaded liposomes to the surface of T cells dramatically increases their proliferation resulting in complete cancer regression and improving survival in this study.
Figure 9.

Lung and bone marrow tumors were established by tail vein injection of 1 × 106 extG-luc–expressing B16F10 cells into C57BL/6 mice. Tumor-bearing mice were treated after 1 week by sublethal irradiation followed by i.v. infusion of 1 × 107 CBR-luc–expressing Vβ13+CD8+ Pmel-1 T cells. One group of mice received Pmel-1 T cells conjugated with 100 nanoparticles per cell carrying a total dose of 5 μg IL-15Sa and IL-21 (4.03 μg IL-15Sa + 0.93 μg IL-21); control groups received unmodified Pmel-1 T cells and a single systemic injection of the same doses of IL-15Sa and IL-21 or Pmel-1 T cells alone. (a) Dual longitudinal in vivo bioluminescence imaging of extG-luc–expressing B16F10 tumors and CBR-luc–expressing Pmel-1 T cells. (b) Frequencies of Vβ13+CD8+ Pmel-1 T cells recovered from pooled lymph nodes of representative mice 16 d after T cell transfer. (c) CBR-luc T cell signal intensities from sequential bioluminescence imaging every 2 d after T cell transfer. Every line represents one mouse, with each dot showing the whole-mouse photon count. (d) Survival of mice after T cell therapy illustrated by Kaplan-Meier curves. Shown are six mice per treatment group pooled from three independent experiments. Reproduced with permission.[176] Copyright 2010, Nature Publishing Group.
4.4. Dendritic Cell Based Immunotherapy
Dendritic cell (DC) based immunotherapy involves the use of a DC vaccine (a DC loaded with antigen)[177] which then must migrate to lymph nodes to present antigens to T cells to activate an immune response. A typical treatment consists of the ex vivo loading of antigen into autologous DCs which are then injected back into the patient. This pathway for cancer immunotherapy can be very robust since very few active DCs can elicit a strong immune response.[178] In fact, a number of clinical trials using DC immunotherapy against cancer have shown feasibility and effectiveness, but only in a limited proportion of patients.[179] The application of nanotechnology imaging strategies to DC based therapies could accelerate their widespread translation into the clinic by enabling real-time tracking to better elucidate pitfalls and to monitor response.[163, 180]
Insight into the failure of many patients to respond to DC based immunotherapies will help direct the development of next generation therapies. A large number of studies have labeled DCs with radioisotopes ex vivo to monitor migration of DCs from the injection site to target lymph nodes using scintigraphic imaging. However, the poor spatial resolution associated with scintigraphic imaging does not provide the anatomical information necessary to track DCs to specific regions of the body. DCs labeled with magnetic iron oxide NPs and 111In can be monitored and tracked using MRI and scintigraphic imaging in melanoma patients, highlighting the drawbacks of single modality scintigraphic imaging.[181] DCs are injected directly into lymph nodes using ultrasound guidance. However, only ~50% of cases are successful in DC injection directly into lymph nodes as shown by MRI, whereas scintigraphic imaging does not provide the necessary spatial resolution to draw an accurate conclusion. In many cases, DCs are injected into the tissue surrounding the lymph node (Figure 10) which could explain why a large number of patients do not respond favorably to DC vaccines.[181] These limitations and costs associated with ex vivo culture and labeling of autologous DCs has prompted the development of alternative DC vaccination strategies.[182]
Figure 10.

MRI images of lymph nodes. (a) MRI before vaccination; the inguinal lymph node to be injected is indicated with a black arrow. (b) MRI after injection showing that the dendritic cells were not accurately delivered into the inguinal lymph node (black arrow) but in the vicinity, in the subcutaneous fat (white arrow). Reproduced with permission.[181] Copyright 2005, Nature Publishing Group.
An alternative strategy to ex vivo loading and maturation of DCs involves in vivo targeting of an antigen to DCs.[182] This involves conjugating an antigen to a DC targeting molecule, or loading an NP targeted to DCs with antigen or DNA encoding antigens. However, a critical parameter in the generation of a strong immune response is the migration of activated DCs to lymph nodes to present antigen to T cells. Tagging DCs in vivo with NPs allows for real-time tracking of DC migration to and accumulation in lymph nodes to ensure that a sufficient immune response is generated. A magnetovaccine that consists of iron oxide NP loaded irradiated tumor cells injected into the hind feet of mice offers the ability to monitor the movement of the NPs after capture by DCs.[183] As the NPs are captured by DCs from irradiated cells at the injection site, DCs migrate to lymph nodes and can be imaged by MRI. This provides information on the frequency of antigen-bearing DCs migrating from the vaccine site to lymph nodes, which ranges from 5,000 to 40,000 in this study.[183]
In order to gain further information about the NP uptake by DCs and subsequent migration with in vivo DC targeting, multimodal imaging must be utilized since even MRI cannot provide information of the intracellular localization of NPs. Dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN) antibody targeted NPs comprising fluorescein isothiocyanate (FITC) conjugated iron oxide NPs loaded into poly(lactic-co-glycolic acid (PLGA) show selective tagging of DCs in whole blood and allow for the monitoring of DC migration in human tissue mimetic collagen scaffolds using MRI.[184] Furthermore, subcellular tracking can be observed using confocal fluorescence imaging of the loaded FITC. These NPs offer a platform for the future development of next generation smart vaccines.[184]
4.5. NP-Mediated Cancer Vaccination
Cancer vaccination aims to enlist the body’s natural defenses to attack cancer cells, as with T cell and DC immunotherapies described above. The tumor microenvironment plays an active role in the suppression of immune system so that the tumor can grow unabated.[185] Both stromal cells, such as tumor associated macrophages, and cells recruited to the tumor, such as regulatory T cells, play a role in immune suppression.[186] Ideally, upon activation, the body’s immune cells would recognize cancer cells as foreign and destroy them as they would with bacteria or other foreign invaders. NPs can be used for vaccination by loading antigens or epitopes on the surface of the NP for uptake by antigen presenting cells eliciting an immunostimulatory cascade.[187]
An effective delivery vehicle for epitope vaccination must meet several key requirements (Figure 11): 1) be constructed as a particle of 20–100 nm in diameter, 2) have a highly repetitive and ordered structure, 3) have the ability to display epitopes for activation of innate immunity, and 4) localize in specific areas of the body for efficient immune response.[188, 189] This has led to the development of virus-like particles (VLPs), i.e., particles assembled from virus components, for vaccination against hepatitis B and human papillomavirus. Furthermore, VLPs are currently being explored for vaccination against other viruses and diseases such as arthritis, Alzheimer’s, and cancer.[190] However, the outcome in use of a VLP as epitope delivery vehicle remains unpredictable due to undesirable structural perturbations caused by the viral coat protein or epitope leading to diminished function.[191] Alternatively, nanotechnology provides an opportunity to develop safer, more effective, and readily modifiable epitope delivery vehicles for cancer vaccination.
Figure 11.
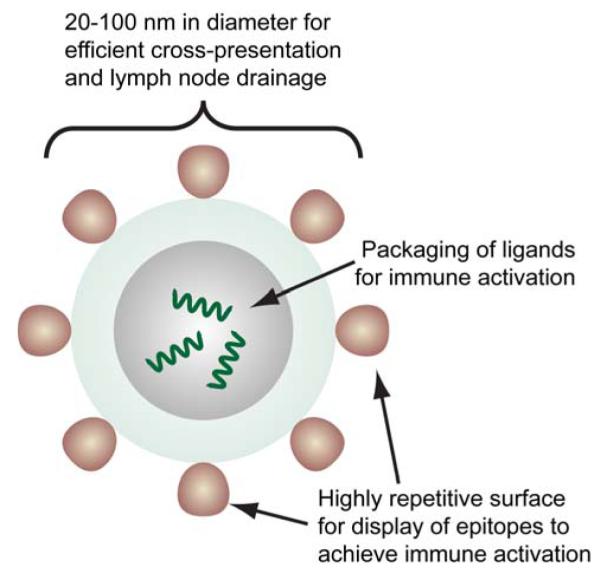
Structure of a virus-like particle (VLP) presenting antigens for immune activation. The size of the VLP ensures efficient trafficking to and recognition by the immune system for activation. Its ability to package ligands and its highly repetitive surface display of epitopes enable efficient activation of both complement and innate immunity. Adapted from.[189]
Using nanotechnology strategies extreme size and shape restraints can be exerted on materials to match the design parameters imposed by biological systems.[192] Specifically, synthesis parameters can be adjusted to create a highly symmetrical nanovector with a specific size and shape,[193] and the surface of the nanovector can contain multiple functional groups[194] for attachment of multiple epitopes that interact with immune cells, and of targeting molecules to direct the delivery to specific locations in the body.[195]
Early epitope delivery vehicles were liposome-based formulations. These studies revealed that the surface organization of the epitope is critical for specific antibody response.[196] More recent work has utilized this knowledge to produce nanostructures with highly organized epitope displays. For example, epitopes assembled onto nanofibers made of the short fibrillizing peptide, Q11, enhanced antibody response in immunized mice.[197] The epitope organization on the fibers provide a high-density display to immune cells for efficient activation. Similarly, a self-assembling peptide based nanoparticle displaying a tandem repeat of a malaria epitope produces a high-titer, long-lasting, high-avidity antibody response in immunized mice.[198] Significantly, this immunization provides protection against an initial challenge of malaria parasites for up to 6 months, and up to 15 months upon a second challenge. Solid gold NPs, which are optically active, have also been used for epitope delivery. The peptide epitope of latent membrane protein-2 from the Epstein-Barr virus organized onto the surface of gold NPs elicits a significantly stronger IFNγ response compared to free epitope in vitro.[199] Furthermore, dendritic cells treated with these epitope-loaded NPs effect CD8+ T cell activation for epitope-specific cytotoxic T lymphocyte killing responses in vitro.
Another strategy for NP-mediated cancer vaccination is to act as an adjuvant for complement activation, a pathway that NPs are usually designed to bypass with medical implants and devices to avoid rejection and/or clearance. Ovalbumin conjugated, polyhydroxylated NPs are able to induce complement activation after lymphatic transport to induce cellular immunity.[200] This lymph node-resident dendritic cell targeting is potentially a new strategy for vaccination and could be applied for cancer vaccines. The hydroxyl groups on the surface of the pluronic-stabilized polypropylene sulfide NPs are thought to bind to exposed thioester of C3b to activate complement by the alternative pathway (Figure 12). This work has been expanded to use polypropylene sulfide NPs showing that surface engineering of NPs has a dramatic effect on complement activation.[201]
Figure 12.

Polyhydroxylated nanoparticle surfaces activate complement. (a) Synthesis and stabilization with two different forms of Pluronic allowed the generation of polyhydroxylated- or polymethoxylated-nanoparticles. (b) The, -terminal OH groups on Pluronic could be converted to OCH3 groups. (c) The proposed mechanism where OH groups on the polyhydroxylated nanoparticles can bind to the exposed thioester of C3b to activate complement by the alternative pathway. (d) Nanoparticle-induced complement activation, as measured through C3a presence in human serum after incubation with nanoparticles, was demonstrated to be high with polyhydroxylated nanoparticles but low with polymethoxylated nanoparticles (OH- and CH3O-NPs, respectively). Results are normalized to control of serum incubation with PBS. Values are means of three independent experiments; error bars correspond to standard error of mean, s.e.m. Reproduced with permission.[200] Copyright 2007, Nature Publishing Group.
5. Applications of Targeted Therapies
The application of these targeted therapies varies significantly between types of cancers. Solid primary tumors behave and respond differently to therapies than blood tumors such as leukemia and lymphoma, metastases, and infiltrative tumors such as glioblastoma multiforme (GBM). Furthermore, cancer stem cells are an ideal target in pre-clinical drug development. Various nanotheranostic strategies have been studied for the targeted treatment of cancer including gene delivery nanovehicles and chemotherapy nano-reservoirs to overcome multiple drug resistance (MDR). Engineering of nanomaterial size and shape in order to directly inhibit target cell function can also provide targeted treatment.
5.1. Solid Tumors
Most cancers form a solid tumor in tissue in which they arose such as in the lung, liver, pancreas, breast, or skin. Once the tumor reaches a critical size, diffusion can no longer provide access to inner tumor cells.[202] Therefore, solid tumors require therapies to actively penetrate deeply into the tumor in order to affect a large proportion of cancer cells. Furthermore, antiangiogenic strategies have been used to cut off blood supply to the tumor so that its metabolic needs cannot be met.[203] Theranostic NPs can help deliver therapeutics deep into the tumor to treat a higher proportion of the cells.
Nanotechnology provides a unique advantage in cancer therapy since the size scale is on the order of the proteins used for cell function. The size and shape of NPs can be tuned to exert a desired therapeutic response on a specific target. In a study comparing CNTs and fullerene conjugated to doxorubicin, it was found that CNTs exerted a pro-angiogenic effect in murine tumors whereas fullerene conjugates inhibited endothelial cell proliferation.[204] Through a variation in shape of carbon-based nanomaterials, their therapeutic role can be drastically altered. CNTs both attenuate the therapeutic function of doxorubicin and promote clustering of integrins which activates Akt through downstream signaling, resulting in angiogenesis, whereas fullerenes provide an antiangiogenic effect. Furthermore, as discussed above, controlling the size of NPs can enable their passive accumulation in the tumor site to enhance tumor uptake of delivered drug. This strategy has been utilized for doxorubicin bound iron oxide NPs to improve the delivery of the drug to xenografts of Lewis lung carcinoma.[205] Delivery of these NPs can be monitored using MRI and results in significant reduction in tumor growth as compared to delivery of the free drug.
The multifunctionality of NPs can enable the targeted delivery of therapies to solid tumors. A liposomal formulation of doxorubicin targeted to H460 lung cancer xenografts in severe combined immunodeficiency (SCID) mice through a single chain variable fragment antibody to c-Met slows tumor growth to 19% of the untreated controls.[206] Inclusion of an imaging moiety within the NP can allow for treatment monitoring ensuring that sufficient drug reaches the tumor site. Iron oxide NPs coated with oleic acid and an amphiphilic block copolymer of polyethylene oxide (PEO) and poly(p-phenylene oxide) (PPO) are able to load large amounts of chemotherapy drug in their hydrophobic layer and be monitored using MRI.[207, 208] Mice with flank orthotopic tumors of human breast cancer MCF-7 cells and injected intravenously with NPs through the tail vein can be imaged at various time points to monitor NP uptake. Figure 13 shows how the tumor uptake of these NPs can be monitored in real-time to provide quantitative information on drug delivery to the tumor. Comparing therapeutic response to quantitative drug accumulation in the tumor could allow the physician to eventually stop treatment once a sufficient dose reaches the tumor, or to continue treatment until an effective dose is achieved. Furthermore, imaging could be used to monitor off-target uptake of drug so treatment could be stopped once a maximum tolerated concentration reaches an off-target organ.
Figure 13.

T2-weighted image of tumor-bearing mouse injected with pluronic F127-modifed magnetic NPs. Enhanced contrast in the tumor (denoted by arrow) is apparent 4 min after the initial injection and is more pronounced at 68 min after a second injection of the MNPs. Images were analyzed for signal intensity in the tumor with Amira software (Visage Imaging, Inc., San Diego, CA). Reproduced with permission.[208] Copyright 2009 Elsevier.
This concept of predictive therapeutic response has been tested.[209] For example, temperature sensitive liposomes have been loaded with doxorubicin as a chemotherapy drug and Gd-DTPA (diethylenetriaminepentaacetic acid chelated Gd) as an MRI contrast agent for image-guided therapy of a xenograft mouse model of mammary carcinoma.[210] The amount of drug uptake in the tumor correlates linearly with the change in MRI contrast (ΔR1) in the tumor, allowing for non-invasive, quantitative analysis of drug uptake. Furthermore, the inhibition of tumor growth can be correlated with ΔR1, the change in relaxivity between before and after NP treatment, which allows for predictive response to therapy so that dosing can be adjusted accordingly.
Various other NP formulations have been used for image guided delivery of chemotherapies. A multifunctional oil-in-water nanoemulsion comprising iron oxide NPs for MRI, the Cy7 fluorophore for optical imaging, and the glucocorticoid prednisolone acetate valerate as an anticancer therapeutic has been developed for image-guided delivery of chemotherapy.[211] Upon intravenous injection into mice with xenograft tumors of colon cancer, these theranostic NPs can be visualized with optical imaging and MRI, and induce a significant reduction in tumor growth (Figure 14).
Figure 14.

Multifunctional nanoemulsion used as a nanotheranostic NP provides image guided therapy in a mouse model of colon cancer. The oil-in-water nanoemulsion comprises iron oxide NPs for MRI, Cy7 fluorophore for optical near-infrared fluorophore (NIRF) imaging, and PAV for therapy. Reproduced with permission.[211] Copyright 2011, American Chemical Society.
The delivery of gene therapeutic agents provides an additional layer of targeting since the delivered nucleic acid is designed to have a selective therapeutic effect on cancer cells. DNA delivery aims to replace a damaged gene with a functional counterpart to restore normal cell function, and siRNA delivery aims to knockdown the expression of oncogenes. Many theranostic NP formulations have been developed for the delivery of DNA and siRNA.[137, 139, 143, 212] Magnetic NPs targeted to breast adenocarcinomas using the EPPT peptide which binds the tumor-specific antigen underglycosylated mucin-1 (uMUC-1) were loaded with anti-baculoviral inhibitor of apoptosis repeat-containing 5 (BIRC5) siRNA to induce apoptosis and Cy5.5 for optical imaging.[213] The delivery of these NPs to xenograft tumors can be monitored using both MRI and optical imaging, and result in a significant reduction in tumor growth due to increased cancer cell apoptosis as compared to scrambled siRNA delivery.
Thermotherapy can be achieved with gold NPs heated by absorption of light resulting in heat-induced cell death. This photothermal therapy has been investigated using gold NPs of various shapes.[214] Hollow gold nanospheres conjugated with a targeting agent for melanoma have been studied for photothermal therapy.[215] Mice with xenograft tumor of melanoma and injected intravenously with these NPs are irradiated with near infrared lasers 4 hours after injection to heat the NPs. Tumors in mice receiving both NPs and laser treatment show significantly increased necrotic area, and reduced uptake of [18F]fluorodeoxyglucose, a radiotracer used to observe metabolism suggesting efficient cell kill.
The superparamagnetic properties associated with nanosized magnetic NPs can also be used for thermotherapy in hyperthermia, as introduced in section 2.5. Significant work has been done in this field in treating brain tumors with ferrofluids.[48, 216] However, the very high concentrations of NPs needed (2 mol/L) require direct injection of NPs into the tumor site. Nevertheless, the potential use of NPs delivered systemically for hyperthermia has been demonstrated in a mouse xenograft model of human breast cancer.[217] Dextran-coated iron oxide NPs conjugated with a ChL6 antibody against breast cancer and delivered intravenously to xenograft mouse models of breast cancer show a significant reduction in tumor growth upon application of a rapidly alternating magnetic field. Additionally, NSCs have been used to deliver NPs to tumors for magnetic hyperthermia. NSCs loaded with core/shell iron/iron oxide (Fe/Fe3O4) NPs are able to deliver the NPs to xenograft tumors of melanoma. Upon application of a rapidly alternating magnetic field, mice receiving NSCs loaded with NPs show a significantly slower tumor growth.[166]
Magnetic hyperthermia can also be used to selectively release drug from NPs which circumvents the need for high local NP concentrations, and provides the advantage of selective drug release to minimize off-target side-effects. To achieve this, mesoporous silica NPs loaded with doxorubicin and iron oxide NPs are capped with heat labile molecules.[218] In normal physiological conditions, little drug is released from these NPs, but in the presence of a rapidly alternating magnetic field the capping molecule is released allowing doxorubicin to diffuse out of the NP.
5.2. Metastases
Metastasis of cancer involves the migration of a cancer cell from the bulk tumor into the surrounding tissue, intravasating into the blood, extravasating from the blood into tissue elsewhere in the body, and forming a secondary tumor. Metastatic cancer accounts for at least 90% of all cancer related deaths, and thus its prevention and therapy could dramatically improve survival.[2, 219] Metastatic cancer is much less responsive to standard chemotherapies and thus novel therapeutic formulations must be explored.
Various methods for inhibiting cell invasion, which is a major component in the metastatic process, have been investigated.[220] These include engineering the shape and size of nanomaterials to specifically interact with and inhibit cell surface proteins involved in cell invasion. For example, carbon nanotubes have been engineered to specifically interact with cell surface ion channels to inhibit ion transport.[221] Furthermore, modeling studies have revealed fullerenes have the potential to specifically inhibit these cell surface ion channels in a similar manner.[222] Surface engineering of NPs has also proven useful to inhibit cell invasion through the multivalent effect where a larger portion of the cell membrane that contains proteins involved in cell invasion is caused to be internalized preventing proper function of these proteins.[223] However, unless the disease is caught early enough, inhibition of invasion is no longer a viable strategy; metastases must be directly treated.
Metastatic ovarian cancer is such a disease where metastases are often present at the time of diagnosis.[224] While Doxil improves the time of survival of patients with metastatic ovarian cancer, the prognosis is still poor with a median survival of 108 weeks.[225] Targeted therapy using multifunctional NPs should dramatically improve the prognosis of these patients. Polymeric NPs loaded with paclitaxel targeted to metastatic ovarian cancer using the HER2 antibody are able to significantly improve the survival in animal models of metastatic ovarian cancer developed by intraperitoneal injection of SKOC-3 cells.[226] This improved survival is due to the increased target cell uptake of paclitaxel bound to NPs.
Metastases can also be treated with targeted gene therapies. A liposome-polycation-hyaluronic acid NP targeted to lung metastases of melanoma using a single-chain antibody fragment can deliver siRNA against tumor promoting genes c-Myc, murine double minute 2 (MDM2), and VEGF, and micro RNA (miRNA)-34a to induce apoptosis.[227] These NPs are able to significantly inhibit metastases in the lung upon intravenous injection (Figure 15).
Figure 15.

Tumor growth/metastasis inhibition by nanoparticles containing siRNA and miRNA. (a) Images of the B16F10 tumor-bearing lungs on day 19 after two consecutive i.v. injections of siRNAs or miRNA in different formulations. (b) Luciferase activity in the tumor-bearing lungs on day 19 after two consecutive i.v. injections on days 8 and 9 of siRNAs and miRNA in different formulations. n = 5–6. ***P < 0.001. Formulations: untreated control (1), combined siRNAs and control miRNA in the GC4-targeted nanoparticles (2), control siRNA and miR-34a in the GC4-targeted nanoparticles (3), combined siRNAs and miR-34a in the control-targeted nanoparticles (4), and combined siRNAs and miR-34a in the GC4-targeted nanoparticles (5). Dose = 0.6 mg total RNA/kg. Combined siRNAs = c-Myc:MDM2:VEGF (1:1:1), siRNA:miRNA = 1:1, weight ratios. Reproduced with permission.[227] Copyright 2010, Nature Publishing Group.
5.3. Hematologic Cancers
Hematologic cancers include those that reside in the blood, bone marrow, or peripheral lymphoid organs and include leukemia, lymphoma, and myeloma. They impose a significant challenge for therapies as compared to solid tumors. The tumor microenvironment of these cancers is vastly different than with solid tumors and provides growth factors for cancer growth and survival resulting in de novo drug resistance, and as a result, cells will not respond to first-line chemotherapies.[228] Furthermore, treated cells can lyse and release their components directly into the blood which leads to potentially lethal electrolyte and metabolic disturbances, and is called tumor lysis syndrome.[229] Therefore, theranostic NPs should aim to induce apoptosis so that cellular components will be packaged into apoptotic bodies for macrophage uptake rather than directly released into the circulation.
Leukemia is the most common cancer in children, but affects people of all ages. Leukemia is a cancer of white blood cells and affects bone marrow. Self-assembling antibody nanorings containing the anti-CD3 antibody show leukemia cell specific uptake in vitro and could be used as leukemia specific drug delivery vehicles.[230] Gene therapy can provide a specific treatment of leukemia by affecting aberrant cell signaling. Transferrin conjugated pH-sensitive lipopolyplex NPs can be used as targeted delivery vehicles for antisense oligodeoxynucleotides to leukemia cell lines and primary cells.[231] Knockdown of the expression of ribonucleotide reductase with these targeted NPs results in increased sensitivity to the chemotherapy agent cytarabine. Drug loaded liposomes have also shown promise in reducing recurrence of de novo acute lymphoblastic leukemia.[232]
Lymphoma is a cancer of the cells of the lymphoid system. Hodgkin lymphomas are solid tumors that form in lymph nodes, whereas non-Hodgkin lymphomas are blood cancers. Hodgkin lymphomas are essentially curable with standard chemotherapy treatments of doxorubicin, bleomycin, vinblastin, and darcarbazine, but improvements can still be made in reducing off-target side effects.[233] Non-Hodgkin lymphomas have historically been treated with standard chemotherapy of cyclophosphamide, doxorubicin, vincristine, and prednisone.[234] Antibody based therapy with Rituximab has been shown to improve response in both first-line treatments and in patients with relapsed or refractory cancers.[235] NPs of n-(2-hydroxypropyl)methacrylamide (HPMA) copolymers loaded with docetaxel used to treat a mouse model of lymphoma can achieve complete remission of the cancer in mice.[236]
Although great strides have been made in the therapy of multiple myeloma, the prognosis remains dismal.[237] Multiple myeloma is a cancer of white blood cells that form lesions in bone marrow and prevents the normal production of blood cells. Interestingly, gold NPs can selectively inhibit proliferation in multiple myeloma cells in vitro.[238] It is thought that the nanosize of these NPs specifically inhibits the function of heparin-binding growth factors which then disrupts cell proliferation though cell cycle arrest.[238] This is another example of how engineering materials at this size scale can have significant effects on biological molecules in the same size range. The use of albumin NPs has also shown promise in improving multiple myeloma therapy.[239, 240] Albumin naturally carries hydrophobic molecules such as vitamins and hormones, and was first used commercially in oncology after US Food and Drug Administration (FDA) approval in 2005 as 130-nanometer albumin-bound paclitaxel, or Abraxane.[241] For multiple myeloma therapy, the albumin helps solubilize water-insoluble rapamycin, and provides a means for transcytosis across endothelial cells to increase drug concentration at the tumor site. In combination with perifosine, albumin bound rapamycin treatment of mice with xenograft tumors of multiple myeloma provides significant tumor growth inhibition and increased survival.[239]
5.4. Overcoming MDR
Multidrug resistance (MDR) is an acquired phenotype in cancer cells, characterized by the overexpression of ATP-binding cassette (ABC) transporters which actively pump chemotherapy drugs out of cancer cells.[3] MDR is a significant problem in cancer chemotherapy since while many chemotherapeutic agents are effective debulking agents (i.e., they are able to significantly reduce the tumor burden), a small proportion of cells that are resistant to the therapy can survive to form a resistant tumor. This acquired drug resistance is different than the microenvironment-mediated de novo drug resistance associated with hematologic cancers discussed above. MDR is a result of the overexpression of ATP-binding cassette (ABC) transporters which increase the efflux of a broad class of hydrophobic drugs from cancer cells, with the most common being ABCB1, or P-glycoprotein (P-gp).[3, 242] Many strategies to inhibit these ABC transporters have been tested but clinical application has been hindered due to low efficacy and high toxicity.[3, 243] Many of these strategies involve the use of excipients that reduce the function of ABC transporters, or chemical inhibitors. Other strategies include attaching the chemotherapy drug to larger molecules or to proteins that are not substrates for ABC transporter mediated efflux. Therefore the drug can be released once inside the target cell to elicit a therapeutic function before it is recognized and effluxed. This strategy can provide sustained intracellular drug concentrations to ensure a therapeutic response (Figure 16Error! Reference source not found.Error! Reference source not found.).[244] Early studies showed that doxorubicin loaded into polymeric nanospheres are able to overcome MDR in multiple models of drug resistant cancers in vitro, and prolonged survival in mice.[245] However, a similar study suggests that simple encapsulation of drug into polymeric NPs is not sufficient to overcome MDR; paclitaxel released from PLGA NPs intracellularly is still a substrate for P-gp and does not overcome MDR.[246]
Figure 16.

Time course study of intracellular retention of fluorescent-labeled Tx in MCF-7 cells. Cells were treated with drug in solution (Tx-Sol) or unconjugated drug-loaded NPs (Tx-NPs) or Tf-conjugated NPs (Tx-NPs-Tf) (dose = 10 ng/mL) in the growth medium. Cells treated with Tx-Sol showed a decrease in green fluorescence intensity of the drug with incubation time whereas Tx-NPs and Tx-NPs-Tf demonstrated an increase, with Tf-conjugated NPs demonstrating the fluorescence of the drug lasting up to 8 days. N = nucleus. Reproduced with permission.[244] Copyright 2005, American Chemical Society.
Doxorubicin conjugated to stearic acid-g-chitosan micelles is able to overcome MDR in a breast cancer model both in vitro and in vivo by improving the intracellular doxorubicin concentration.[247] Doxorubicin conjugated to the polymer prevents its efflux from the cell, and drug is released from the polymeric micelle once inside the cell. Furthermore, doxorubicin conjugated to iron oxide NPs (NP-DOX) is able to overcome MDR in glioma cells in vitro through a similar mechanism.[248] The NP-DOX conjugate selectively releases doxorubicin in an acidic pH environment and thus a higher intracellular doxorubicin concentration is maintained. The iron oxide NP core provides a means for treatment tracking through MRI. Biodegradable microcapsules loaded with doxorubicin or paclitaxel have been used to overcome MDR in colorectal cancer cells through improved intracellular delivery.[249] These nanocapsules are able to selectively release drug intracellularly so that the drug can reach its intracellular site of action before being pumped out by efflux pumps.
Another strategy to improve intracellular accumulation of anticancer drugs is to deliver efflux pump inhibitors by NPs along with drug. This can overcome the systemic toxicity associated with P-gp inhibitors and increase the effectiveness of chemotherapy in the tumor since NPs deliver both inhibitor and chemotherapy drug to the same cell at the same time. Polymeric NPs loaded with tariquidar, a P-gp inhibitor, and paclitaxel are able to slow tumor growth using significantly lower concentrations of paclitaxel than is needed without tariquidar.[250]
ATP has also been a target for reduction of ABC mediated drug efflux. Diminishing intracellular ATP inhibits the function of ABC transporters. Liposomes containing the Brij 78 surfactant can deplete ATP in drug-resistant cells, which results in increased sensitivity towards delivered drug.[251] These drug loaded liposomes are able to increase resistant-cell sensitivity to paclitaxel and doxorubicin by 1) increasing intracellular drug concentration by bypassing ABC transporters and 2) inhibiting ABC transporter function through depletion of ATP by Brij 78.
NPs loaded with siRNA against P-gp are able to reverse MDR in osteosarcoma through knockdown of P-gp expression.[252] With lipid modified dextran NPs carrying siRNA and the subsequent treatment with doxorubicin resulted in a greater sensitivity in NP treated cells, which was attributed to greater intracellular retention of drug. A similar approach using polymeric NPs carrying both siRNA against P-gp and paclitaxel provides the advantage of drug being delivered to the same cells that receive siRNA treatment.[253] Knockdown of P-gp using the siRNA and paclitaxel loaded NPs increases the intracellular accumulation of paclitaxel in vitro and slows the growth of mammary adenocarcinoma xenograft tumors in vivo. Polymeric NPs loaded with plasmid DNA encoding the anti-P-gp short hairpin RNA (shRNA) are able to significantly reduce the expression of this efflux pump in drug resistance breast cancer cells.[254] This results in a marked increase in cell sensitivity to doxorubicin treatment both in vitro and in vivo owing to a decrease in drug efflux.
An alternative strategy to delivery of siRNA to knockdown the expression of efflux pumps is to utilize an antibody against the efflux pump to inhibit its function. Doxorubicin loaded CNTs activated with a P-gp antibody are able to deliver a therapeutic dose of drug to MDR leukemia cells.[255] The CNTs are able to deliver a high dose of doxorubicin into the cell while the P-gp antibody inhibits efflux pump function.
5.5. Treating Cancer Stem Cells
Cancer stem cells (CSCs) are cancer cells that are capable of self-renewal and drive the growth of a tumor which consists of both CSCs and non-CSCs.[256, 257] CSCs were first found in acute myeloid leukemia, confirming the early hypothesis of their existence.[258] Subsequent findings of CSCs in many other tumor types indicated that these CSCs are present in a broad variety of tumor types, and signaled a drastic paradigm shift in cancer treatment.[259] The CSC concept has garnered significant attention since it explains many of the current difficulties in oncology such as metastases and MDR (both discussed above). Furthermore, standard therapies that eradicate the bulk of a tumor may leave surviving CSCs that would cause relapse. Interestingly, CSCs may be the reason for the failure of many anti-angiogenesis therapies since CSCs have been shown to differentiate into endothelial cells in the tumor microenvironment.[260] Thus, the ability to selectively treat the CSC population (Figure 17) could have a profound impact on cancer therapy, preventing relapse and metastasis.[259] This motivation has led to the use of many CSC-specific therapies that inhibit pathways such as Wnt, Notch, and Hedgehog,[261] but nanotechnology has yet to make a significant impact in this field.
Figure 17.
Treatment of cancer stem cells (CSCs). Standard chemotherapy agents debulk the tumor, but can leave residual drug resistant CSCs which will lead to relapse. A CSC targeted therapy that can selectively and efficiently kill the CSCs will leave a benign mass.
Polymeric NPs of curcumin, a polyphenolic compound derived from turmeric, decreases growth in brain cancer cells, and reduces the CSC population.[262] Inhibition of the Hedgehog pathway likely accounts for the selective reduction in the CSC population. Similarly, γ-secretase inhibitors block Notch signaling but their clinical use is hindered by severe side-effects.[263] Mesoporous silica NPs can deliver this class of inhibitors to tumors to inhibit the Notch pathway.[263] The sensitivity of CSCs to standard chemotherapies has been increased through delivery of siRNA against efflux pumps using biodegradable lipid NPs.[264] These NPs are able to knockdown expression of the efflux pump P-gp in the CSC enriched population of CHOK1 cells, which increases their sensitivity to subsequent paclitaxel treatment.
NPs are expected to advance the CSC-specific therapies by improving their delivery to their target site of action. Various cell surface markers distinguish CSCs from non-CSCs[257, 265] which could be used to direct theranostic NPs to CSCs and inhibit their function.[266] For example, brain cancer CSCs express the stem cell marker CD133 on their surface, and CD133 antibodies are commonly used for fluorescence-activated cell sorting (FACS) enrichment of the CD133+ population of cells.[267] Furthermore, CD133 is expressed on the surface of CSCs from breast cancer,[268] prostate cancer,[269] lung cancer,[270] colon cancer,[271] pancreatic cancer,[272] ovarian cancer,[273] and liver cancer.[274] However, use of these stem cell markers for targeted drug delivery could also affect normal stem and progenitor cells in which long-term effects are still unknown.
The minimal amount of nanotechnology-based research on CSCs may be due to the lack of good CSC models. The development of in vitro models of CSCs should help promote future development of nanomedicine approaches to treatment of CSCs.
6. Conclusions and Outlook
Cancer is a devastating disease that affects millions of people yearly. Although a vast amount of research has increased our knowledge of cancer biology and therapy which has improved survival of cancer patients, the disease remains lethal. The limitations in current cancer therapies signal the need for novel therapies that attack these specific issues. Nanotechnology provides a unique opportunity to combat cancer on the molecular scale through careful engineering of nanomedicines to specifically interact with cancer cells and inhibit cancer cell function. Various NP formulations have already made their way into the clinic and have become the standard of care in some cancer patients.
Moving forward, priority should be placed on NPs that directly address the current limitations in cancer therapy including MDR, metastases, and CSCs, as they could have an immediate and direct impact in patient survival. They will provide additional treatment options to patients who no longer respond to current treatment regimens. Furthermore, directing nanomedicine development for these patients could accelerate their clinical translation as these are the patients who are generally enrolled in new clinical trials, and are often a last treatment option. Cancers of the pancreas, brain, and liver are some of the most devastating and have a very low survival rate. Also, metastases, drug resistant cells, and cancer stem cells represent the populations of cancer cells that show significantly reduced response to therapy.
Beyond use of NPs as nanomedicines, more focus should be placed on use of NPs as tools to learn more about cancer biology and failure of treatments. The ability to monitor biodistribution of treatments, migration of cells throughout the body, and tumor development and evolution in real-time can elucidate new pathways cancer cells rely on. This information will be invaluable for improvement of therapy. This will require the development and improvement of imaging modalities that have high spatial and temporal resolution combined with high sensitivity for cellular and molecular tracking.
Most exciting are the theranostic nanomedicines that combine imaging and treatment into a single NP formulation. Although the bench-top development of these NPs is tortuous and expensive owing to the complexity of the formulations, their theranostic properties should allow for rapid pre-clinical development since specific information about pharmacokinetics and treatment efficacy can be obtained simultaneously. Furthermore, we envision these NPs being used for real-time monitoring of drug delivery so that dosing and type of therapeutics can be adjusted based on tumor and off-target tissue accumulation. This information can ensure patients are neither undertreated nor overtreated. This will require studies that determine quantitatively the amount of tumor uptake that results in sufficient treatment, and the amount of off-target uptake that results in harmful side-effects.
Despite the significant advancements that have been made, nanotechnology is still a relatively young field, and little is known about the long-term effects of exposure to nanomaterials, especially in clearance organs such as the liver, spleen, and kidneys. Furthermore, the potential toxicity associated with the wide variety of nanomaterials available ranges from completely inert to highly toxic, which could slow their advancement into the clinic. In order for this promising field to rapidly progress, focus must be placed on elucidating the safety of these novel materials. This will rely on the development of better characterization tools and methodology, and more reproducible synthesis strategies so that accurate and broadly applicable conclusions can be drawn. This includes the size, shape, and surface charge of NPs, as well as the number of functional groups, drug loading capacity and releasing mechanism.
The exponential increase in research publications in the field of nanomedicine over the past 5 years suggests the clinical translation of many new, more effective, therapies is on the horizon. Soon, we may see cancer nanotheranostics revolutionize the treatment of cancer.
Table 1.
Ligands utilized for exploiting active transport across the BBB.
| Ligand | NP formulation | NP hydrodynami c size |
NP zeta potential |
Model | BBB integrity confirmation |
Reference |
|---|---|---|---|---|---|---|
| Myristoylated polyarginine peptide |
Cross-linked dextran coated superparamagnetic iron oxide (CLIO) |
40 nm | Not reported |
Intracranial implant of U-87 MG cells into mice |
Intracranial implant of U-87 MG cells into mice |
[87] |
| Chlorotoxin | Chitosan-g-PEG coated iron oxide NPs |
33 nm | 4.2 mV | Transgenic model of medulloblastom a |
Brain tumor uptake compared to control NPs. Gd-DTPA used to confirm BBB integrity. |
[96] |
| RI7217 (rat anti-mouse CD71 clone) |
Liposome of EPC, cholesterol, EPG, and MPB-PE |
116 nm | −14.8 mV |
Balb/c mice | Brain uptake compared to control and other ligandcoupled liposomes |
[89] |
| Lactoferrin | DMSA modified superparamagnetic iron oxide NPs |
74.8 nm | −11.1 mV |
Intracranial implant of C6 cells into rats |
Brain tumor uptake compared to control NPs |
[94] |
| Leptin30 | Dendrigraft poly-l- lysine: DNA complexes |
141 nm | 1.15 mV | Nude mice | Permeability across BCEC monolayer assessed in vitro. Brain uptake compared to control NPs. |
[277] |
| p- aminophenyl- α-D-manno- pyranoside |
Daunorubicin loaded, Tf modified liposomes |
122.8 nm | −6.37 mV |
Intracranial implant of C6 cells into rats |
Permeability across BMVEC monolayer assessed in vitro. Brain tumor uptake compared to control NPs. |
[278] |
| OX26 monoclonal antibody |
Chitosan-g-PEG NPs |
636.69 nm | 18.23 mV |
Swiss albino mice |
Brain uptake compared to control NPs. |
[279] |
| OX26 monoclonal antibody |
6-coumarin or NC- 1900 loaded PEG- PCL polymersomes |
100 nm | −21.3 mV |
Sprague– Dawley rats |
Brain uptake compared to control NPs and NPs with varying amounts of OX26. |
[280] |
| TAT | FITC or QD loaded cholesterol- terminated poly(ethylene glycol) NPs |
190 nm | 22 mV | Sprague– Dawley rats |
Brain uptake compared to free FITC or QD |
[281] |
| Cationic albumin |
PEG-PLA NPs | 82.1 nm | −12.19 mV |
In vitro only | Permeability across BCEC |
[282] |
Acknowledgements
We acknowledge the support of the NIH grants R01CA134213, R01EB006043 and T32CA138312, and Kyocera Professor Endowment.
Biography

Forrest Kievit received his Bachelor’s degree in 2007 from the Bioengineering Department at the University of Washington in Seattle, WA. He is currently a Ph.D. student advised by Prof. Zhang in the Department of Materials Science & Engineering at the University of Washington. His research focuses on the application of nanomaterials for cancer therapy.

Miqin Zhang is Kyocera Professor of Materials Science & Engineering and Adjunct Professor in the Departments of Neurological Surgery, Radiology, and Orthopaedics & Sports Medicine at the University of Washington (UW). She joined the faculty of the UW in 1999 after receiving her Ph.D. from University of California at Berkley. Her research focuses on nanomedicine for cancer diagnosis and treatment, biodegradable scaffolds for tissue engineering, and biosensors for detection of chemical and biological agents.
References
- [1].World Health Organization; Geneva: 2011. [Google Scholar]
- [2].Mehlen P, Puisieux A. Nat Rev Cancer. 2006;6:449. doi: 10.1038/nrc1886. [DOI] [PubMed] [Google Scholar]
- [3].Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Nat Rev Drug Discov. 2006;5:219. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- [4].Cho KJ, Wang X, Nie SM, Chen Z, Shin DM. Clin. Cancer Res. 2008;14:1310. doi: 10.1158/1078-0432.CCR-07-1441. [DOI] [PubMed] [Google Scholar]
- [5].Brindle K. Nat. Rev. Cancer. 2008;8:94. doi: 10.1038/nrc2289. [DOI] [PubMed] [Google Scholar]
- [6].Jain RK, Stylianopoulos T. Nat. Rev. Clin. Oncol. 2010;7:653. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]; Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nat. Nanotechnol. 2007;2:751. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- [7].Davis ME, Chen Z, Shin DM. Nat. Rev. Drug Discov. 2008;7:771. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- [8].Gradishar WJ, Tjulandin S, Davidson N, Shaw H, Desai N, Bhar P, Hawkins M, O’Shaughnessy J. J Clin Oncol. 2005;23:7794. doi: 10.1200/JCO.2005.04.937. [DOI] [PubMed] [Google Scholar]
- [9].Fang C, Zhang M, Control J. Release. 2010;146:2. doi: 10.1016/j.jconrel.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; Xie J, Lee S, Chen XY. Adv. Drug Deliv. Rev. 2010;62:1064. doi: 10.1016/j.addr.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Barreto JA, O’Malley W, Kubeil M, Graham B, Stephan H, Spiccia L. Adv. Mater. 2011;23:H18. doi: 10.1002/adma.201100140. [DOI] [PubMed] [Google Scholar]; Rozhkova EA. Adv. Mater. 2011;23:H136. doi: 10.1002/adma.201004714. [DOI] [PubMed] [Google Scholar]; Minelli C, Lowe SB, Stevens MM. Small. 2010;6:2336. doi: 10.1002/smll.201000523. [DOI] [PubMed] [Google Scholar]
- [11].Weissleder R, Pittet MJ. Nature. 2008;452:580. doi: 10.1038/nature06917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Malam Y, Loizidou M, Seifalian AM. Trends Pharmacol. Sci. 2009;30:592. doi: 10.1016/j.tips.2009.08.004. [DOI] [PubMed] [Google Scholar]
- [13].Torchilin VP. Pharm. Res. 2007;24:1. doi: 10.1007/s11095-006-9132-0. [DOI] [PubMed] [Google Scholar]
- [14].Petersen AL, Binderup T, Rasmussen P, Henriksen JR, Elema DR, Kjaer A, Andresen TL. Biomaterials. 2011;32:2334. doi: 10.1016/j.biomaterials.2010.11.059. [DOI] [PubMed] [Google Scholar]
- [15].Chang Y-J, Chang C-H, Yu C-Y, Chang T-J, Chen L-C, Chen M-H, Lee T-W, Ting G. Nuclear Medicine and Biology. 2010;37:95. doi: 10.1016/j.nucmedbio.2009.08.006. [DOI] [PubMed] [Google Scholar]
- [16].Fattahi H, Laurent S, Liu FJ, Arsalani N, Elst LV, Muller RN. Nanomedicine. 2011;6:529. doi: 10.2217/nnm.11.14. [DOI] [PubMed] [Google Scholar]
- [17].Aime S, Castelli DD, Crich SG, Gianolio E, Terreno E. Accounts Chem. Res. 2009;42:822. doi: 10.1021/ar800192p. [DOI] [PubMed] [Google Scholar]
- [18].Lammers T, Subr V, Ulbrich K, Hennink WE, Storm G, Kiessling F. Nano Today. 2010;5:197. [Google Scholar]
- [19].Lanza GM, Winter PM, Caruthers SD, Hughes MS, Hu G, Schmieder AH, Wickline SA. Angiogenesis. 2010;13:189. doi: 10.1007/s10456-010-9166-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; Akers WJ, Zhang ZR, Berezin M, Ye YP, Agee A, Guo K, Fuhrhop RW, Wickline SA, Lanza GM, Achilefu S. Nanomedicine. 2010;5:715. doi: 10.2217/nnm.10.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ferrara KW, Borden MA, Zhang H. Accounts Chem. Res. 2009;42:881. doi: 10.1021/ar8002442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Jain PK, Huang XH, El-Sayed IH, El-Sayed MA. Accounts Chem. Res. 2008;41:1578. doi: 10.1021/ar7002804. [DOI] [PubMed] [Google Scholar]; Jain PK, El-Sayed IH, El-Sayed MA. Nano Today. 2007;2:18. [Google Scholar]; Huang XH, Neretina S, El-Sayed MA. Adv. Mater. 2009;21:4880. doi: 10.1002/adma.200802789. [DOI] [PubMed] [Google Scholar]; Giljohann DA, Seferos DS, Daniel WL, Massich MD, Patel PC, Mirkin CA. Angew. Chem.-Int. Edit. 2010;49:3280. doi: 10.1002/anie.200904359. [DOI] [PMC free article] [PubMed] [Google Scholar]; Millstone JE, Hurst SJ, Metraux GS, Cutler JI, Mirkin CA. Small. 2009;5:646. doi: 10.1002/smll.200801480. [DOI] [PubMed] [Google Scholar]
- [22].Eustis S, El-Sayed M. J. Phys. Chem. B. 2005;109:16350. doi: 10.1021/jp052951a. [DOI] [PubMed] [Google Scholar]
- [23].Wu X, Ming T, Wang X, Wang PN, Wang JF, Chen JY. ACS Nano. 2010;4:113. doi: 10.1021/nn901064m. [DOI] [PubMed] [Google Scholar]
- [24].Wang HF, Huff TB, Zweifel DA, He W, Low PS, Wei A, Cheng JX. Proc. Natl. Acad. Sci. U. S. A. 2005;102:15752. doi: 10.1073/pnas.0504892102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sonnichsen C, Franzl T, Wilk T, von Plessen G, Feldmann J, Wilson O, Mulvaney P. Phys. Rev. Lett. 2002;88:4. doi: 10.1103/PhysRevLett.88.077402. [DOI] [PubMed] [Google Scholar]
- [26].Jain PK, El-Sayed MA. Nano Lett. 2007;7:2854. doi: 10.1021/nl071496m. [DOI] [PubMed] [Google Scholar]; Chen JY, Wiley B, Li ZY, Campbell D, Saeki F, Cang H, Au L, Lee J, Li XD, Xia YN. Adv. Mater. 2005;17:2255. [Google Scholar]
- [27].Wang H, Zheng LF, Peng C, Guo R, Shen MW, Shi XY, Zhang GX. Biomaterials. 2011;32:2979. doi: 10.1016/j.biomaterials.2011.01.001. [DOI] [PubMed] [Google Scholar]; Kim D, Park S, Lee JH, Jeong YY, Jon S. J. Am. Chem. Soc. 2007;129:7661. doi: 10.1021/ja071471p. [DOI] [PubMed] [Google Scholar]; Popovtzer R, Agrawal A, Kotov NA, Popovtzer A, Balter J, Carey TE, Kopelman R. Nano Lett. 2008;8:4593. doi: 10.1021/nl8029114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Boisselier E, Astruc D. Chem. Soc. Rev. 2009;38:1759. doi: 10.1039/b806051g. [DOI] [PubMed] [Google Scholar]
- [29].Pan DP, Pramanik M, Senpan A, Yang XM, Song KH, Scott MJ, Zhang HY, Gaffney PJ, Wickline SA, Wang LV, Lanza GM. Angew. Chem.-Int. Edit. 2009;48:4170. doi: 10.1002/anie.200805947. [DOI] [PMC free article] [PubMed] [Google Scholar]; Levi J, Kothapalli SR, Ma TJ, Hartman K, Khuri-Yakub BT, Gambhir SS. J. Am. Chem. Soc. 2010;132:11264. doi: 10.1021/ja104000a. [DOI] [PMC free article] [PubMed] [Google Scholar]; Pan DPJ, Pramanik M, Senpan A, Ghosh S, Wickline SA, Wang LV, Lanza GM. Biomaterials. 2010;31:4088. doi: 10.1016/j.biomaterials.2010.01.136. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kim C, Favazza C, Wang LHV. Chem. Rev. 2010;110:2756. doi: 10.1021/cr900266s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Smith AM, Nie S. Acc Chem Res. 2010;43:190. doi: 10.1021/ar9001069. [DOI] [PMC free article] [PubMed] [Google Scholar]; Peng X. Acc Chem Res. 2010;43:1387. doi: 10.1021/ar100025m. [DOI] [PubMed] [Google Scholar]
- [31].Yong KT, Ding H, Roy I, Law WC, Bergey EJ, Maitra A, Prasad PN. Acs Nano. 2009;3:502. doi: 10.1021/nn8008933. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zrazhevskiy P, Sena M, Gao XH. Chem. Soc. Rev. 2010;39:4326. doi: 10.1039/b915139g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].So M-K, Xu C, Loening AM, Gambhir SS, Rao J. Nat Biotech. 2006;24:339. doi: 10.1038/nbt1188. [DOI] [PubMed] [Google Scholar]
- [33].Lacerda L, Bianco A, Prato M, Kostarelos K. Adv. Drug Deliv. Rev. 2006;58:1460. doi: 10.1016/j.addr.2006.09.015. [DOI] [PubMed] [Google Scholar]; Kostarelos K, Bianco A, Prato M. Nat. Nanotechnol. 2009;4:627. doi: 10.1038/nnano.2009.241. [DOI] [PubMed] [Google Scholar]; Prato M, Kostarelos K, Bianco A. Accounts Chem. Res. 2008;41:60. doi: 10.1021/ar700089b. [DOI] [PubMed] [Google Scholar]
- [34].Tasis D, Tagmatarchis N, Bianco A, Prato M. Chem. Rev. 2006;106:1105. doi: 10.1021/cr050569o. [DOI] [PubMed] [Google Scholar]
- [35].Krishna V, Singh A, Sharma P, Iwakuma N, Wang Q, Zhang Q, Knapik J, Jiang H, Grobmyer SR, Koopman B, Moudgil B. Small. 2010;6:2236. doi: 10.1002/smll.201000847. [DOI] [PubMed] [Google Scholar]
- [36].Burke A, Ding XF, Singh R, Kraft RA, Levi-Polyachenko N, Rylander MN, Szot C, Buchanan C, Whitney J, Fisher J, Hatcher HC, D’Agostino R, Kock ND, Ajayan PM, Carroll DL, Akman S, Torti FM, Torti SV. Proc. Natl. Acad. Sci. U. S. A. 2009;106:12897. doi: 10.1073/pnas.0905195106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Boczkowski J, Lanone S. Nanomedicine. 2007;2:407. doi: 10.2217/17435889.2.4.407. [DOI] [PubMed] [Google Scholar]; Kaiser JP, Roesslein M, Buerki-Thurnherr T, Wick P. Curr. Med. Chem. 2011;18:2115. doi: 10.2174/092986711795656171. [DOI] [PubMed] [Google Scholar]; Donaldson K, Murphy F, Schinwald A, Duffin R, Poland CA. Nanomedicine. 2011;6:143. doi: 10.2217/nnm.10.139. [DOI] [PubMed] [Google Scholar]
- [38].Hao R, Xing R, Xu Z, Hou Y, Gao S, Sun S. Adv Mater. 2010;22:2729. doi: 10.1002/adma.201000260. [DOI] [PubMed] [Google Scholar]
- [39].Sun C, Lee JSH, Zhang MQ. Adv. Drug Deliv. Rev. 2008;60:1252. doi: 10.1016/j.addr.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; Veiseh O, Gunn JW, Zhang M. Adv Drug Deliv Rev. 2010;62:284. doi: 10.1016/j.addr.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Stephen ZR, Kievit FM, Zhang M. Materials Today. 2011;14:274. doi: 10.1016/S1369-7021(11)70163-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Sun C, Du K, Fang C, Bhattarai N, Veiseh O, Kievit F, Stephen Z, Lee D, Ellenbogen RG, Ratner B, Zhang M. ACS Nano. 2010;4:2402. doi: 10.1021/nn100190v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Weissleder R, Stark DD, Engelstad BL, Bacon BR, Compton CC, White DL, Jacobs P, Lewis J. AJR Am. J. Roentgenol. 1989;152:167. doi: 10.2214/ajr.152.1.167. [DOI] [PubMed] [Google Scholar]
- [43].Lee JH, Huh YM, Jun YW, Seo JW, Jang JT, Song HT, Kim S, Cho EJ, Yoon HG, Suh JS, Cheon J. Nat. Med. 2007;13:95. doi: 10.1038/nm1467. [DOI] [PubMed] [Google Scholar]; Jun Y.-w., Huh Y-M, Choi J.-s., Lee J-H, Song H-T, KimKim S, Yoon K-S, Kim J-S, Shin J-S, Suh J, Cheon J. Am. Chem. Soc. 2005;127:5732. doi: 10.1021/ja0422155. [DOI] [PubMed] [Google Scholar]; Lu AH, Salabas EL, Schuth F. Angew Chem Int Ed Engl. 2007;46:1222. doi: 10.1002/anie.200602866. [DOI] [PubMed] [Google Scholar]
- [44].Branca RT, Cleveland ZI, Fubara B, Kumar CSSR, Maronpot RR, Leuschner C, Warren WS, Driehuys B. Proceedings of the National Academy of Sciences. 2011;107:3693. doi: 10.1073/pnas.1000386107. [DOI] [PMC free article] [PubMed] [Google Scholar]; Na HB, Song IC, Hyeon T. Adv. Mater. 2009;21:2133. [Google Scholar]
- [45].Kim T, Momin E, Choi J, Yuan K, Zaidi H, Kim J, Park M, Lee N, McMahon MT, Quinones-Hinojosa A, Bulte JWM, Hyeon T, Gilad AA. J. Am. Chem. Soc. 2011;133:2955. doi: 10.1021/ja1084095. [DOI] [PMC free article] [PubMed] [Google Scholar]; Gilad AA, Walczak P, McMahon MT, Bin H, Na, Lee JH, An K, Hyeon T, van Zijl PCM, Bulte JWM. Magn. Reson. Med. 2008;60:1. doi: 10.1002/mrm.21622. [DOI] [PMC free article] [PubMed] [Google Scholar]; Na HB, Lee JH, An KJ, Park YI, Park M, Lee IS, Nam DH, Kim ST, Kim SH, Kim SW, Lim KH, Kim KS, Kim SO, Hyeon T. Angew. Chem.-Int. Edit. 2007;46:5397. doi: 10.1002/anie.200604775. [DOI] [PubMed] [Google Scholar]; Yang H, Zhuang YM, Hu H, Du XX, Zhang CX, Shi XY, Wu HX, Yang SP. Adv. Funct. Mater. 2010;20:1733. [Google Scholar]
- [46].McDonald MA, Watkin KL. Acad. Radiol. 2006;13:421. doi: 10.1016/j.acra.2005.11.005. [DOI] [PubMed] [Google Scholar]; Fortin MA, Petoral RM, Soderlind F, Klasson A, Engstrom M, Veres T, Kall PO, Uvdal K. Nanotechnology. 2007;18 [Google Scholar]; Bridot JL, Faure AC, Laurent S, Riviere C, Billotey C, Hiba B, Janier M, Josserand V, Coll JL, Vander Elst L, Muller R, Roux S, Perriat P, Tillement O. J. Am. Chem. Soc. 2007;129:5076. doi: 10.1021/ja068356j. [DOI] [PubMed] [Google Scholar]
- [47].Senpan A, Caruthers SD, Rhee I, Mauro NA, Pan DPJ, Hu G, Scott MJ, Fuhrhop RW, Gaffney PJ, Wickline SA, Lanzat GM. Acs Nano. 2009;3:3917. doi: 10.1021/nn900819y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Jordan A, Scholz R, Wust P, Fahling H, Felix R. J. Magn. Magn. Mater. 1999;201:413. [Google Scholar]
- [49].Meek ST, Greathouse JA, Allendorf MD. Adv Mater. 2011;23:249. doi: 10.1002/adma.201002854. [DOI] [PubMed] [Google Scholar]
- [50].Taylor KML, Rieter WJ, Lin WB. J. Am. Chem. Soc. 2008;130:14358. doi: 10.1021/ja803777x. [DOI] [PubMed] [Google Scholar]; Taylor KML, Jin A, Lin WB. Angew. Chem.-Int. Edit. 2008;47:7722. doi: 10.1002/anie.200802911. [DOI] [PubMed] [Google Scholar]; Hatakeyama W, Sanchez TJ, Rowe MD, Serkova NJ, Liberatore MW, Boyes SG. ACS Appl. Mater. Interfaces. 2011;3:1502. doi: 10.1021/am200075q. [DOI] [PubMed] [Google Scholar]
- [51].Horcajada P, Chalati T, Serre C, Gillet B, Sebrie C, Baati T, Eubank JF, Heurtaux D, Clayette P, Kreuz C, Chang JS, Hwang YK, Marsaud V, Bories PN, Cynober L, Gil S, Ferey G, Couvreur P, Gref R. Nat. Mater. 2010;9:172. doi: 10.1038/nmat2608. [DOI] [PubMed] [Google Scholar]
- [52].Mueller U, Schubert M, Teich F, Puetter H, Schierle-Arndt K, Pastre J. J. Mater. Chem. 2006;16:626. [Google Scholar]
- [53].Wang F, Banerjee D, Liu YS, Chen XY, Liu XG. Analyst. 2010;135:1839. doi: 10.1039/c0an00144a. [DOI] [PubMed] [Google Scholar]
- [54].Yu XF, Sun ZB, Li M, Xiang Y, Wang QQ, Tang FF, Wu YL, Cao ZJ, Li WX. Biomaterials. 2010;31:8724. doi: 10.1016/j.biomaterials.2010.07.099. [DOI] [PubMed] [Google Scholar]
- [55].Xiong LQ, Chen ZG, Yu MX, Li FY, Liu C, Huang CH. Biomaterials. 2009;30:5592. doi: 10.1016/j.biomaterials.2009.06.015. [DOI] [PubMed] [Google Scholar]
- [56].Carpenter CM, Sun C, Pratx G, Rao R, Xing L. Med. Phys. 2010;37:4011. doi: 10.1118/1.3457332. [DOI] [PMC free article] [PubMed] [Google Scholar]; Pratx G, Carpenter CM, Sun C, Xing L. IEEE Trans. Med. Imaging. 2010;29:1992. doi: 10.1109/TMI.2010.2055883. [DOI] [PMC free article] [PubMed] [Google Scholar]; Pratx G, Carpenter CM, Sun C, Rao RP, Xing L. Opt. Lett. 2010;35:3345. doi: 10.1364/OL.35.003345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Sun C, Pratx G, Carpenter CM, Liu H, Cheng Z, Gambhir SS, Xing L. Adv. Mater. 2011;23:H195. doi: 10.1002/adma.201100919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Swierczewska M, Seulki L, Xiaoyuan C. Molecular Imaging. 2011;10:3. [PMC free article] [PubMed] [Google Scholar]
- [59].Townsend DW, Beyer T, Blodgett TM. Semin. Nucl. Med. 2003;33:193. doi: 10.1053/snuc.2003.127314. [DOI] [PubMed] [Google Scholar]; Blodgett TM, Meltzer CC, Townsend DW. Radiology. 2007;242:360. doi: 10.1148/radiol.2422051113. [DOI] [PubMed] [Google Scholar]; Townsend DW. Phys. Med. Biol. 2008;53:R1. doi: 10.1088/0031-9155/53/4/R01. [DOI] [PubMed] [Google Scholar]
- [60].Morales-Avila E, Ferro-Flores G, Ocampo-Garcia BE, De Leon-Rodriguez LM, Santos-Cuevas CL, Garcia-Becerra R, Medina LA, Gomez-Olivan L. Bioconjugate Chem. 2011;22:913. doi: 10.1021/bc100551s. [DOI] [PubMed] [Google Scholar]; Zhou M, Zhang R, Huang M, Lu W, Song S, Melancon MP, Tian M, Liang D, Li C. J. Am. Chem. Soc. 2010;132:15351. doi: 10.1021/ja106855m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Veiseh O, Sun C, Gunn J, Kohler N, Gabikian P, Lee D, Bhattarai N, Ellenbogen R, Sze R, Hallahan A, Olson J, Zhang M. Nano Lett. 2005;5:1003. doi: 10.1021/nl0502569. [DOI] [PubMed] [Google Scholar]
- [62].van Schooneveld MM, Cormode DP, Koole R, van Wijngaarden JT, Calcagno C, Skajaa T, Hilhorst J, t Hart DC, Fayad ZA, Mulder WJ, Meijerink A. Contrast Media Mol Imaging. 2010;5:231. doi: 10.1002/cmmi.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kievit FM, Zhang M. Acc Chem Res. 2011 doi: 10.1021/ar2000277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Jia G, Cao Z, Xue H, Xu Y, Jiang S. Langmuir. 2009 doi: 10.1021/la803737c. [DOI] [PubMed] [Google Scholar]; Otsuka H, Nagasaki Y, Kataoka K. Adv. Drug. Deliv. Rev. 2003;55:403. doi: 10.1016/s0169-409x(02)00226-0. [DOI] [PubMed] [Google Scholar]; Ma M, Liu WF, Hill PS, Bratlie KM, Siegwart DJ, Chin J, Park M, Guerreiro J, Anderson DG. Adv. Mater. 2011;23:H189. doi: 10.1002/adma.201100513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Fang C, Bhattarai N, Sun C, Zhang MQ. Small. 2009;5:1637. doi: 10.1002/smll.200801647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Shubayev VI, Pisanic TR, Jin SH. Adv. Drug Deliv. Rev. 2009;61:467. doi: 10.1016/j.addr.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; Gupta AK, Gupta M. Biomaterials. 2005;26:3995. doi: 10.1016/j.biomaterials.2004.10.012. [DOI] [PubMed] [Google Scholar]
- [67].Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Ipe BI, Bawendi MG, Frangioni JV. Nat. Biotechnol. 2007;25:1165. doi: 10.1038/nbt1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Jang SH, Wientjes MG, Lu D, Au JLS. Pharm. Res. 2003;20:1337. doi: 10.1023/a:1025785505977. [DOI] [PubMed] [Google Scholar]
- [69].Klibanov AL, Maruyama K, Torchilin VP, Huang L. FEBS Lett. 1990;268:235. doi: 10.1016/0014-5793(90)81016-h. [DOI] [PubMed] [Google Scholar]; Liu DF, Wu W, Ling JJ, Wen S, Gu N, Zhang XZ. Adv. Funct. Mater. 2011;21:1498. [Google Scholar]
- [70].Abbott NJ, Ronnback L, Hansson E. Nat. Rev. Neurosci. 2006;7:41. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- [71].Steeg PS, Camphausen KA, Smith QR. Nat Rev Cancer. 2011;11:352. doi: 10.1038/nrc3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Lockman PR, Mittapalli RK, Taskar KS, Rudraraju V, Gril B, Bohn KA, Adkins CE, Roberts A, Thorsheim HR, Gaasch JA, Huang S, Palmieri D, Steeg PS, Smith QR. Clin. Cancer Res. 2010;16:5664. doi: 10.1158/1078-0432.CCR-10-1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Kroll RA, Pagel MA, Muldoon LL, Roman-Goldstein S, Fiamengo SA, Neuwelt EA. Neurosurgery. 1998;43:879. doi: 10.1097/00006123-199810000-00090. [DOI] [PubMed] [Google Scholar]
- [74].Mykhaylyk O, Cherchenko A, Ilkin A, Dudchenko N, Ruditsa V, Novoseletz M, Zozulya Y. J. Magn. Magn. Mater. 2001;225:241. [Google Scholar]
- [75].Hynynen K, McDannold N, Vykhodtseva N, Raymond S, Weissleder R, Jolesz FA, Sheikov N. J Neurosurg. 2006;105:445. doi: 10.3171/jns.2006.105.3.445. [DOI] [PubMed] [Google Scholar]
- [76].Liu HL, Hua MY, Yang HW, Huang CY, Chu PC, Wu JS, Tseng IC, Wang JJ, Yen TC, Chen PY, Wei KC. Proc Natl Acad Sci U S A. 2010;107:15205. doi: 10.1073/pnas.1003388107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Prados MD, Schold SC, Fine HA, Jaeckle K, Hochberg F, Mechtler L, Fetell MR, Phuphanich S, Feun L, Janus TJ, Ford K, Graney W. Neuro-Oncology. 2003;5:96. doi: 10.1093/neuonc/5.2.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Campbell M, Kiang AS, Kenna PF, Kerskens C, Blau C, O’Dwyer L, Tivnan A, Kelly JA, Brankin B, Farrar GJ, Humphries P. J. Gene. Med. 2008;10:930. doi: 10.1002/jgm.1211. [DOI] [PubMed] [Google Scholar]
- [79].Bobo RH, Laske DW, Akbasak A, Morrison PF, Dedrick RL, Oldfield EH. Proc. Natl. Acad. Sci. U. S. A. 1994;91:2076. doi: 10.1073/pnas.91.6.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Vinchon-Petit S, Jarnet D, Paillard A, Benoit JP, Garcion E, Menei P. J. Neuro-Oncol. 2010;97:195. doi: 10.1007/s11060-009-0012-4. [DOI] [PubMed] [Google Scholar]; Allard E, Passirani C, Benoit JP. Biomaterials. 2009;30:2302. doi: 10.1016/j.biomaterials.2009.01.003. [DOI] [PubMed] [Google Scholar]
- [81].Hadjipanayis CG, Machaidze R, Kaluzova M, Wang L, Schuette AJ, Chen H, Wu X, Mao H. Cancer Res. 2010;70:6303. doi: 10.1158/0008-5472.CAN-10-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Bidros DS, Liu JK, Vogelbaum MA. Future Oncol. 2010;6:117. doi: 10.2217/fon.09.135. [DOI] [PubMed] [Google Scholar]
- [83].Voges J, Reszka R, Gossmann A, Dittmar C, Richter R, Garlip G, Kracht L, Coenen HH, Sturm V, Wienhard K, Heiss WD, Jacobs AH. Ann. Neurol. 2003;54:479. doi: 10.1002/ana.10688. [DOI] [PubMed] [Google Scholar]
- [84].Prabhu SS, Broaddus WC, Gillies GT, Loudon WG, Chen ZJ, Smith B. Surg. Neurol. 1998;50:367. doi: 10.1016/s0090-3019(97)00361-3. [DOI] [PubMed] [Google Scholar]
- [85].Huynh GH, Ozawa T, Deen DF, Tihan T, Szoka FC. Brain Res. 2007;1128:181. doi: 10.1016/j.brainres.2006.10.041. [DOI] [PubMed] [Google Scholar]; Motion JPM, Huynh GH, Szoka FC, Siegel RA. Pharm. Res. 2011;28:472. doi: 10.1007/s11095-010-0296-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Wang P, Olbricht WL. Ann. Biomed. Eng. 2010;38:2512. doi: 10.1007/s10439-010-0022-6. [DOI] [PubMed] [Google Scholar]
- [87].Kumar M, Medarova Z, Pantazopoulos P, Dai GP, Moore A. Magn. Reson. Med. 2010;63:617. doi: 10.1002/mrm.22216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Smith MW, Gumbleton M. J. Drug Target. 2006;14:191. doi: 10.1080/10611860600650086. [DOI] [PubMed] [Google Scholar]
- [89].van Rooy I, Mastrobattista E, Storm G, Hennink WE, Schiffelers RM. J. Control. Release. 2011;150:30. doi: 10.1016/j.jconrel.2010.11.014. [DOI] [PubMed] [Google Scholar]
- [90].Bickel U, Yoshikawa T, Pardridge WM. Adv. Drug Deliv. Rev. 2001;46:247. doi: 10.1016/s0169-409x(00)00139-3. [DOI] [PubMed] [Google Scholar]
- [91].Huwyler J, Wu D, Pardridge WM. Proc Natl Acad Sci U S A. 1996;93:14164. doi: 10.1073/pnas.93.24.14164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Cerletti A, Drewe J, Fricker G, Eberle AN, Huwyler J. J Drug Target. 2000;8:435. doi: 10.3109/10611860008997919. [DOI] [PubMed] [Google Scholar]
- [93].Herz J, Bock HH. Annu. Rev. Biochem. 2002;71:405. doi: 10.1146/annurev.biochem.71.110601.135342. [DOI] [PubMed] [Google Scholar]; Talukder MJR, Takeuchi T, Harada E. J. Vet. Med. Sci. 2003;65:957. doi: 10.1292/jvms.65.957. [DOI] [PubMed] [Google Scholar]
- [94].Xie H, Zhu YH, Jiang WL, Zhou Q, Yang H, Gu N, Zhang Y, Xu HB, Xu HB, Yang XL. Biomaterials. 2011;32:495. doi: 10.1016/j.biomaterials.2010.09.024. [DOI] [PubMed] [Google Scholar]
- [95].Huang RQ, Ke WL, Liu Y, Jiang C, Pei YY. Biomaterials. 2008;29:238. doi: 10.1016/j.biomaterials.2007.09.024. [DOI] [PubMed] [Google Scholar]
- [96].Veiseh O, Sun C, Fang C, Bhattarai N, Gunn J, Kievit F, Du K, Pullar B, Lee D, Ellenbogen RG, Olson J, Zhang M. Cancer Res. 2009;69:6200. doi: 10.1158/0008-5472.CAN-09-1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Kesavan K, Ratliff J, Johnson EW, Dahlberg W, Asara JM, Misra P, Frangioni JV, Jacoby DB. Journal of Biological Chemistry. 2010;285:4366. doi: 10.1074/jbc.M109.066092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Hatton BA, Villavicencio EH, Tsuchiya KD, Pritchard JI, Ditzler S, Pullar B, Hansen S, Knoblaugh SE, Lee D, Eberhart CG, Hallahan AR, Olson JM. Cancer Res. 2008;68:1768. doi: 10.1158/0008-5472.CAN-07-5092. [DOI] [PubMed] [Google Scholar]
- [99].Salinas S, Schiavo G, Kremer EJ. Nat. Rev. Microbiol. 2010;8:645. doi: 10.1038/nrmicro2395. [DOI] [PubMed] [Google Scholar]
- [100].Veiseh O, Kievit FM, Mok H, Ayesh J, Clark C, Fang C, Leung M, Arami H, Park JO, Zhang M. Biomaterials. 2011;32:5717. doi: 10.1016/j.biomaterials.2011.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Li WJ, Nicol F, Szoka FC. Adv. Drug Deliv. Rev. 2004;56:967. doi: 10.1016/j.addr.2003.10.041. [DOI] [PubMed] [Google Scholar]; Liu BR, Huang YW, Winiarz JG, Chiang HJ, Lee HJ. Biomaterials. 2011;32:3520. doi: 10.1016/j.biomaterials.2011.01.041. [DOI] [PubMed] [Google Scholar]; Schmidt N, Mishra A, Lai GH, Wong GCL. FEBS Lett. 2010;584:1806. doi: 10.1016/j.febslet.2009.11.046. [DOI] [PubMed] [Google Scholar]
- [102].Doherty GJ, McMahon HT. Annu. Rev. Biochem. 2009;78:857. doi: 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]; Iversen T-G, Skotland T, Sandvig K. Nano Today. 2011;6:176. [Google Scholar]
- [103].Hillaireau H, Couvreur P. Cell. Mol. Life Sci. 2009;66:2873. doi: 10.1007/s00018-009-0053-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Kaneda MM, Sasaki Y, Lanza GM, Milbrandt J, Wickline SA. Biomaterials. 2010;31:3079. doi: 10.1016/j.biomaterials.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Ferrari A, Pellegrini V, Arcangeli C, Fittipaldi A, Giacca M, Beltram F. Mol. Ther. 2003;8:284. doi: 10.1016/s1525-0016(03)00122-9. [DOI] [PubMed] [Google Scholar]
- [106].Dausend J, Musyanovych A, Dass M, Walther P, Schrezenmeier H, Landfester K, Mailander V. Macromol. Biosci. 2008;8:1135. doi: 10.1002/mabi.200800123. [DOI] [PubMed] [Google Scholar]
- [107].Jiang W, Kim BYS, Rutka JT, Chan WCW. Nat. Nanotechnol. 2008;3:145. doi: 10.1038/nnano.2008.30. [DOI] [PubMed] [Google Scholar]; Jin H, Heller DA, Sharma R, Strano MS. Acs Nano. 2009;3:149. doi: 10.1021/nn800532m. [DOI] [PubMed] [Google Scholar]; Chithrani BD, Ghazani AA, Chan WCW. Nano Lett. 2006;6:662. doi: 10.1021/nl052396o. [DOI] [PubMed] [Google Scholar]
- [108].Chithrani BD, Chan WCW. Nano Lett. 2007;7:1542. doi: 10.1021/nl070363y. [DOI] [PubMed] [Google Scholar]
- [109].Gao H, Shi W, Freund LB. Proc. Natl. Acad. Sci. U. S. A. 2005;102:9469. doi: 10.1073/pnas.0503879102. [DOI] [PMC free article] [PubMed] [Google Scholar]; Bao G, Bao XR. Proc. Natl. Acad. Sci. U. S. A. 2005;102:9997. doi: 10.1073/pnas.0504555102. [DOI] [PMC free article] [PubMed] [Google Scholar]; Deserno M, Gelbart WM. The Journal of Physical Chemistry B. 2002;106:5543. [Google Scholar]
- [110].Mintzer MA, Simanek EE. Chem Rev. 2009;109:259. doi: 10.1021/cr800409e. [DOI] [PubMed] [Google Scholar]
- [111].Wagner E. Adv. Drug Deliv. Rev. 1999;38:279. doi: 10.1016/s0169-409x(99)00033-2. [DOI] [PubMed] [Google Scholar]
- [112].Pack DW, Hoffman AS, Pun S, Stayton PS. Nat Rev Drug Discov. 2005;4:581. doi: 10.1038/nrd1775. [DOI] [PubMed] [Google Scholar]
- [113].Rajendran L, Knolker HJ, Simons K. Nat. Rev. Drug Discov. 2010;9:29. doi: 10.1038/nrd2897. [DOI] [PubMed] [Google Scholar]
- [114].Godbey WT, Wu KK, Mikos AG. Proc Natl Acad Sci U S A. 1999;96:5177. doi: 10.1073/pnas.96.9.5177. [DOI] [PMC free article] [PubMed] [Google Scholar]; Clamme JP, Krishnamoorthy G, Mely Y. Biochim Biophys Acta. 2003;1617:52. doi: 10.1016/j.bbamem.2003.09.002. [DOI] [PubMed] [Google Scholar]
- [115].Sebestyen MG, Ludtke JJ, Bassik MC, Zhang GF, Budker V, Lukhtanov EA, Hagstrom JE, Wolff JA. Nature Biotechnology. 1998;16:80. doi: 10.1038/nbt0198-80. [DOI] [PubMed] [Google Scholar]; Dang CV, Lee WM. Mol Cell Biol. 1988;8:4048. doi: 10.1128/mcb.8.10.4048. [DOI] [PMC free article] [PubMed] [Google Scholar]; Gharakhanian E, Takahashi J, Kasamatsu H. Virology. 1987;157:440. doi: 10.1016/0042-6822(87)90286-8. [DOI] [PubMed] [Google Scholar]; Kleinschmidt JA, Seiter A. Embo J. 1988;7:1605. doi: 10.1002/j.1460-2075.1988.tb02986.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lyons RH, Ferguson BQ, Rosenberg M. Mol Cell Biol. 1987;7:2451. doi: 10.1128/mcb.7.7.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kaneda Y, Iwai K, Uchida T. Science. 1989;243:375. doi: 10.1126/science.2911748. [DOI] [PubMed] [Google Scholar]
- [116].Ng CP, Goodman TT, Park IK, Pun SH. Biomaterials. 2009;30:951. doi: 10.1016/j.biomaterials.2008.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Jordan MA, Wilson L. Nat. Rev. Cancer. 2004;4:253. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- [118].Kang BH, Plescia J, Dohi T, Rosa J, Doxsey SJ, Altieri DC. Cell. 2007;131:257. doi: 10.1016/j.cell.2007.08.028. [DOI] [PubMed] [Google Scholar]; Kang BH, Plescia J, Song HY, Meli M, Colombo G, Beebe K, Scroggins B, Neckers L, Altieri DC. J. Clin. Invest. 2009;119:454. doi: 10.1172/JCI37613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Torchilin VP. Adv. Drug Deliv. Rev. 2006;58:1532. doi: 10.1016/j.addr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- [120].Bailon P, Won CY. Expert Opin. Drug Deliv. 2009;6:1. doi: 10.1517/17425240802650568. [DOI] [PubMed] [Google Scholar]; Maruyama K. Adv. Drug Deliv. Rev. 2011;63:161. doi: 10.1016/j.addr.2010.09.003. [DOI] [PubMed] [Google Scholar]
- [121].O’Brien ME, Wigler N, Inbar M, Rosso R, Grischke E, Santoro A, Catane R, Kieback DG, Tomczak P, Ackland SP, Orlandi F, Mellars L, Alland L, Tendler C. Ann Oncol. 2004;15:440. doi: 10.1093/annonc/mdh097. [DOI] [PubMed] [Google Scholar]
- [122].Primeau AJ, Rendon A, Hedley D, Lilge L, Tannock IF. Clin Cancer Res. 2005;11:8782. doi: 10.1158/1078-0432.CCR-05-1664. [DOI] [PubMed] [Google Scholar]; Minchinton AI, Tannock IF. Nat Rev Cancer. 2006;6:583. doi: 10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- [123].Lukyanov AN, Elbayoumi TA, Chakilam AR, Torchilin VP. J Control Release. 2004;100:135. doi: 10.1016/j.jconrel.2004.08.007. [DOI] [PubMed] [Google Scholar]
- [124].Nakatsura T, Yoshitake Y, Senju S, Monji M, Komori H, Motomura Y, Hosaka S, Beppu T, Ishiko T, Kamohara H, Ashihara H, Katagiri T, Furukawa Y, Fujiyama S, Ogawa M, Nakamura Y, Nishimura Y. Biochem. Biophys. Res. Commun. 2003;306:16. doi: 10.1016/s0006-291x(03)00908-2. [DOI] [PubMed] [Google Scholar]
- [125].Park JO, Stephen Z, Sun C, Veiseh O, Kievit FM, Fang C, Leung M, Mok H, Zhang MQ. Molecular Imaging. 2011;10:69. [PMC free article] [PubMed] [Google Scholar]
- [126].Arruebo M, Valladares M, Gonzalez-Fernandez A. J. Nanomater. 2009 [Google Scholar]
- [127].Veiseh M, Gabikian P, Bahrami SB, Veiseh O, Zhang M, Hackman RC, Ravanpay AC, Stroud MR, Kusuma Y, Hansen SJ, Kwok D, Munoz NM, Sze RW, Grady WM, Greenberg NM, Ellenbogen RG, Olson JM. Cancer Res. 2007;67:6882. doi: 10.1158/0008-5472.CAN-06-3948. [DOI] [PubMed] [Google Scholar]
- [128].Lee MJE, Veiseh O, Bhattarai N, Sun C, Hansen SJ, Ditzler S, Knoblaugh S, Lee D, Ellenbogen R, Zhang MQ, Olson JM. Plos One. 2010;5:8. doi: 10.1371/journal.pone.0009536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Sun C, Veiseh O, Gunn J, Fang C, Hansen S, Lee D, Sze R, Ellenbogen RG, Olson J, Zhang M. Small. 2008;4:372. doi: 10.1002/smll.200700784. [DOI] [PMC free article] [PubMed] [Google Scholar]; Meng XX, Wan JQ, Jing M, Zhao SG, Cai W, Liu EZ. Acta Pharmacol Sin. 2007;28:2019. doi: 10.1111/j.1745-7254.2007.00661.x. [DOI] [PubMed] [Google Scholar]
- [130].Sun C, Sze R, Zhang M. J. Biomed. Mater. Res. A. 2006;78:550. doi: 10.1002/jbm.a.30781. [DOI] [PubMed] [Google Scholar]
- [131].Meier R, Henning TD, Boddington S, Tavri S, Arora S, Piontek G, Rudelius M, Corot C, Daldrup-Link HE. Radiology. 2010;255:527. doi: 10.1148/radiol.10090050. [DOI] [PubMed] [Google Scholar]
- [132].Gunn J, Park SI, Veiseh O, Press OW, Zhang MQ. Mol. Biosyst. 2011;7:742. doi: 10.1039/c005154c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Devaraj NK, Upadhyay R, Hatin JB, Hilderbrand SA, Weissleder R. Angew. Chem.-Int. Edit. 2009;48:7013. doi: 10.1002/anie.200903233. [DOI] [PMC free article] [PubMed] [Google Scholar]; Devaraj NK, Weissleder R, Hilderbrand SA. Bioconjugate Chem. 2008;19:2297. doi: 10.1021/bc8004446. [DOI] [PMC free article] [PubMed] [Google Scholar]; Blackman ML, Royzen M, Fox JM. J. Am. Chem. Soc. 2008;130:13518. doi: 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]; Rossin R, Renart Verkerk P, van den Bosch SM, Vulders RCM, Verel I, Lub J, Robillard MS. Angewandte Chemie International Edition. 2010;49:3375. doi: 10.1002/anie.200906294. [DOI] [PubMed] [Google Scholar]
- [134].Haun JB, Devaraj NK, Hilderbrand SA, Lee H, Weissleder R. Nat. Nanotechnol. 2010;5:660. doi: 10.1038/nnano.2010.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Kirpotin DB, Drummond DC, Shao Y, Shalaby MR, Hong K, Nielsen UB, Marks JD, Benz CC, Park JW. Cancer Res. 2006;66:6732. doi: 10.1158/0008-5472.CAN-05-4199. [DOI] [PubMed] [Google Scholar]; Bartlett DW, Su H, Hildebrandt IJ, Weber WA, Davis ME. Proc Natl Acad Sci U S A. 2007;104:15549. doi: 10.1073/pnas.0707461104. [DOI] [PMC free article] [PubMed] [Google Scholar]; Choi CH, Alabi CA, Webster P, Davis ME. Proc Natl Acad Sci U S A. 2010;107:1235. doi: 10.1073/pnas.0914140107. [DOI] [PMC free article] [PubMed] [Google Scholar]; Pirollo KF, Chang EH. Trends Biotechnol. 2008;26:552. doi: 10.1016/j.tibtech.2008.06.007. [DOI] [PubMed] [Google Scholar]
- [136].Wang X, Li J, Wang Y, Cho KJ, Kim G, Gjyrezi A, Koenig L, Giannakakou P, Shin HJ, Tighiouart M, Nie S, Chen ZG, Shin DM. ACS Nano. 2009;3:3165. doi: 10.1021/nn900649v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Kievit FM, Veiseh O, Fang C, Bhattarai N, Lee D, Ellenbogen RG, Zhang M. ACS Nano. 2010;4:4587. doi: 10.1021/nn1008512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Veiseh O, Kievit FM, Gunn JW, Ratner BD, Zhang M. Biomaterials. 2009;30:649. doi: 10.1016/j.biomaterials.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Veiseh O, Kievit FM, Fang C, Mu N, Jana S, Leung MC, Mok H, Ellenbogen RG, Park JO, Zhang M. Biomaterials. 2010;31:8032. doi: 10.1016/j.biomaterials.2010.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Fang C, Veiseh O, Kievit F, Bhattarai N, Wang F, Stephen Z, Li C, Lee D, Ellenbogen RG, Zhang M. Nanomedicine (Lond) 2010;5:1357. doi: 10.2217/nnm.10.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Kim B, Han G, Toley BJ, Kim CK, Rotello VM, Forbes NS. Nat. Nanotechnol. 2010;5:465. doi: 10.1038/nnano.2010.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Mok H, Park JW, Park TG. Bioconjugate Chem. 2008;19:797. doi: 10.1021/bc700464m. [DOI] [PubMed] [Google Scholar]; Zhou ZX, Shen YQ, Tang JB, Fan MH, Van Kirk EA, Murdoch WJ, Radosz M. Adv. Funct. Mater. 2009;19:3580. [Google Scholar]; Xu PS, Van Kirk EA, Zhan YH, Murdoch WJ, Radosz M, Shen YQ. Angew. Chem.-Int. Edit. 2007;46:4999. doi: 10.1002/anie.200605254. [DOI] [PubMed] [Google Scholar]
- [143].Mok H, Veiseh O, Fang C, Kievit FM, Wang FY, Park JO, Zhang M. Mol. Pharm. 2010 doi: 10.1021/mp100221h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Lee ES, Gao ZG, Kim D, Park K, Kwon IC, Bae YH. Journal of Controlled Release. 2008;129:228. doi: 10.1016/j.jconrel.2008.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Jiang T, Olson ES, Nguyen QT, Roy M, Jennings PA, Tsien RY. Proc Natl Acad Sci U S A. 2004;101:17867. doi: 10.1073/pnas.0408191101. [DOI] [PMC free article] [PubMed] [Google Scholar]; Aguilera TA, Olson ES, Timmers MM, Jiang T, Tsien RY. Integr. Biol. 2009;1:371. doi: 10.1039/b904878b. [DOI] [PMC free article] [PubMed] [Google Scholar]; Olson ES, Aguilera TA, Jiang T, Ellies LG, Nguyen QT, Wong EH, Gross LA, Tsien RY. Integr Biol (Camb) 2009;1:382. doi: 10.1039/b904890a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Nguyen QT, Olson ES, Aguilera TA, Jiang T, Scadeng M, Ellies LG, Tsien RY. Proc. Natl. Acad. Sci. U. S. A. 2010;107:4317. doi: 10.1073/pnas.0910261107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Grainger SJ, Serna JV, Sunny S, Zhou Y, Deng CX, El-Sayed MEH. Mol. Pharm. 2010;7:2006. doi: 10.1021/mp100280b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Grantab R, Sivananthan S, Tannock IF. Cancer Res. 2006;66:1033. doi: 10.1158/0008-5472.CAN-05-3077. [DOI] [PubMed] [Google Scholar]
- [149].Foy SP, Manthe RL, Foy ST, Dimitrijevic S, Krishnamurthy N, Labhasetwar V. ACS Nano. 2010;4:5217. doi: 10.1021/nn101427t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Pouponneau P, Leroux J-C, Soulez G, Gaboury L, Martel S. Biomaterials. 2011;32:3481. doi: 10.1016/j.biomaterials.2010.12.059. [DOI] [PubMed] [Google Scholar]; Mathieu JB, Martel S. Biomed Microdevices. 2007;9:801. doi: 10.1007/s10544-007-9092-0. [DOI] [PubMed] [Google Scholar]; Mathieu JB, Martel S. Magn Reson Med. 2010;63:1336. doi: 10.1002/mrm.22279. [DOI] [PubMed] [Google Scholar]
- [151].Bulte JWM, Douglas T, Witwer B, Zhang SC, Strable E, Lewis BK, Zywicke H, Miller B, van Gelderen P, Moskowitz BM, Duncan ID, Frank JA. Nat. Biotechnol. 2001;19:1141. doi: 10.1038/nbt1201-1141. [DOI] [PubMed] [Google Scholar]
- [152].Dai L-J, Moniri MR, Zeng Z-R, Zhou JX, Rayat J, Warnock GL. Cancer Lett. 2011;305:8. doi: 10.1016/j.canlet.2011.02.012. [DOI] [PubMed] [Google Scholar]
- [153].El-Haibi CP, Karnoub AE. J. Mammary Gland Biol. Neoplasia. 2010;15:399. doi: 10.1007/s10911-010-9196-7. [DOI] [PubMed] [Google Scholar]
- [154].Hu YL, Fu YH, Tabata Y, Gao JQ. J. Control. Release. 2010;147:154. doi: 10.1016/j.jconrel.2010.05.015. [DOI] [PubMed] [Google Scholar]
- [155].Stagg J, Lejeune L, Paquin A, Galipeau J. Hum. Gene Ther. 2004;15:597. doi: 10.1089/104303404323142042. [DOI] [PubMed] [Google Scholar]; Chen X, Lin X, Zhao J, Shi W, Zhang H, Wang Y, Kan B, Du L, Wang B, Wei Y, Liu Y, Zhao X. Mol. Ther. 2008;16:749. doi: 10.1038/mt.2008.3. [DOI] [PubMed] [Google Scholar]; Nakamizo A, Marini F, Amano T, Khan A, Studeny M, Gumin J, Chen J, Hentschel S, Vecil G, Dembinski J, Andreeff M, Lang FF. Cancer Res. 2005;65:3307. doi: 10.1158/0008-5472.CAN-04-1874. [DOI] [PubMed] [Google Scholar]; Studeny M, Marini FC, Champlin RE, Zompetta C, Fidler IJ, Andreeff M. Cancer Res. 2002;62:3603. [PubMed] [Google Scholar]
- [156].Grisendi G, Bussolari R, Cafarelli L, Petak I, Rasini V, Veronesi E, De Santis G, Spano C, Tagliazzucchi M, Barti-Juhasz H, Scarabelli L, Bambi F, Frassoldati A, Rossi G, Casali C, Morandi U, Horwitz EM, Paolucci P, Conte P, Dominici M. Cancer Res. 2010;70:3718. doi: 10.1158/0008-5472.CAN-09-1865. [DOI] [PubMed] [Google Scholar]; Loebinger MR, Eddaoudi A, Davies D, Janes SM. Cancer Res. 2009;69:4134. doi: 10.1158/0008-5472.CAN-08-4698. [DOI] [PMC free article] [PubMed] [Google Scholar]; Sasportas LS, Kasmieh R, Wakimoto H, Hingtgen S, van de Water J, Mohapatra G, Figueiredo JL, Martuza RL, Weissleder R, Shah K. Proc. Natl. Acad. Sci. U. S. A. 2009;106:4822. doi: 10.1073/pnas.0806647106. [DOI] [PMC free article] [PubMed] [Google Scholar]; Mohr A, Albarenque SM, Deedigan L, Yu R, Reidy M, Fulda S, Zwacka RM. Stem Cells. 2010;28:2109. doi: 10.1002/stem.533. [DOI] [PubMed] [Google Scholar]; Yang BJ, Wu X, Mao Y, Bao WM, Gao L, Zhou P, Xie R, Zhou LF, Zhu JH. Neurosurgery. 2009;65:610. doi: 10.1227/01.NEU.0000350227.61132.A7. [DOI] [PubMed] [Google Scholar]; Luetzkendorf J, Mueller LP, Mueller T, Caysa H, Nerger K, Schmoll HJ. J. Cell. Mol. Med. 2010;14:2292. doi: 10.1111/j.1582-4934.2009.00794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; Menon LG, Kelly K, Yang HW, Kim SK, Black PM, Carroll RS. Stem Cells. 2009;27:2320. doi: 10.1002/stem.136. [DOI] [PubMed] [Google Scholar]; Choi SA, Hwang SK, Wang KC, Cho BK, Phi JH, Lee JY, Jung HW, Lee DH, Kim SK. Neuro-Oncology. 2011;13:61. doi: 10.1093/neuonc/noq147. [DOI] [PMC free article] [PubMed] [Google Scholar]; Loebinger MR, Sage EK, Davies D, Janes SM. Br. J. Cancer. 2010;103:1692. doi: 10.1038/sj.bjc.6605952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Stoff-Khalili MA, Rivera AA, Mathis JM, Banerjee NS, Moon AS, Hess A, Rocconi RP, Numnum TM, Everts M, Chow LT, Douglas JT, Siegal GP, Zhu ZB, Bender HG, Dall P, Stoff A, Pereboeva L, Curiel DT. Breast Cancer Res. Treat. 2007;105:157. doi: 10.1007/s10549-006-9449-8. [DOI] [PubMed] [Google Scholar]; Mader EK, Maeyama Y, Lin Y, Butler GW, Russell HM, Galanis E, Russell SJ, Dietz AB, Peng KW. Clin. Cancer Res. 2009;15:7246. doi: 10.1158/1078-0432.CCR-09-1292. [DOI] [PMC free article] [PubMed] [Google Scholar]; Komarova S, Kawakami Y, Stoff-Khalili MA, Curiel DT, Pereboeva L. Mol. Cancer Ther. 2006;5:755. doi: 10.1158/1535-7163.MCT-05-0334. [DOI] [PubMed] [Google Scholar]
- [158].Kucerova L, Matuskova M, Pastorakova A, Tyciakova S, Jakubikova J, Bohovic R, Altanerova V, Altaner C. J. Gene. Med. 2008;10:1071. doi: 10.1002/jgm.1239. [DOI] [PubMed] [Google Scholar]; Matuskova M, Hlubinova K, Pastorakova A, Hunakova L, Altanerova V, Altaner C, Kucerova L. Cancer Lett. 2010;290:58. doi: 10.1016/j.canlet.2009.08.028. [DOI] [PubMed] [Google Scholar]; Amano S, Li SY, Gu CY, Gao Y, Koizumi S, Yamamoto S, Terakawa S, Namba H. Int. J. Oncol. 2009;35:1265. doi: 10.3892/ijo_00000443. [DOI] [PubMed] [Google Scholar]
- [159].Frank RT, Najbauer J, Aboody KS. Stem Cells. 2010;28:2084. doi: 10.1002/stem.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Roger M, Clavreul A, Venier-Julienne MC, Passirani C, Montero-Menei C, Menei P. Biomaterials. 2011;32:2106. doi: 10.1016/j.biomaterials.2010.11.056. [DOI] [PubMed] [Google Scholar]
- [161].Roger M, Clavreul A, Venier-Julienne MC, Passirani C, Sindji L, Schiller P, Montero-Menei C, Menei P. Biomaterials. 2010;31:8393. doi: 10.1016/j.biomaterials.2010.07.048. [DOI] [PubMed] [Google Scholar]; Tang C, Russell PJ, Martiniello-Wilks R, Rasko JEJ, Khatri A. Stem Cells. 2010;28:1686. doi: 10.1002/stem.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Rosen AB, Kelly DJ, Schuldt AJT, Lu J, Potapova IA, Doronin SV, Robichaud KJ, Robinson RB, Rosen MR, Brink PR, Gaudette GR, Cohen IS. Stem Cells. 2007;25:2128. doi: 10.1634/stemcells.2006-0722. [DOI] [PubMed] [Google Scholar]; Wu X, Hu J, Zhou LF, Mao Y, Yang B, Gao L, Xie R, Xu F, Zhang D, Liu J, Zhu JH. J. Neurosurg. 2008;108:320. doi: 10.3171/JNS/2008/108/2/0320. [DOI] [PubMed] [Google Scholar]
- [163].Srinivas M, Aarntzen E, Bulte JWM, Oyen WJ, Heerschap A, de Vries IJM, Figdor CG. Adv. Drug Deliv. Rev. 2010;62:1080. doi: 10.1016/j.addr.2010.08.009. [DOI] [PubMed] [Google Scholar]
- [164].Loebinger MR, Kyrtatos PG, Turmaine M, Price AN, Pankhurst Q, Lythgoe MF, Janes SM. Cancer Res. 2009;69:8862. doi: 10.1158/0008-5472.CAN-09-1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Aboody KS, Brown A, Rainov NG, Bower KA, Liu S, Yang W, Small JE, Herrlinger U, Ourednik V, Black PM, Breakefield XO, Snyder EY. Proc Natl Acad Sci U S A. 2000;97:12846. doi: 10.1073/pnas.97.23.12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Rachakatla RS, Balivada S, Seo GM, Myers CB, Wang HW, Samarakoon TN, Dani R, Pyle M, Kroh FO, Walker B, Leaym XX, Koper OB, Chikan V, Bossmann SH, Tamura M, Troyer DL. ACS Nano. 2010;4:7093. doi: 10.1021/nn100870z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Fahmy TM, Fong PM, Park J, Constable T, Saltzman WM. Aaps Journal. 2007;9:E171. doi: 10.1208/aapsj0902019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Schreiber RD, Old LJ, Smyth MJ. Science. 2011;331:1565. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]; Kim R, Emi M, Tanabe K, Arihiro K. Cancer Res. 2006;66:5527. doi: 10.1158/0008-5472.CAN-05-4128. [DOI] [PubMed] [Google Scholar]
- [169].Disis ML, Bernhard H, Jaffee EM. Lancet. 2009;373:673. doi: 10.1016/S0140-6736(09)60404-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME, Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, Zheng ZL, Nahvi A, de Vries CR, Rogers-Freezer LJ, Mavroukakis SA, Rosenberg SA. Nat. Rev. Cancer. Science. 2008;2006;8314:299, 126. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, Robinson MR, Raffeld M, Duray P, Seipp CA, Rogers-Freezer L, Morton KE, Mavroukakis SA, White DE, Rosenberg SA. Science. 2002;298:850. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]; Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, Greenberg PD. Proc. Natl. Acad. Sci. U. S. A. 2002;99:16168. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Kennedy L, Bear A, Young J, Lewinski N, Kim J, Foster A, Drezek R. Nanoscale Research Letters. 2011;6:283. doi: 10.1186/1556-276X-6-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Bernhard H, Neudorfer J, Gebhard K, Conrad H, Hermann C, Nahrig J, Fend F, Weber W, Busch DH, Peschel C. Cancer Immunol. Immunother. 2008;57:271. doi: 10.1007/s00262-007-0355-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Kircher MF, Allport JR, Graves EE, Love V, Josephson L, Lichtman AH, Weissleder R. Cancer Res. 2003;63:6838. [PubMed] [Google Scholar]; Gunn J, Wallen H, Veiseh O, Sun C, Fang C, Cao JH, Yee C, Zhang MQ. Small. 2008;4:712. doi: 10.1002/smll.200701103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Pittet MJ, Swirski FK, Reynolds F, Josephson L, Weissleder R. Nat. Protoc. 2006;1:73. doi: 10.1038/nprot.2006.11. [DOI] [PubMed] [Google Scholar]
- [175].Smirnov P, Poirier-Quinot M, Wilhelm C, Lavergne E, Ginefri JC, Combadiere B, Clement O, Darrasse L, Gazeau F. Magn. Reson. Med. 2008;60:1292. doi: 10.1002/mrm.21812. [DOI] [PubMed] [Google Scholar]
- [176].Stephan MT, Moon JJ, Um SH, Bershteyn A, Irvine DJ. Nat Med. 2010;16:1035. doi: 10.1038/nm.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Figdor CG, de Vries IJM, Lesterhuis WJ, Melief CJM. Nat. Med. 2004;10:475. doi: 10.1038/nm1039. [DOI] [PubMed] [Google Scholar]
- [178].Verdijk P, Aarntzen E, Lesterhuis WJ, Boullart ACI, Kok E, van Rossum MM, Strijk S, Eijckeler F, Bonenkamp JJ, Jacobs JFM, Blokx W, van Krieken J, Joosten I, Boerman OC, Oyen WJG, Adema G, Punt CJA, Figdor CG, de Vries IJM. Clin. Cancer Res. 2009;15:2531. doi: 10.1158/1078-0432.CCR-08-2729. [DOI] [PubMed] [Google Scholar]
- [179].Nestle FO, Banchereau J, Hart D. Nat. Med. 2001;7:761. doi: 10.1038/89863. [DOI] [PubMed] [Google Scholar]
- [180].Baumjohann D, Hess A, Budinsky L, Brune K, Schuler G, Lutz MB. Eur. J. Immunol. 2006;36:2544. doi: 10.1002/eji.200535742. [DOI] [PubMed] [Google Scholar]; Verdijk P, Scheenen TWJ, Lesterhuis WJ, Gambarota G, Veltien AA, Walczak P, Scharenborg NM, Bulte JWM, Punt CJA, Heerschap A, Figdorl CG, de Vries IJM. Int. J. Cancer. 2007;120:978. doi: 10.1002/ijc.22385. [DOI] [PubMed] [Google Scholar]; Schimmelpfennig CH, Schulz S, Arber C, Baker J, Tarner I, McBride J, Contag CH, Negrin RS. Am. J. Pathol. 2005;167:1321. doi: 10.1016/S0002-9440(10)61219-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; Olasz EB, Lang LX, Seidel J, Green MV, Eckelman WC, Katz SI. J. Immunol. Methods. 2002;260:137. doi: 10.1016/s0022-1759(01)00528-2. [DOI] [PubMed] [Google Scholar]
- [181].de Vries IJM, Lesterhuis WJ, Barentsz JO, Verdijk P, van Krieken JH, Boerman OC, Oyen WJG, Bonenkamp JJ, Boezeman JB, Adema GJ, Bulte JWM, Scheenen TWJ, Punt CJA, Heerschap A, Figdor CG. Nat. Biotechnol. 2005;23:1407. doi: 10.1038/nbt1154. [DOI] [PubMed] [Google Scholar]
- [182].Tacken PJ, de Vries IJM, Torensma R, Figdor CG. Nat. Rev. Immunol. 2007;7:790. doi: 10.1038/nri2173. [DOI] [PubMed] [Google Scholar]
- [183].Long CM, van Laarhoven HWM, Bulte JWM, Levitsky HI. Cancer Res. 2009;69:3180. doi: 10.1158/0008-5472.CAN-08-3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Cruz LJ, Tacken PJ, Bonetto F, Buschow SI, Croes HJ, Wijers M, de Vries IJ, Figdor CG. Mol. Pharm. 2011;8:520. doi: 10.1021/mp100356k. [DOI] [PubMed] [Google Scholar]
- [185].Whiteside TL. Oncogene. 2008;27:5904. doi: 10.1038/onc.2008.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Wang HY, Wang RF. Curr. Opin. Immunol. 2007;19:217. doi: 10.1016/j.coi.2007.02.004. [DOI] [PubMed] [Google Scholar]; Mantovani A, Schioppa T, Porta C, Allavena P, Sica A. Cancer Metastasis Rev. 2006;25:315. doi: 10.1007/s10555-006-9001-7. [DOI] [PubMed] [Google Scholar]; Whiteside TL. Semin. Cancer Biol. 2006;16:3. doi: 10.1016/j.semcancer.2005.07.008. [DOI] [PubMed] [Google Scholar]
- [187].Krishnamachari Y, Geary SM, Lemke CD, Salem AK. Pharm. Res. 2011;28:215. doi: 10.1007/s11095-010-0241-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Chackerian B. Expert Rev Vaccines. 2007;6:381. doi: 10.1586/14760584.6.3.381. [DOI] [PubMed] [Google Scholar]; Ludwig C, Wagner R. Curr Opin Biotechnol. 2007;18:537. doi: 10.1016/j.copbio.2007.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Jennings GT, Bachmann MF. Biol Chem. 2008;389:521. doi: 10.1515/bc.2008.064. [DOI] [PubMed] [Google Scholar]
- [190].Peek LJ, Middaugh CR, Berkland C. Adv Drug Deliv Rev. 2008;60:915. doi: 10.1016/j.addr.2007.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Kratz PA, Bottcher B, Nassal M. Proc Natl Acad Sci U S A. 1999;96:1915. doi: 10.1073/pnas.96.5.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]; Tissot AC, Renhofa R, Schmitz N, Cielens I, Meijerink E, Ose V, Jennings GT, Saudan P, Pumpens P, Bachmann MF. PLoS One. 2010;5:e9809. doi: 10.1371/journal.pone.0009809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Davis ME, Chen ZG, Shin DM. Nat Rev Drug Discov. 2008;7:771. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- [193].Yin Y, Alivisatos AP. Nature. 2005;437:664. doi: 10.1038/nature04165. [DOI] [PubMed] [Google Scholar]
- [194].Torchilin VP. Adv Drug Deliv Rev. 2006;58:1532. doi: 10.1016/j.addr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- [195].Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nat Nanotechnol. 2007;2:751. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- [196].Guan HH, Budzynski W, Koganty RR, Krantz MJ, Reddish MA, Rogers JA, Longenecker BM. J. Samuel, Bioconjug Chem. 1998;9:451. doi: 10.1021/bc970183n. [DOI] [PubMed] [Google Scholar]; Nakano Y, Mori M, Nishinohara S, Takita Y, Naito S, Kato H, Taneichi M, Komuro K, Uchida T. Bioconjug Chem. 2001;12:391. doi: 10.1021/bc0001185. [DOI] [PubMed] [Google Scholar]
- [197].Rudra JS, Tian YF, Jung JP, Collier JH. Proc Natl Acad Sci U S A. 2010;107:622. doi: 10.1073/pnas.0912124107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Kaba SA, Brando C, Guo Q, Mittelholzer C, Raman S, Tropel D, Aebi U, Burkhard P, Lanar DE. J Immunol. 2009;183:7268. doi: 10.4049/jimmunol.0901957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Cheung WH, Chan VS, Pang HW, Wong MK, Guo ZH, Tam PK, Che CM, Lin CL, Yu WY. Bioconjug Chem. 2009;20:24. doi: 10.1021/bc800167q. [DOI] [PubMed] [Google Scholar]
- [200].Reddy ST, van der Vlies AJ, Simeoni E, Angeli V, Randolph GJ, O’Neill CP, Lee LK, Swartz MA, Hubbell JA. Nat. Biotechnol. 2007;25:1159. doi: 10.1038/nbt1332. [DOI] [PubMed] [Google Scholar]
- [201].Thomas SN, van der Vlies AJ, O’Neil CP, Reddy ST, Yu SS, Giorgio TD, Swartz MA, Hubbell JA. Biomaterials. 2011;32:2194. doi: 10.1016/j.biomaterials.2010.11.037. [DOI] [PubMed] [Google Scholar]
- [202].Carmeliet P, Jain RK. Nature. 2000;407:249. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- [203].Folkman J. N Engl J Med. 1971;285:1182. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- [204].Chaudhuri P, Harfouche R, Soni S, Hentschel DM, Sengupta S. Acs Nano. 2010;4:574. doi: 10.1021/nn901465h. [DOI] [PubMed] [Google Scholar]
- [205].Yu MK, Jeong YY, Park J, Park S, Kim JW, Min JJ, Kim K, Jon S. Angew Chem Int Ed Engl. 2008;47:5362. doi: 10.1002/anie.200800857. [DOI] [PubMed] [Google Scholar]
- [206].Lu RM, Chang YL, Chen MS, Wu HC. Biomaterials. 2011;32:3265. doi: 10.1016/j.biomaterials.2010.12.061. [DOI] [PubMed] [Google Scholar]
- [207].Jain TK, Richey J, Strand M, Leslie-Pelecky DL, Flask CA, Labhasetwar V. Biomaterials. 2008;29:4012. doi: 10.1016/j.biomaterials.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Jain TK, Foy SP, Erokwu B, Dimitrijevic S, Flask CA, Labhasetwar V. biomaterials. 2009;30:6748. doi: 10.1016/j.biomaterials.2009.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Ponce AM, Viglianti BL, Yu D, Yarmolenko PS, Michelich CR, Woo J, Bally MB, Dewhirst MW. J. Natl. Cancer Inst. 2007;99:53. doi: 10.1093/jnci/djk005. [DOI] [PubMed] [Google Scholar]; Viglianti BL, Ponce AM, Michelich CR, Yu D, Abraham SA, Sanders L, Yarmolenko PS, Schroeder T, MacFall JR, Barboriak DP, Colvin OM, Bally MB, Dewhirst MW. Magn. Reson. Med. 2006;56:1011. doi: 10.1002/mrm.21032. [DOI] [PubMed] [Google Scholar]; de Smet M, Heijman E, Langereis S, Hijnen NM, Grull H. J. Control. Release. 2011;150:102. doi: 10.1016/j.jconrel.2010.10.036. [DOI] [PubMed] [Google Scholar]; Neubauer AM, Sim H, Winter PM, Caruthers SD, Williams TA, Robertson JD, Sept D, Lanza GM, Wickline SA. Magn. Reson. Med. 2008;60:1353. doi: 10.1002/mrm.21795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Tagami T, Foltz WD, Ernsting MJ, Lee CM, Tannock IF, May JP, Li S-D. Biomaterials. 2011;32:6570. doi: 10.1016/j.biomaterials.2011.05.029. [DOI] [PubMed] [Google Scholar]
- [211].Gianella A, Jarzyna PA, Mani V, Ramachandran S, Calcagno C, Tang J, Kann B, Dijk WJR, Thijssen VL, Griffioen AW, Storm G, Fayad ZA, Mulder WJM. ACS Nano. 2011;5:4422. doi: 10.1021/nn103336a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Kievit FM, Veiseh O, Bhattarai N, Fang C, Gunn JW, Lee D, Ellenbogen RG, Olson JM, Zhang M. Adv Funct Mater. 2009;19:2244. doi: 10.1002/adfm.200801844. [DOI] [PMC free article] [PubMed] [Google Scholar]; Namgung R, Zhang Y, Fang QL, Singha K, Lee HJ, Kwon IK, Jeong YY, Park IK, Son SJ, Kim WJ. Biomaterials. 2011;32:3042. doi: 10.1016/j.biomaterials.2010.12.018. [DOI] [PubMed] [Google Scholar]; Zhang P, Liu WG. Biomaterials. 2010;31:3087. doi: 10.1016/j.biomaterials.2010.01.007. [DOI] [PubMed] [Google Scholar]; Zhang P, Yang JH, Li WC, Wang W, Liu CJ, Griffith M, Liu WG. J. Mater. Chem. 2011;21:7755. [Google Scholar]; Liu G, Swierczewska M, Lee S, Chen XY. Nano Today. 2010;5:524. doi: 10.1016/j.nantod.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Kumar M, Yigit M, Dai G, Moore A, Medarova Z. Cancer Res. 2010;70:7553. doi: 10.1158/0008-5472.CAN-10-2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Skrabalak SE, Au L, Lu XM, Li XD, Xia YN. Nanomedicine. 2007;2:657. doi: 10.2217/17435889.2.5.657. [DOI] [PubMed] [Google Scholar]; Huang XH, El-Sayed IH, Qian W, El-Sayed MA. Nano Lett. 2007;7:1591. doi: 10.1021/nl070472c. [DOI] [PubMed] [Google Scholar]; O’Neal DP, Hirsch LR, Halas NJ, Payne JD, West JL. Cancer Lett. 2004;209:171. doi: 10.1016/j.canlet.2004.02.004. [DOI] [PubMed] [Google Scholar]; Gobin AM, Lee MH, Halas NJ, James WD, Drezek RA, West JL. Nano Lett. 2007;7:1929. doi: 10.1021/nl070610y. [DOI] [PubMed] [Google Scholar]
- [215].Lu W, Xiong CY, Zhang GD, Huang Q, Zhang R, Zhang JZ, Li C. Clin. Cancer Res. 2009;15:876. doi: 10.1158/1078-0432.CCR-08-1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Jordan A, Scholz R, Maier-Hauff K, van Landeghem FKH, Waldoefner N, Teichgraeber U, Pinkernelle J, Bruhn H, Neumann F, Thiesen B, von Deimling A, Felix R. J. Neuro-Oncol. 2006;78:7. doi: 10.1007/s11060-005-9059-z. [DOI] [PubMed] [Google Scholar]; Jordan A, Wust P, Fahling H, John W, Hinz A, Felix R. Int. J. Hyperthermia. 1993;9:51. doi: 10.3109/02656739309061478. [DOI] [PubMed] [Google Scholar]; Maier-Hauff K, Rothe R, Scholz R, Gneveckow U, Wust P, Thiesen B, Feussner A, von Deimling A, Waldoefner N, Felix R, Jordan A. J Neurooncol. 2007;81:53. doi: 10.1007/s11060-006-9195-0. [DOI] [PubMed] [Google Scholar]
- [217].DeNardo SJ, DeNardo GL, Natarajan A, Miers LA, Foreman AR, Gruettner C, Adamson GN, Ivkov R. J. Nucl. Med. 2007;48:437. [PubMed] [Google Scholar]
- [218].Thomas CR, Ferris DP, Lee JH, Choi E, Cho MH, Kim ES, Stoddart JF, Shin JS, Cheon J, Zink JI. J. Am. Chem. Soc. 2010;132:10623. doi: 10.1021/ja1022267. [DOI] [PubMed] [Google Scholar]
- [219].Ma L, Reinhardt F, Pan E, Soutschek J, Bhat B, Marcusson EG, Teruya-Feldstein J, Bell GW, Weinberg RA. Nat Biotechnol. 2010;28:341. doi: 10.1038/nbt.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]; Weigelt B, Peterse JL, van ’t Veer LJ. Nat Rev Cancer. 2005;5:591. doi: 10.1038/nrc1670. [DOI] [PubMed] [Google Scholar]
- [220].Veiseh O, Kievit FM, Ellenbogen RG, Zhang M. Adv Drug Deliv Rev. 2011;63:582. doi: 10.1016/j.addr.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Park KH, Chhowalla M, Iqbal Z, Sesti F. J Biol Chem. 2003;278:50212. doi: 10.1074/jbc.M310216200. [DOI] [PubMed] [Google Scholar]; Chhowalla M, Unalan HE, Wang Y, Iqbal Z, Park K, Sesti F. Nanotechnology. 2005;16:2982. [Google Scholar]; Xu H, Bai J, Meng J, Hao W, Xu H, Cao JM. Nanotechnology. 2009;20:285102. doi: 10.1088/0957-4484/20/28/285102. [DOI] [PubMed] [Google Scholar]
- [222].Kraszewski S, Tarek M, Treptow W, Ramseyer C. ACS Nano. 2010;4:4158. doi: 10.1021/nn100723r. [DOI] [PubMed] [Google Scholar]
- [223].Veiseh O, Gunn JW, Kievit FM, Sun C, Fang C, Lee JS, Zhang M. Small. 2009;5:256. doi: 10.1002/smll.200800646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Cannistra SA. N Engl J Med. 2004;351:2519. doi: 10.1056/NEJMra041842. [DOI] [PubMed] [Google Scholar]
- [225].Gordon AN, Fleagle JT, Guthrie D, Parkin DE, Gore ME, Lacave AJ. J Clin Oncol. 2001;19:3312. doi: 10.1200/JCO.2001.19.14.3312. [DOI] [PubMed] [Google Scholar]
- [226].Cirstoiu-Hapca A, Buchegger F, Lange N, Bossy L, Gurny R, Delie F. J. Control. Release. 2010;144:324. doi: 10.1016/j.jconrel.2010.02.026. [DOI] [PubMed] [Google Scholar]
- [227].Chen YC, Zhu XD, Zhang XJ, Liu B. L. Huang, Mol. Ther. 2010;18:1650. doi: 10.1038/mt.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Li Z-W, Dalton WS. Blood Reviews. 2006;20:333. doi: 10.1016/j.blre.2005.08.003. [DOI] [PubMed] [Google Scholar]; Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Nat Rev Cancer. 2007;7:585. doi: 10.1038/nrc2189. [DOI] [PubMed] [Google Scholar]
- [229].Howard SC, Jones DP, Pui CH. N. Engl. J. Med. 2011;364:1844. doi: 10.1056/NEJMra0904569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Li Q, So CR, Fegan A, Cody V, Sarikaya M, Vallera DA, Wagner CR. J. Am. Chem. Soc. 2010;132:17247. doi: 10.1021/ja107153a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Jin Y, Liu SJ, Yu B, Golan S, Koh CG, Yang JT, Huynh L, Yang XJ, Pang JX, Muthusamy N, Chan KK, Byrd JC, Talmon Y, Lee LJ, Lee RJ, Marcucci G. Mol. Pharm. 2010;7:196. doi: 10.1021/mp900205r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Thomas DA, Kantarjian HM, Stock W, Heffner LT, Faderl S, Garcia-Manero G, Ferrajoli A, Wierda W, Pierce S, Lu BA, Deitcher SR, O’Brien S. Cancer. 2009;115:5490. doi: 10.1002/cncr.24632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Connors JM. J. Clin. Oncol. 2005;23:6400. doi: 10.1200/JCO.2005.05.016. [DOI] [PubMed] [Google Scholar]
- [234].Azim HA, Santoro L, Bociek RG, Gandini S, Malek RA, Azim HA. Ann. Oncol. 2010;21:1064. doi: 10.1093/annonc/mdp425. [DOI] [PubMed] [Google Scholar]
- [235].Plosker GL, Figgitt DP. Drugs. 2003;63:803. doi: 10.2165/00003495-200363080-00005. [DOI] [PubMed] [Google Scholar]; Schulz H, Bohlius JF, Trelle S, Skoetz N, Reiser M, Kober T, Schwarzer G, Herold M, Dreyling M, Hallek M, Engert A. J. Natl. Cancer Inst. 2007;99:706. doi: 10.1093/jnci/djk152. [DOI] [PubMed] [Google Scholar]
- [236].Etrych T, Sirova M, Starovoytova L, Rihova B, Ulbrich K. Mol. Pharm. 2010;7:1015. doi: 10.1021/mp100119f. [DOI] [PubMed] [Google Scholar]
- [237].Palumbo A, Anderson K. N Engl J Med. 2011;364:1046. doi: 10.1056/NEJMra1011442. [DOI] [PubMed] [Google Scholar]
- [238].Bhattacharya R, Patra CR, Verma R, Kumar S, Greipp PR, Mukherjee P. Adv. Mater. 2007;19:711. [Google Scholar]
- [239].Cirstea D, Hideshima T, Rodig S, Santo L, Pozzi S, Vallet S, Ikeda H, Perrone G, Gorgun G, Patel K, Desai N, Sportelli P, Kapoor S, Vali S, Mukherjee S, Munshi NC, Anderson KC, Raje N. Mol. Cancer Ther. 2010;9:963. doi: 10.1158/1535-7163.MCT-09-0763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Kratz F. J. Control. Release. 2008;132:171. doi: 10.1016/j.jconrel.2008.05.010. [DOI] [PubMed] [Google Scholar]
- [241].Hawkins MJ, Soon-Shiong P, Desai N. Adv. Drug Deliv. Rev. 2008;60:876. doi: 10.1016/j.addr.2007.08.044. [DOI] [PubMed] [Google Scholar]
- [242].Fletcher JI, Haber M, Henderson MJ, Norris MD. Nat. Rev. Cancer. 2010;10:147. doi: 10.1038/nrc2789. [DOI] [PubMed] [Google Scholar]
- [243].Bansal T, Akhtar N, Jaggi M, Khar RK, Talegaonkar S. Drug Discov Today. 2009;14:1067. doi: 10.1016/j.drudis.2009.07.010. [DOI] [PubMed] [Google Scholar]
- [244].Sahoo SK, Labhasetwar V. Mol. Pharm. 2005;2:373. doi: 10.1021/mp050032z. [DOI] [PubMed] [Google Scholar]
- [245].Cuvier C, Roblottreupel L, Millot JM, Lizard G, Chevillard S, Manfait M, Couvreur P, Poupon MF. Biochem. Pharmacol. 1992;44:509. doi: 10.1016/0006-2952(92)90443-m. [DOI] [PubMed] [Google Scholar]
- [246].Chavanpatil MD, Patil Y, Panyam J. Int. J. Pharm. 2006;320:150. doi: 10.1016/j.ijpharm.2006.03.045. [DOI] [PubMed] [Google Scholar]
- [247].Hu F-Q, Liu L-N, Du Y-Z, Yuan H. Biomaterials. 2009;30:6955. doi: 10.1016/j.biomaterials.2009.09.008. [DOI] [PubMed] [Google Scholar]
- [248].Kievit FM, Wang FY, Fang C, Mok H, Wang K, Silber JR, Ellenbogen RG, Zhang M. J Control Release. 2011 doi: 10.1016/j.jconrel.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Yan Y, Ochs CJ, Such GK, Heath JK, Nice EC, Caruso F. Adv. Mater. 2010;22:5398. doi: 10.1002/adma.201003162. [DOI] [PubMed] [Google Scholar]
- [250].Patil Y, Sadhukha T, Ma LN, Panyam J. Journal of Controlled Release. 2009;136:21. doi: 10.1016/j.jconrel.2009.01.021. [DOI] [PubMed] [Google Scholar]
- [251].Dong XW, Mattingly CA, Tseng MT, Cho MJ, Liu Y, Adams VR, Mumper RJ. Cancer Res. 2009;69:3918. doi: 10.1158/0008-5472.CAN-08-2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Susa M, Iyer AK, Ryu K, Choy E, Hornicek FJ, Mankin H, Milane L, Amiji MM, Duan Z. Plos One. 2010;5 doi: 10.1371/journal.pone.0010764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Patil YB, Swaminathan SK, Sadhukha T, Ma LA, Panyam J. Biomaterials. 2010;31:358. doi: 10.1016/j.biomaterials.2009.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].Gao Y, Chen LL, Zhang ZW, Chen Y, Li YP. Biomaterials. 2011;32:1738. doi: 10.1016/j.biomaterials.2010.11.001. [DOI] [PubMed] [Google Scholar]
- [255].Li RB, Wu R, Zhao L, Wu MH, Yang L, Zou HF. ACS Nano. 2010;4:1399. doi: 10.1021/nn9011225. [DOI] [PubMed] [Google Scholar]
- [256].Dick JE. Blood. 2008;112:4793. doi: 10.1182/blood-2008-08-077941. [DOI] [PubMed] [Google Scholar]; Jordan CT, Guzman ML, Noble M. N Engl J Med. 2006;355:1253. doi: 10.1056/NEJMra061808. [DOI] [PubMed] [Google Scholar]; Clevers H. Nat. Med. 2011;17:313. doi: 10.1038/nm.2304. [DOI] [PubMed] [Google Scholar]; Rosen JM, Jordan CT. Science. 2009;324:1670. doi: 10.1126/science.1171837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].O’Brien CA, Kreso A, Jamieson CH. Clin Cancer Res. 2010;16:3113. doi: 10.1158/1078-0432.CCR-09-2824. [DOI] [PubMed] [Google Scholar]
- [258].Bonnet D, Dick JE. Nat Med. 1997;3:730. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]; Fialkow PJ, Gartler SM, Yoshida A. Proc Natl Acad Sci U S A. 1967;58:1468. doi: 10.1073/pnas.58.4.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]; Hamburger AW, Salmon SE. Science. 1977;197:461. doi: 10.1126/science.560061. [DOI] [PubMed] [Google Scholar]; Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. Nature. 1994;367:645. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- [259].Zhou BB, Zhang H, Damelin M, Geles KG, Grindley JC, Dirks PB. Nat Rev Drug Discov. 2009;8:806. doi: 10.1038/nrd2137. [DOI] [PubMed] [Google Scholar]
- [260].Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, Maira G, Parati EA, Stassi G, Larocca LM, De Maria R. Nature. 2010;468:824. doi: 10.1038/nature09557. [DOI] [PubMed] [Google Scholar]; Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A, Fligelman B, Leversha M, Brennan C, Tabar V. Nature. 2010;468:829. doi: 10.1038/nature09624. [DOI] [PubMed] [Google Scholar]
- [261].Takebe N, Harris PJ, Warren RQ, Ivy SP. Nat. Rev. Clin. Oncol. 2011;8:97. doi: 10.1038/nrclinonc.2010.196. [DOI] [PubMed] [Google Scholar]
- [262].Lim KJ, Bisht S, Bar EE, Maitra A, Eberhart CG. Cancer Biol. Ther. 2011;11:464. doi: 10.4161/cbt.11.5.14410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [263].Mamaeva V, Rosenholm JM, Bate-Eya LT, Bergman L, Peuhu E, Duchanoy A, Fortelius LE, Landor S, Toivola DM, Linden M, Sahlgren C. Mol Ther. 2011 doi: 10.1038/mt.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [264].Liu C, Zhao G, Liu J, Ma N, Chivukula P, Perelman L, Okada K, Chen Z, Gough D, Yu L. J. Control. Release. 2009;140:277. doi: 10.1016/j.jconrel.2009.08.013. [DOI] [PubMed] [Google Scholar]
- [265].Hermann PC, Bhaskar S, Cioffi M, Heeschen C. Semin. Cancer Biol. 2010;20:77. doi: 10.1016/j.semcancer.2010.03.004. [DOI] [PubMed] [Google Scholar]
- [266].Keysar SB, Jimeno A. Mol. Cancer Ther. 2010;9:2450. doi: 10.1158/1535-7163.MCT-10-0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [267].Singh SK, Clarke ID, Hide T, Dirks PB. Oncogene. 2004;23:7267. doi: 10.1038/sj.onc.1207946. [DOI] [PubMed] [Google Scholar]
- [268].Wright MH, Calcagno AM, Salcido CD, Carlson MD, Ambudkar SV, Varticovski L. Breast Cancer Res. 2008;10 doi: 10.1186/bcr1855. [DOI] [PMC free article] [PubMed] [Google Scholar]; Hwang-Verslues WW, Kuo WH, Chang PH, Pan CC, Wang HH, Tsai ST, Jeng YM, Shew JY, Kung JT, Chen CH, Lee E, Chang KJ, Lee WH. Plos One. 2009;4 doi: 10.1371/journal.pone.0008377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [269].Miki J, Furusato B, Li HZ, Gu YP, Takahashi H, Egawa S, Sesterhenn IA, McLeod DG, Srivastava S, Rhim JS. Cancer Res. 2007;67:3153. doi: 10.1158/0008-5472.CAN-06-4429. [DOI] [PubMed] [Google Scholar]
- [270].Bertolini G, Roz L, Perego P, Tortoreto M, Fontanella E, Gatti L, Pratesi G, Fabbri A, Andriani F, Tinelli S, Roz E, Caserini R, Lo Vullo S, Camerini T, Mariani L, Delia D, Calabro E, Pastorino U, Sozzi G. Proc. Natl. Acad. Sci. U. S. A. 2009;106:16281. doi: 10.1073/pnas.0905653106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [271].O’Brien CA, Pollett A, Gallinger S, Dick JE. Nature. 2007;445:106. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]; Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De Maria R. Nature. 2007;445:111. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- [272].Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C. Cell Stem Cell. 2007;1:313. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- [273].Curley MD, Therrien VA, Cummings CL, Sergent PA, Koulouris CR, Friel AM, Roberts DJ, Seiden MV, Scadden DT, Rueda BR, Foster R. Stem Cells. 2009;27:2875. doi: 10.1002/stem.236. [DOI] [PubMed] [Google Scholar]
- [274].Ma S, Chan KW, Hu L, Lee TKW, Wo JYH, Ng IL, Zheng BJ, Guan XY. Gastroenterology. 2007;132:2542. doi: 10.1053/j.gastro.2007.04.025. [DOI] [PubMed] [Google Scholar]
- [275].Schluep T, Hwang J, Hildebrandt IJ, Czernin J, Choi CHJ, Alabi CA, Mack BC, Davis ME. Proceedings of the National Academy of Sciences. 2009;106:11394. doi: 10.1073/pnas.0905487106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Jang B, Park JY, Tung CH, Kim IH, Choi Y. Acs Nano. 2011;5:1086. doi: 10.1021/nn102722z. [DOI] [PubMed] [Google Scholar]
- [277].Liu Y, Li J, Shao K, Huang R, Ye L, Lou J, Jiang C. Biomaterials. 2010;31:5246. doi: 10.1016/j.biomaterials.2010.03.011. [DOI] [PubMed] [Google Scholar]
- [278].Ying X, Wen H, Lu W-L, Du J, Guo J, Tian W, Men Y, Zhang Y, Li R-J, Yang T-Y, Shang D-W, Lou J-N, Zhang L-R, Zhang Q. J. Control. Release. 2010;141:183. doi: 10.1016/j.jconrel.2009.09.020. [DOI] [PubMed] [Google Scholar]
- [279].Aktas Y, Yemisci M, Andrieux K, Gursoy RN, Alonso MJ, Fernandez-Megia E, Novoa-Carballal R, Quinoa E, Riguera R, Sargon MF, Celik HH, Demir AS, Hincal AA, Dalkara T, Capan Y, Couvreur P. Bioconjugate Chem. 2005;16:1503. doi: 10.1021/bc050217o. [DOI] [PubMed] [Google Scholar]
- [280].Pang Z, Lu W, Gao H, Hu K, Chen J, Zhang C, Gao X, Jiang X, Zhu C. J. Control. Release. 2008;128:120. doi: 10.1016/j.jconrel.2008.03.007. [DOI] [PubMed] [Google Scholar]
- [281].Liu L, Guo K, Lu J, Venkatraman SS, Luo D, Ng KC, Ling E-A, Moochhala S, Yang Y-Y. Biomaterials. 2008;29:1509. doi: 10.1016/j.biomaterials.2007.11.014. [DOI] [PubMed] [Google Scholar]
- [282].Lu W, Tan Y-Z, Hu K-L, Jiang X-G. Int. J. Pharm. 2005;295:247. doi: 10.1016/j.ijpharm.2005.01.043. [DOI] [PubMed] [Google Scholar]