

Abstract
Congenital myasthenic syndromes are a heterogeneous group of inherited disorders that arise from impaired signal transmission at the neuromuscular synapse. They are characterized by fatigable muscle weakness. We performed whole-exome sequencing to determine the underlying defect in a group of individuals with an inherited limb-girdle pattern of myasthenic weakness. We identify DPAGT1 as a gene in which mutations cause a congenital myasthenic syndrome. We describe seven different mutations found in five individuals with DPAGT1 mutations. The affected individuals share a number of common clinical features, including involvement of proximal limb muscles, response to treatment with cholinesterase inhibitors and 3,4-diaminopyridine, and the presence of tubular aggregates in muscle biopsies. Analyses of motor endplates from two of the individuals demonstrate a severe reduction of endplate acetylcholine receptors. DPAGT1 is an essential enzyme catalyzing the first committed step of N-linked protein glycosylation. Our findings underscore the importance of N-linked protein glycosylation for proper functioning of the neuromuscular junction. Using the DPAGT1-specific inhibitor tunicamycin, we show that DPAGT1 is required for efficient glycosylation of acetylcholine-receptor subunits and for efficient export of acetylcholine receptors to the cell surface. We suggest that the primary pathogenic mechanism of DPAGT1 mutations is reduced levels of acetylcholine receptors at the endplate region. These individuals share clinical features similar to those of congenital myasthenic syndrome due to GFPT1 mutations, and their disorder might be part of a larger subgroup comprising the congenital myasthenic syndromes that result from defects in the N-linked glycosylation pathway and that manifest through impaired neuromuscular transmission.
Main Text
Congenital myasthenic syndromes (CMSs) are inherited disorders of neuromuscular transmission.1,2 They are a heterogeneous group of disorders in which the safety margin for neuromuscular transmission is compromised as a result of mutations in a series of different genes encoding proteins at the neuromuscular synapse. These disorders are characterized by fatigable muscle weakness, and the most commonly affected muscles are ocular, bulbar, and limb muscles. The age of onset is variable, although most cases present with the disorder in infancy or early childhood. To date, mutations in 15 different genes have been shown to lead to impaired neuromuscular transmission, although some are limited to single case reports.1,2 Whereas most CMS-associated genes have a defined function at the neuromuscular junction (NMJ), the recently described GFPT1 encodes glutamine-fructose-6-phosphate transaminase 1, which is ubiquitously expressed and is involved in the synthesis of UDP-N-acetylglucosamine, a saccharide that serves as a building block for protein and lipid glycosylation. Although the exact role of GFPT1 in NMJ function is unknown, it is possible that when mutated, it impairs glycosylation and, consequently, the function of one or more component proteins of the NMJ.3
There remain a number of CMS subtypes for which the underlying mutations have not been identified. Individuals with a predominant limb-girdle pattern of muscle weakness have been found to have mutations in DOK74 (MIM 610285) and GFPT1 (MIM 138292).3 Although these cases share several phenotypic features, muscle biopsy has shown that the majority of individuals with GFPT1 mutations have tubular aggregates, which are not seen in muscle biopsies from individuals with DOK7-associated CMS.5 However, tubular aggregates have been identified in muscle biopsies of additional cases who do not have GFPT1 mutations. Here, we performed whole-exome capture and high-throughput sequencing to identify a second CMS-associated mutation that underlies a limb-girdle-type congenital myasthenia with tubular aggregates in muscle biopsy. Ethical approval for studies on CMSs was obtained from Oxfordshire Research Ethics Committees B (04.OXB.017) and C (09/H0606/74).
Initially, we studied two unrelated individuals (cases 1 and 2) with tubular aggregates in muscle biopsies and without GFPT1 mutations. We performed whole-exome capture from 3 μg of genomic DNA by using Agilent SureSelect Human All Exon Kit v.2 according to the manufacturer's protocol. We sequenced the captured libraries by using 51 bp paired-end reads on Illumina HiSeq or Genome Analyzer IIx platforms. We mapped sequence data to human genome build hg19 by using Novoalign software (Novocraft Technologies). The duplicate reads generated as a result of PCR amplification were filtered out, and only reads that mapped uniquely to the genome were used for further analysis. Aligned sequence data was visualized with GBrowse6 and the UCSC genome browser.7 Variants were called with either Samtools8 or Platypus9 programs. Variants were filtered out if they were present in dbSNP13210 (unless they were annotated as medically associated SNPs). This filtering narrowed the list of variants to 1,574 and 1,287 variants per exome for cases 1 and 2, respectively (see Table S1, available online). Functional annotation of the variants with ANNOVAR software11 allowed us to separate nonsynonymous substitutions, splicing mutations, and mutations in 3′ UTRs or 5′ UTRs, further limiting the number of interesting variants to 377 and 300 per exome for cases 1 and 2, respectively.
CMSs are commonly inherited in an autosomal-recessive manner. Thus, we focused on the genes that had either homozygous variants or contained two or more heterozygous variants. Among these, 34 genes had potential mutations in both analyzed individuals. Further filtering of these variants with our in-house database of 14 exomes from cases with unrelated disorders allowed us to eliminate all but one gene—DPAGT1 (RefSeq NM_001382.3), which encodes dolichyl-phosphate (UDP-N-acetylglucosamine) N-acetylglucosaminephosphotransferase 1. Case 1 had two heterozygous nonsynonymous mutations, c.324G>C (p.Met108Ile) and c.349G>A (p.Val117Ile), whereas case 2 had a heterozygous frameshift single-nucleotide duplication, c.699dup (p.Thr234Hisfs∗116), along with the nonsynonymous mutation c.349G>A (p.Val117Ile).
We confirmed the presence of all four mutations in the genome of analyzed individuals by using Sanger sequencing. To determine whether DPAGT1 mutations are present in other CMS cases, we performed Sanger sequencing of DPAGT1 on a cohort of 31 unrelated cases of suspected CMS with varying phenotypic features, and we identified two more individuals who had two heterozygous nonsynonymous mutations in this gene. Case 3 had a combination of mutations c.358C>A (p.Leu120Met) and c.791T>G (p.Val264Gly), whereas case 4 had mutations c.478G>A (p.Gly160Ser) and c.574G>A (p.Gly192Ser) (Table 1). All four individuals with DPAGT1 mutations were genetically screened for possible mutations in GFPT1 or in other known CMS-associated genes, and all proved to be negative.
Table 1.
Clinical Details of Cases with DPAGT1 Mutations
| Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | |
|---|---|---|---|---|---|
| Gender | male | female | male | female | female |
| Current age | 43 years | 57 years | 25 years | 58 years | 6 years |
| Age when assessed | 43 years | 53 years | 17 years | 48 years | 6 years |
| Mutations | c.324G>C (p.Met108Ile) and c.349G>A (p.Val117Ile) | c.349G>A (p.Val117Ile) and c.699dup (p.Thr234Hisfs∗116) | c.358C>A (p.Leu120Met) and c.791T>G (p.Val264Gly) | c.478G>A (p.Gly160Ser) and c.574G>A (p.Gly192Ser) | c.358C>A (p.Leu120Met) and c.791T>G (p.Val264Gly) |
| Progression | stable but with long-term fluctuations | slowly progressive | some improvement during teenage years | slowly progressive | stable in infancy and small improvement in childhood |
| Age of onset | 2.5 years | 7 years | 0.5 years | 2 years | in first year |
| Symptoms at onset | episodic difficulty walking | falls and is unable to keep up with friends | hypotonia and poor head control | abnormal gait | hypotonia and delayed motor development |
| Ptosis | − | − | + (mild and fatigable) | − | − |
| Ophthalmoplegia | − | − | − | − | − |
| Wasting | − | − | − (although thin) | sternocleidomastoid | − |
| Contractures | − | − | − | − | − |
| Spine | − | scoliosis | scoliosis | − | − |
| Decrement on RNS | + | + | + | + | + |
| Muscle biopsy | tubular aggregates | tubular aggregates | tubular aggregates | tubular aggregates | ND |
| Positive response to treatment | P, D, Sa | P, D | P | P, D | P |
| MRC Muscle Power Grade | |||||
| Face | 5 | 5− | 5 | 5 | 5− |
| Neck flexion/extension | 5/4 | 4+/4+ | 4/5 | 4/5 | 4/4 |
| Shoulder abduction | 4− | 3− | 4− | 3 | 4 |
| Elbow flexion/extension | 4−/4− | 4/4 | 4+/4− | 4+/3+ | 4/4 |
| Wrist/finger extension | 4−/ND | 4+/4+ | 5/4 | ND/ND | 5−/5− |
| Finger/thumb abduction | 4/ND | 4+/4+ | 4/ND | ND/ND | 5/5 |
| Hip flexion/extension | 4/4+ | 4/4+ | 4−/ND | 4−/ND | 4+/4+ |
| Knee flexion/extension | 4+/4 | 4+/4+ | 4+/4 | 4/4 | 4+/4+ |
| Ankle dorsiflexion | 4− | 4+ | 4 | 4 | 5− |
| Single-Fiber Electromyography | |||||
| Abnormal jitter | + | + | + | + | ND |
| Blocking | + | + | + | + | ND |
Bold text indicates ongoing treatment. The following abbreviations are used: ND, not done; RNS, repetitive nerve stimulation; P, pyridostigmine; D, 3, 4-diaminopyridine; Sa, Salbutamol; and MRC, Medical Research Council.
To analyze whether DPAGT1 mutations segregate with disease, we performed Sanger sequencing of DPAGT1 in family members. In all available samples, the mutations displayed a perfect Mendelian inheritance pattern and segregated with the disease (Figure 1). For case 1, no material from family members was available for analysis. However, because these two mutations were less than 51 nucleotides (the length of each read in our sequencing experiment) apart, we could see that each sequencing read contained only one or the other of these mutations. No reads contained both or none of the mutations, indicating that these two mutations were present on two different chromosomes and were thus most likely inherited from two different parents. For case 2, genomic DNA was available from four unaffected siblings. Two of the siblings were heterozygous for the frameshift mutation, one was heterozygous for c.349G>A, and one sibling had neither mutation. For case 3, each parent was heterozygous for one of the mutations. Additionally, case 3 had one affected sibling and one unaffected sibling. The affected sibling (case 5) had the same DPAGT1 mutations as case 3, whereas the unaffected sibling carried only one of the mutations. For case 4, family members were not available for analysis. Thus, the segregation of the DPAGT1 mutations is consistent with the hypothesis that the mutations underlie the CMS phenotype.
Figure 1.
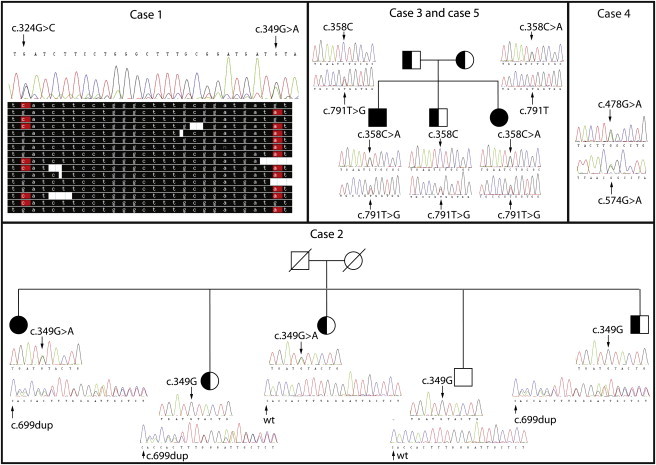
Segregation of DPAGT1 Mutations
Pedigrees of the families of all affected individuals are shown together with Sanger-sequencing verification of variants for each family member. For case 1, alignment of several representative reads generated by next-generation sequencing is shown to demonstrate that two mutations are always present in different reads and never occur in the same read.
None of the DPAGT1 variants described above were present in dbSNP 13510 or the 1000 Genomes12 database. Additionally, we checked the Exome Variant Server13 and found that this database lists one of our mutations, c.324G>C, but the frequency of this mutation is very low—0.0186% (or 2 in 10,758 alleles) in the general population or 0.0142% (1 in 7,020 alleles) in the European American population. All identified individuals are of European origin, and thus, the frequencies described in the Exome Variant Server are valid for our study. Such low frequency of the mutation incidence suggests that the mutation is present in the population in a heterozygous state and is consistent with the notion of the mutation being pathogenic. Thus, it is unlikely that any of the described DPAGT1 variants are common polymorphisms; however, they are likely to cause disease.
The duplication mutation c.699dup causes a shift in the reading frame in the middle of DPAGT1 after residue 234 and a premature termination codon 116 amino acids after the duplication. It is unlikely that any functional protein can be produced from this allele. The premature stop codon is introduced into the penultimate exon 8 (out of 9 exons in DPAGT1). It might be that the mRNA of this allele will be subject to nonsense-mediated decay. In support of this, no detectable protein was produced in HEK 293 cells transfected with a pcDNA3.1-hygro plasmid containing the DPAGT1 c.699dup mutant (Figure 3D). All six single-amino-acid substitutions in DPAGT1 affect residues present in conserved regions of the protein (Figure 2) and thus might affect the wild-type function of the protein, although Met108, Val117, and Gly160 vary in C. elegans and yeast. To further assess whether the mutations are likely to disrupt the structure and function of the protein, we used PolyPhen2,14 which showed that all six missense substitutions were likely to be damaging (Table S2). Interestingly, all identified individuals had at least one mutation in exon 3 of DPAGT1, suggesting that the part of the protein that this exon encodes might be especially important for a function related to neuromuscular transmission.
Figure 3.
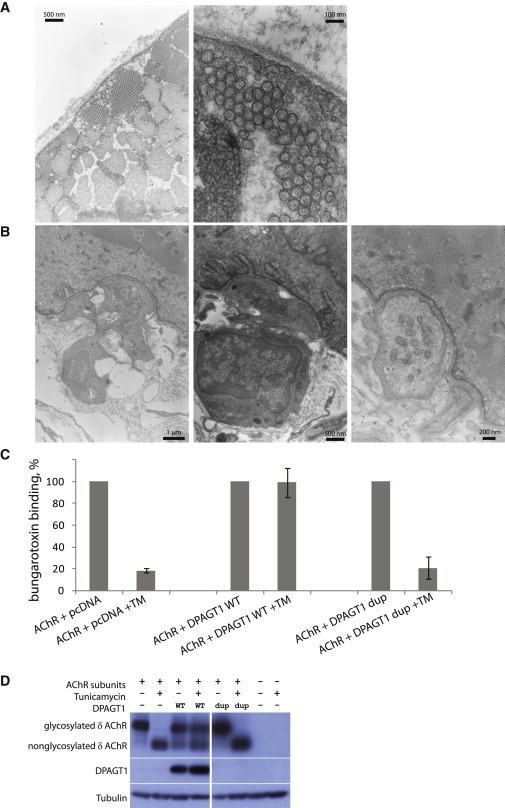
DPAGT1 Is Required for AChR-Subunit Glycosylation and Receptor Export
(A) The electron micrograph from the muscle biopsy from case 1 shows the presence of multiple tubular aggregates.
(B) The electron micrograph from motor-endplate regions in case 1 shows a severe reduction of postsynaptic folding. Various magnifications are shown.
(C) Assay for the level of α-BuTx binding to the surface of HEK 293 cells. Cells were transfected with 1.5 μg of expression plasmids with AChR subunits (2α, 1β, 1δ, and 1ε) and with 4.5 μg of an empty pcDNA vector, a pcDNA-DPAGT1-WT plasmid, or a pcDNA-DPAGT1-c.699dup plasmid. Sixteen hours after transfection, cells were incubated with or without 1 μg/ml tunicamycin. Binding of α-BuTx was normalized to surface expression in the absence of tunicamycin (100%). Error bars represent the standard error of the mean.
(D) Immunoblot of cell extracts from the α-BuTx cell-surface-binding experiment (above). As described, cells were transfected with 1.5 μg of plasmids with AChR subunits (2α, 1β, 1δ, and 1ε) and with 4.5 μg of an empty pcDNA vector (−), a pcDNA-DPAGT1-WT (“WT”) plasmid, or a pcDNA-DPAGT1-c.699dup (“dup”) plasmid. Sixteen hours after transfection, cells were incubated with ± 1 μg/ml tunicamycin. Cell extracts were subject to immunoblotting and probed with antibodies against the AChR δ subunit, DPAGT1 (raised to amino acids 27–57 of DPAGT1), or α-tubulin as a loading control. The levels of endogenous DPAGT1 were too low to be detected at the exposure conditions shown.
Figure 2.
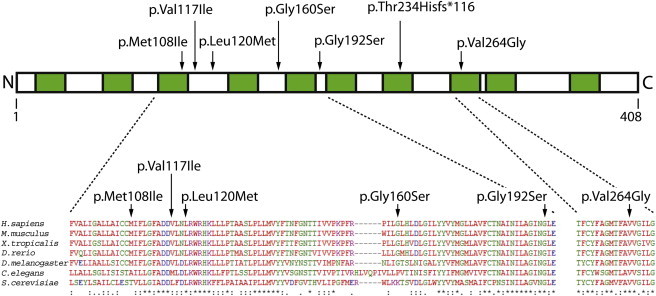
DPAGT1 Structure and Conservation
Predicted transmembrane regions are shown in green. Protein alignment was performed in ClustalW2 (see Web Resources). Positions of amino acid substitutions are shown with arrows.
All five cases with mutations in DPAGT1 show a characteristic CMS phenotype. All had neurophysiological features that indicate a disorder of neuromuscular transmission on electromyography (EMG); such features were a decrement on 3 Hz repetitive nerve stimulation and jitter and blocking on single-fiber EMG (Table 1). They share a number of clinical features that might be used for distinguishing this form of CMS. They have minimal involvement of facial, ocular, and bulbar muscles—these muscle groups can be frequently affected in many other forms of CMS. By contrast, the most prominently affected muscles in cases with DPAGT1 mutations are proximal limb muscles, although some distal muscle groups can be affected (Table 1). As is seen for GFPT1-associated CMS,15 the age of onset tends to be later than for many other CMSs; it occurs in childhood rather than at birth or early infancy. To date, all analyzed cases have had tubular aggregates present in their muscle biopsies (Table 1, Figure 3A; see discussion below). In terms of treatment, all cases showed a beneficial response to anticholinesterase medication, and two benefited from taking 3,4-diaminopyridine, which increases acetylcholine release from the nerve terminal. Thus, individuals with mutations in DPAGT1 have clinical features (which show similarities to GFPT1-associated CMS)15 that should help distinguish them from individuals with other forms of CMS.
DPAGT1 encodes dolichyl-phosphate (UDP-N-acetylglucosamine) N-acetylglucosaminephosphotransferase 1. This enzyme is essential for N-linked protein glycosylation.16 In eukaryotes, N-linked protein glycosylation occurs in the endoplasmic reticulum (ER) and starts with the assembly of the core glycan Glc3Man9GlcNAc2 on the lipid dolichol; the oligosaccharide is subsequently transferred from the lipid onto the asparagine residue of nascent proteins. DPAGT1 is a transmembrane ER protein that catalyzes the first committed step of the core glycan assembly—addition of GlcNAc-1-P from cytoplasmic UDP-GlcNAc to dolichol-P. It is highly conserved in all eukaryotes and has membrane topology that is predicted to span the ER membrane ten times;17 mice with knocked out Dpagt1 die shortly after implantation, demonstrating the essential role of this gene.18
To date, two reports of mutations in DPAGT1 describe a severe congenital disorder of glycosylation type Ij19,20 (CDG-IJ [MIM 608093]). Three cases with this disorder have been clinically characterized. One individual had mutation c.509A>G (p.Tyr170Cys) along with an unidentified splicing mutation and produced only 12% of the wild-type DPAGT1 level.19 The other two cases were siblings from a consanguineous family and had homozygous mutation c.341C>G (p.Ala114Gly).20 The catalytic activity of the latter mutant was reduced to 18% of that of the wild-type level. All three CDG-IJ cases had very severe clinical manifestations. All had delayed development, microcephaly, and intractable seizures, and one had mental retardation. The siblings with homozygous mutations died within the first year of life, whereas the other case survived beyond six years of age. All three are also reported to have had a severe hypotonia, suggesting that neuromuscular transmission might also have been seriously compromised. The symptoms of the individuals with CMSs described in this paper were limited to neuromuscular function, and the nonmuscle abnormalities characteristic of CDG-IJ were not observed in any of these cases. As of yet, we are not in a position to explain why the symptoms in these CMS cases are different from those of the individuals with the CDG-IJ disorder. One possibility is that in the cases that we describe, the DPAGT1 mutations are less damaging to DPAGT1 function. A residual level of protein activity might be able to provide sufficient function in tissues other than at the NMJ. It is of note that all of our cases have a missense amino acid substitution localized in exon 3, and thus, the region of the protein encoded by this exon could have a role important for function at the NMJ.
Different types of disorders of glycosylation are usually diagnosed with a transferrin glycosylation assay.21 Transferrin is an abundant serum protein that normally has two disialo-biantennary N-glycans attached to two specific asparagine residues. In healthy individuals, the most common form of transferrin is tetrasialotransferrin. However, when glycosylation is deficient, hyposialyated transferrin forms are increased, especially disialotransferrin, asialotransferrin, monosialotransferrin, and trisialotransferrin. The measurement of the concentration of these hypoglycosylated forms of transferrin is often used as a screening tool for different forms of congenital disorders of glycosylation. One of the approaches for measuring the transferrin glycosylation levels is by the use of column anion-exchange separation followed by immunoturbidimetry.21,22 When this assay was performed on the serum samples from two of the DPAGT1 CMS cases (cases 3 and 5), both showed abnormal levels of transferrin glycosylation: Case 3 had 5.98% glycosylation-deficient transferrin, and case 5 had 8.18% deficient transferrin (normal levels are <2.6%). Both displayed an increased presence of asialotransferrin and disialotransferrin. These results demonstrate that these two CMS cases have a generalized defect in glycosylation, and they provide strong supporting evidence that the described variants in DPAGT1 are pathogenic. The described assay for the transferrin glycosylation might be useful for future detection of individuals with similar disorders. However, it is still unclear whether the standard glycosylation assay (with isoelectric focusing) will be sufficiently sensitive to detect the changes in transferrin glycosylation occurring in DPAGT1-associated CMS cases.
The clinical features of case 1 have been briefly described previously23 (subject LGM2). A motor-point muscle biopsy (muscle vastus lateralis) has been extensively characterized as having possible structural and functional abnormalities of the NMJ. Labeling of the NMJs with radiolabeled 125I-α-bungarotoxin (BuTx) revealed that the number of α-BuTx binding sites per NMJ was ten times lower in this subject (0.24 × 107 BuTx per NMJ in the affected subject versus 2.34 × 107 BuTx per NMJ in controls). This was paralleled by a reduction of the amplitude of synaptic potentials and currents.23 Ultrastructural studies of the NMJs showed that the amount of postsynaptic folding in the subject was five times lower than that in the control NMJs: the average total length of the folded membrane at each NMJ was 20.6 μm in the affected subject and 103.5 μm in the control (as illustrated in three separate micrographs in Figure 3B). These features are characteristically seen in acetylcholine receptor (AChR)-deficiency syndrome, in which they contribute to impaired neuromuscular transmission.24–27 The primary defect of reduced postsynaptic AChR decreases the sensitivity of the postsynaptic membrane to the transmitter acetylcholine. A secondary effect of the loss of endplate AChR is loss of postsynaptic folds. This would be likely to increase the threshold for action-potential generation in the muscle fiber by reducing both the number and availability of voltage-gated sodium channels, which are normally concentrated in the folds, and reducing the electrical amplification normally provided by the high electrical resistance of the narrow regions of cytoplasm between the folds.28 In accordance with the results from the muscle biopsy from case 1, a muscle biopsy from case 2 also revealed a reduction in endplate α-BuTx binding (performed in the National Hospital for Neurology and Neurosurgery, London in 1987). Thus, both analyzed biopsies show a reduction of endplate AChR. In keeping with the loss of endplate AChR, all the affected individuals respond to anticholinesterase medication, and two individuals benefit from taking 3,4-diaminopyridine; both of these medications are standard treatments for AChR deficiency.2 These observations are consistent with the hypothesis that DPAGT1 mutations cause a loss of endplate AChR.
Adult AChR is a pentameric receptor consisting of subunits 2α, 1β, 1δ, and 1ε. The pentameric receptor is assembled in the ER before being exported to the plasma membrane.29 All four subunits of AChR are N-glycosylated.30 Glycosylation is required for the insertion of AChRs into the plasma membrane through the regulation of subunit stability, folding, assembly, and intracellular transport.31,32 It is therefore possible that in DPAGT1 mutants, AChR subunits are abnormally glycosylated and thus fail to be exported to the plasma membrane.
To establish whether DPAGT1 is required for AChR export, we used an α-BuTx binding assay to measure levels of AChR expressed on the cell surface. In this experiment, cDNAs of 2α, 1β, 1δ, and 1ε AChR subunits were transfected into HEK 293 cells either with wild-type DPAGT1 or with an empty vector. The next day, the medium was changed either to normal growth medium or to growth medium containing 1 μg/ml tunicamycin. Forty-eight hours after transfection, the surface expression of AChR was measured with radioactively labeled 125I-α-BuTx. Tunicamycin is a specific inhibitor of DPAGT133 and has previously been shown to inhibit AChR-subunit glycosylation and export to the cell surface.34 We tested whether overexpression of wild-type DPAGT1 could rescue this effect of tunicamycin on AChR export. Addition of tunicamycin to the cells had a drastic effect on AChR export in cells transfected with the empty vector; fewer than 20% of the receptors were able to get to the cell surface (Figure 3C). However, cells overexpressing wild-type DPAGT1 were resistant to treatment with tunicamycin and displayed normal levels of AChR at the cell surface. Overexpression of the DPAGT1 c.699dup mutant did not restore normal levels of the receptor that gets inserted into the plasma membrane in the presence of the tunicamycin inhibitor. The result is consistent with the observation that little, if any, functional DPAGT1 was produced from the plasmid with this mutation (Figure 3D). Immunoblot analysis of the cell extract from these experiments showed that glycosylation of the δ AChR subunit (the most heavily glycosylated of the four AChR subunits) was completely lost upon treatment with tunicamycin in cells transfected with the empty vector. However, glycosylation was partially rescued when wild-type DPAGT1, but not the DPAGT1 c.699dup (p.Thr234Hisfs∗116) mutant, was overexpressed in the cells (Figure 3D). All other missense substitutions in DPAGT1 were able to rescue inhibition with tunicamycin (data not shown). This is consistent with previously published papers in which it was shown that even catalytically inactive DPAGT1 can rescue inhibition with tunicamycin because the protein can still bind the inhibitor and thus efficiently buffer it out.35,36 We conclude that DPAGT1 is indeed required for AChR export to the cell surface, and the results are consistent with loss of endplate AChR in the affected individuals.
A feature of the biopsy from case 1 is the presence of numerous tubular aggregates inside the muscle (Figure 3A). Tubular aggregates are commonly found in individuals with GFPT1 mutations but are notably absent from the muscles of individuals with DOK7-associated CMS; these latter individuals have a similar predominantly limb-girdle pattern of muscle weakness. Similar aggregates were observed in each of the analyzed individuals with DPAGT1 mutations (Table 1). The exact structure and physiological significance of tubular aggregates is not clear at present. They are usually characterized as long membranous tubules packed with different misfolded and aggregated membrane proteins.37,38 They are believed to arise from membranes of sarcoplasmic reticulum. Tubular aggregates are rare and have been previously observed in certain cases with myopathies, myotonia, or myotonic dystrophy, in some cases with channelopathies, and in most individuals with CMS caused by mutations in GFPT1.3,39,40 Both DPAGT1 and GFPT1 are required for protein glycosylation, which in turn is essential for correct protein folding and targeting.41 It might be that in the individuals with compromised function of DPAGT1 and GFPT1, certain cellular transmembrane and secreted proteins are not appropriately glycosylated; this will lead to their misfolding and aggregation in the sarcoplasmic reticulum and the subsequent formation of tubular aggregates.
In summary, we identify DPAGT1 as a gene in which mutations can cause a CMS. DPAGT1, similar to GFPT1, is involved in protein glycosylation. However, whereas the pathogenic mechanism of GFPT1 has not been established, we suggest that the primary pathogenic mechanism of DPAGT1 mutations is disruption of AChR-subunit glycosylation, inefficient export of AChR to the cell surface, and consequently reduced levels of endplate AChRs. The fact that two genes involved in the protein glycosylation pathway lead to the development of CMSs suggests that there might be a series of such genes in which mutations might affect function at the NMJ. We do not yet know why the symptoms resulting from the mutations we identified are restricted to muscle and the NMJ in particular. Further study of DPAGT1 mutations will help define the role of glycosylation for structure and function at the neuromuscular synapse and will have potential implications for synaptic transmission in the CNS.
Acknowledgments
We thank the High-Throughput Genomics Group at the Wellcome Trust Centre for Human Genetics (funded by Wellcome Trust grant reference 090532/Z/09/Z and Medical Research Council Hub grant G0900747 91070) for the generation of the sequencing data. We are grateful to Gerton Lunter and Andy Rimmer for providing their program Platypus ahead of its publication. We thank Carol Young for electron-microscopy work. K.B. is a fellow of the Wellcome Trust-funded OXION: Ion Channels and Disease Initiative. S.R.F.T. is supported by a grant from the Wellcome Trust. We are grateful for funding from the Medical Research Council, UK, the Muscular Dystrophy Campaign, and the Myasthenia Gravis Association. We thank the cases with congenital myasthenic syndromes and their families for participating in this study and for their consent, which was obtained with ethical approval from Oxfordshire Research Ethics Committees B (04.OXB.017) and C (09/H0606/74).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org/
ClustalW2, http://www.ebi.ac.uk/Tools/msa/clustalw2/
Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Platypus, http://www.well.ox.ac.uk/platypus
References
- 1.Chaouch A., Beeson D., Hantaï D., Lochmüller H. 186th ENMC International Workshop: Congenital myasthenic syndromes 24-26 June 2011, Naarden, The Netherlands. Neuromuscul. Disord. 2012;22:566–576. doi: 10.1016/j.nmd.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 2.Engel A.G. Current status of the congenital myasthenic syndromes. Neuromuscul. Disord. 2012;22:99–111. doi: 10.1016/j.nmd.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Senderek J., Müller J.S., Dusl M., Strom T.M., Guergueltcheva V., Diepolder I., Laval S.H., Maxwell S., Cossins J., Krause S. Hexosamine biosynthetic pathway mutations cause neuromuscular transmission defect. Am. J. Hum. Genet. 2011;88:162–172. doi: 10.1016/j.ajhg.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beeson D., Higuchi O., Palace J., Cossins J., Spearman H., Maxwell S., Newsom-Davis J., Burke G., Fawcett P., Motomura M. Dok-7 mutations underlie a neuromuscular junction synaptopathy. Science. 2006;313:1975–1978. doi: 10.1126/science.1130837. [DOI] [PubMed] [Google Scholar]
- 5.Palace J., Lashley D., Newsom-Davis J., Cossins J., Maxwell S., Kennett R., Jayawant S., Yamanashi Y., Beeson D. Clinical features of the DOK7 neuromuscular junction synaptopathy. Brain. 2007;130:1507–1515. doi: 10.1093/brain/awm072. [DOI] [PubMed] [Google Scholar]
- 6.Stein L.D., Mungall C., Shu S., Caudy M., Mangone M., Day A., Nickerson E., Stajich J.E., Harris T.W., Arva A., Lewis S. The generic genome browser: A building block for a model organism system database. Genome Res. 2002;12:1599–1610. doi: 10.1101/gr.403602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rimmer, A., Mathieson, I., McVean, G., and Lunter, G. (2012). Platypus program: Integrated Variant Caller. The Wellcome Trust Centre for Human Genetics, University of Oxford (http://www.well.ox.ac.uk/platypus).
- 10.Sherry S.T., Ward M.H., Kholodov M., Baker J., Phan L., Smigielski E.M., Sirotkin K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang K., Li M., Hakonarson H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Exome Variant Server. NHLBI Exome Sequencing Project (ESP), Seattle, WA (http://evs.gs.washington.edu/EVS/).
- 14.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guergueltcheva V., Müller J.S., Dusl M., Senderek J., Oldfors A., Lindbergh C., Maxwell S., Colomer J., Mallebrera C.J., Nascimento A. Congenital myasthenic syndrome with tubular aggregates caused by GFPT1 mutations. J. Neurol. 2011 doi: 10.1007/s00415-011-6262-z. [DOI] [PubMed] [Google Scholar]
- 16.Bretthauer R.K. Structure, expression, and regulation of UDP-GlcNAc: Dolichol phosphate GlcNAc-1-phosphate transferase (DPAGT1) Curr. Drug Targets. 2009;10:477–482. doi: 10.2174/138945009788488369. [DOI] [PubMed] [Google Scholar]
- 17.Zhu X.Y., Lehrman M.A. Cloning, sequence, and expression of a cDNA encoding hamster UDP-GlcNAc: Dolichol phosphate N-acetylglucosamine-1-phosphate transferase. J. Biol. Chem. 1990;265:14250–14255. [PubMed] [Google Scholar]
- 18.Marek K.W., Vijay I.K., Marth J.D. A recessive deletion in the GlcNAc-1-phosphotransferase gene results in peri-implantation embryonic lethality. Glycobiology. 1999;9:1263–1271. doi: 10.1093/glycob/9.11.1263. [DOI] [PubMed] [Google Scholar]
- 19.Wu X., Rush J.S., Karaoglu D., Krasnewich D., Lubinsky M.S., Waechter C.J., Gilmore R., Freeze H.H. Deficiency of UDP-GlcNAc:Dolichol Phosphate N-Acetylglucosamine-1 Phosphate Transferase (DPAGT1) causes a novel congenital disorder of Glycosylation Type Ij. Hum. Mutat. 2003;22:144–150. doi: 10.1002/humu.10239. [DOI] [PubMed] [Google Scholar]
- 20.Würde A.E., Reunert J., Rust S., Hertzberg C., Haverkämper S., Nürnberg G., Nürnberg P., Lehle L., Rossi R., Marquardt T. Congenital disorder of glycosylation type Ij (CDG-Ij, DPAGT1-CDG): Extending the clinical and molecular spectrum of a rare disease. Mol. Genet. Metab. 2012;105:634–641. doi: 10.1016/j.ymgme.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 21.Marklová E., Albahri Z. Screening and diagnosis of congenital disorders of glycosylation. Clin. Chim. Acta. 2007;385:6–20. doi: 10.1016/j.cca.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 22.Pascual-Castroviejo I., Pascual-Pascual S.I., Quijano-Roy S., Gutiérrez-Molina M., Morales M.C., Velázquez-Fragua R., Maties M. [Cerebellar ataxia of Norman-Jaeken. Presentation of seven Spanish patients] Rev. Neurol. 2006;42:723–728. [PubMed] [Google Scholar]
- 23.Slater C.R., Fawcett P.R., Walls T.J., Lyons P.R., Bailey S.J., Beeson D., Young C., Gardner-Medwin D. Pre- and post-synaptic abnormalities associated with impaired neuromuscular transmission in a group of patients with ‘limb-girdle myasthenia’. Brain. 2006;129:2061–2076. doi: 10.1093/brain/awl200. [DOI] [PubMed] [Google Scholar]
- 24.Croxen R., Young C., Slater C., Haslam S., Brydson M., Vincent A., Beeson D. Endplate gamma- and epsilon-subunit mRNA levels in AChR deficiency syndrome due to epsilon-subunit null mutations. Brain. 2001;124:1362–1372. doi: 10.1093/brain/124.7.1362. [DOI] [PubMed] [Google Scholar]
- 25.Ohno K., Quiram P.A., Milone M., Wang H.L., Harper M.C., Pruitt J.N., 2nd, Brengman J.M., Pao L., Fischbeck K.H., Crawford T.O. Congenital myasthenic syndromes due to heteroallelic nonsense/missense mutations in the acetylcholine receptor epsilon subunit gene: identification and functional characterization of six new mutations. Hum. Mol. Genet. 1997;6:753–766. doi: 10.1093/hmg/6.5.753. [DOI] [PubMed] [Google Scholar]
- 26.Ruff R.L. Endplate contributions to the safety factor for neuromuscular transmission. Muscle Nerve. 2011;44:854–861. doi: 10.1002/mus.22177. [DOI] [PubMed] [Google Scholar]
- 27.Slater C.R., Young C., Wood S.J., Bewick G.S., Anderson L.V., Baxter P., Fawcett P.R., Roberts M., Jacobson L., Kuks J. Utrophin abundance is reduced at neuromuscular junctions of patients with both inherited and acquired acetylcholine receptor deficiencies. Brain. 1997;120:1513–1531. doi: 10.1093/brain/120.9.1513. [DOI] [PubMed] [Google Scholar]
- 28.Slater C.R. Reliability of neuromuscular transmission and how it is maintained. Handb. Clin. Neurol. 2008;91:27–101. doi: 10.1016/S0072-9752(07)01502-3. [DOI] [PubMed] [Google Scholar]
- 29.Wanamaker C.P., Christianson J.C., Green W.N. Regulation of nicotinic acetylcholine receptor assembly. Ann. N Y Acad. Sci. 2003;998:66–80. doi: 10.1196/annals.1254.009. [DOI] [PubMed] [Google Scholar]
- 30.Nomoto H., Takahashi N., Nagaki Y., Endo S., Arata Y., Hayashi K. Carbohydrate structures of acetylcholine receptor from Torpedo californica and distribution of oligosaccharides among the subunits. Eur. J. Biochem. 1986;157:233–242. doi: 10.1111/j.1432-1033.1986.tb09661.x. [DOI] [PubMed] [Google Scholar]
- 31.Gehle V.M., Sumikawa K. Site-directed mutagenesis of the conserved N-glycosylation site on the nicotinic acetylcholine receptor subunits. Brain Res. Mol. Brain Res. 1991;11:17–25. doi: 10.1016/0169-328x(91)90016-q. [DOI] [PubMed] [Google Scholar]
- 32.Gehle V.M., Walcott E.C., Nishizaki T., Sumikawa K. N-glycosylation at the conserved sites ensures the expression of properly folded functional ACh receptors. Brain Res. Mol. Brain Res. 1997;45:219–229. doi: 10.1016/s0169-328x(96)00256-2. [DOI] [PubMed] [Google Scholar]
- 33.Lehle L., Tanner W. The specific site of tunicamycin inhibition in the formation of dolichol-bound N-acetylglucosamine derivatives. FEBS Lett. 1976;72:167–170. doi: 10.1016/0014-5793(76)80922-2. [DOI] [PubMed] [Google Scholar]
- 34.Merlie J.P., Sebbane R., Tzartos S., Lindstrom J. Inhibition of glycosylation with tunicamycin blocks assembly of newly synthesized acetylcholine receptor subunits in muscle cells. J. Biol. Chem. 1982;257:2694–2701. [PubMed] [Google Scholar]
- 35.Dal Nogare A.R., Dan N., Lehrman M.A. Conserved sequences in enzymes of the UDP-GlcNAc/MurNAc family are essential in hamster UDP-GlcNAc:dolichol-P GlcNAc-1-P transferase. Glycobiology. 1998;8:625–632. doi: 10.1093/glycob/8.6.625. [DOI] [PubMed] [Google Scholar]
- 36.Dan N., Middleton R.B., Lehrman M.A. Hamster UDP-N-acetylglucosamine: Dolichol-P N-acetylglucosamine-1-P transferase has multiple transmembrane spans and a critical cytosolic loop. J. Biol. Chem. 1996;271:30717–30724. doi: 10.1074/jbc.271.48.30717. [DOI] [PubMed] [Google Scholar]
- 37.Pavlovicová M., Novotová M., Zahradník I. Structure and composition of tubular aggregates of skeletal muscle fibres. Gen. Physiol. Biophys. 2003;22:425–440. [PubMed] [Google Scholar]
- 38.Schiaffino S. Tubular aggregates in skeletal muscle: Just a special type of protein aggregates? Neuromuscul. Disord. 2012;22:199–207. doi: 10.1016/j.nmd.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 39.Meola G., Sansone V., Rotondo G., Mancinelli E. Muscle biopsy and cell cultures: Potential diagnostic tools in hereditary skeletal muscle channelopathies. Eur. J. Histochem. 2003;47:17–28. doi: 10.4081/803. [DOI] [PubMed] [Google Scholar]
- 40.Oh S.J., Park K.S., Ryan H.F., Jr., Danon M.J., Lu J., Naini A.B., DiMauro S. Exercise-induced cramp, myoglobinuria, and tubular aggregates in phosphoglycerate mutase deficiency. Muscle Nerve. 2006;34:572–576. doi: 10.1002/mus.20622. [DOI] [PubMed] [Google Scholar]
- 41.Larkin A., Imperiali B. The expanding horizons of asparagine-linked glycosylation. Biochemistry. 2011;50:4411–4426. doi: 10.1021/bi200346n. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.