

Abstract
Congenital erythropoietic porphyria (CEP) is due to a deficiency in the enzymatic activity of uroporphyrinogen III synthase (UROS); such a deficiency leads to porphyrin accumulation and results in skin lesions and hemolytic anemia. CEP is a candidate for retrolentivirus-mediated gene therapy, but recent reports of insertional leukemogenesis underscore the need for safer methods. The discovery of induced pluripotent stem cells (iPSCs) has opened up new horizons in gene therapy because it might overcome the difficulty of obtaining sufficient amounts of autologous hematopoietic stem cells for transplantation and the risk of genotoxicity. In this study, we isolated keratinocytes from a CEP-affected individual and generated iPSCs with two excisable lentiviral vectors. Gene correction of CEP-derived iPSCs was obtained by lentiviral transduction of a therapeutic vector containing UROS cDNA under the control of an erythroid-specific promoter shielded by insulators. One iPSC clone, free of reprogramming genes, was obtained with a single proviral integration of the therapeutic vector in a genomic safe region. Metabolic correction of erythroblasts derived from iPSC clones was demonstrated by the disappearance of fluorocytes. This study reports the feasibility of porphyria gene therapy with the use of iPSCs.
Introduction
Congenital erythropoietic porphyria (CEP [MIM 263700]) is an autosomal-recessive disorder characterized by a profound deficiency in the enzymatic activity of uroporphyrinogen III synthase (UROS; EC 4.2.1.75), the fourth enzyme of the heme biosynthetic pathway.1,2 The enzymatic defect causes the accumulation of the nonphysiological porphyrin isomer uroporphyrin I. The lack of UROS causes mutilating dermatological lesions resulting from the release of photocatalytic porphyrins and transfusion-dependent hemolytic anemia with massive secondary hypersplenism. The clinical severity of the disease and the lack of specific treatment—apart from when bone marrow (BM) transplantation with a histocompatibility leukocyte antigen (HLA)-compatible donor is available3–8—are strong arguments for gene therapy.2 We, as well as others,9–11 have generated an animal model of CEP in order to study the pathophysiology of the disease and to evaluate the feasibility of stem cell and gene therapy for this disease. Lentivirus-mediated transfer of the human therapeutic UROS cDNA into hematopoietic stem cells (HSCs) from homozygous Urosc.744C>A/c.744C>A-knockin mice (p.Pro248Gln) (the Uros RefSeq accession number is NM_009479.2) resulted in the complete and long-term enzymatic, metabolic, and phenotypic correction of the disease. This correction is favored by the survival advantage of corrected red blood cells.12 This data supports the proof of concept of a successful gene therapy for this disease by transplantation of genetically modified HSCs. Gene-transfer technologies have now been successfully used in clinical gene-therapy applications for many genetic diseases,13–19 but recent reports of proviral insertional leukemogenesis underscore the need for safer methods.20
The discovery of key transcription factors enabling reprogramming of a somatic cell into a pluripotent stem cell, called an induced pluripotent stem cell (iPSC), has provoked an exciting rebound in gene-therapy research.21–24 Gene-therapy model systems based on iPSCs have been created for a wide range of hereditary hematologic disorders, such as Fanconi anemia (MIM 227650),25 sickle-cell anemia (MIM 141900),26 thalassemia (MIM 613985),27 and hemophilia B (MIM 306900).28 Because iPSCs can be maintained indefinitely in vitro, they represent an unlimited source of cells, and this surmounts the difficulty of obtaining sufficient amounts of autologous HSCs for transplantation. Moreover, the clonal approach allowed by iPSCs could overcome the risk of insertional oncogenesis by the characterization of proviral integration sites (ISs) of the therapeutic vectors and the selection of iPSC clones with ISs in a genomic safe harbor (GSH).27,29
The main goal of this project was to evaluate whether iPSCs might provide a key cell resource for gene therapy in a CEP model. To achieve this goal, we derived human iPSCs with epidermal keratinocytes taken from a CEP-affected individual by using excisable lentiviral vectors. Excised iPSC clones, free of reprogramming factors, were corrected (gene additive therapy). All ISs were found in nononcogenic genes, and one IS was found without cancer-related genes nearby. Then, they were differentiated into HSCs. Enzymatic and metabolic corrections were obtained in erythroid cells derived from corrected CEP iPSC clones. This study is a proof of principle of a safe iPSC-mediated gene therapy for CEP.
Material and Methods
Lentiviral Constructions and Productions
Two lentiviral vectors were used for iPSC generation (OSK 1 and Mshp53) (Figure 1B). The polycistronic pKP332 (Lenti-OSK1) vector (OSK 1) was obtained from Addgene (plasmid 21627). OCT4 (POU5F1 [MIM 164177]), SOX2 (MIM 184429), and KLF4 (MIM 602253) cDNAs, linked with porcine teschovirus-1 2A sequences, were under the control of the EF1-α promoter in a self-inactivating (SIN) lentiviral vector containing a Lox P site in the truncated 3' long terminal repeat (LTR).30 We constructed Mshp53 containing MYC (MIM 190080) cDNA and shRNA against TP53 (MIM 191170) as follows: MYC from a lentiviral Cre-Lox pMXs-hc-MYC plasmid (17220 from Addgene) was digested by NotI and inserted into a subcloning vector to flank the cassette with EcoRI and BamH1 restriction sites. The EGFP cassette from pLVTH-siP53 (12239 from Addgene) was removed with EcoRI and BamHI and replaced by the insert of MYC under the control of the EF1- α promoter.
Figure 1.
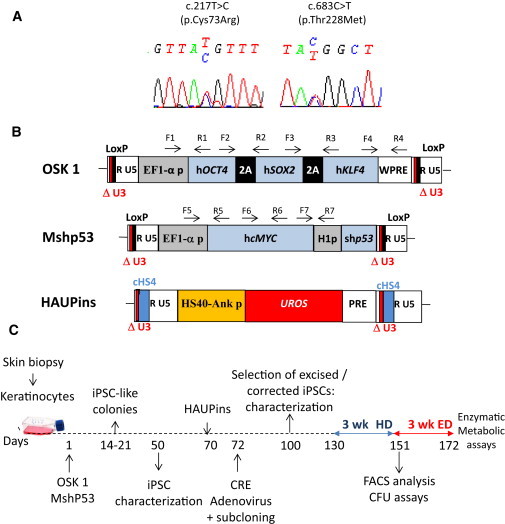
CEP Characterization and Schematic Drawing of iPSC Generation
(A) Genotyping of keratinocytes from a CEP-affected individual compound heterozygous for UROS mutations c.217T>C (p.Cys73Arg) and c.683C>T (p.Thr228Met).
(B) Schematic representation of the proviral form of the lentivectors used. OSK 1 is an excisable single polycistronic vector coexpressing OCT4, SOX2, and KLF4 cDNAs linked with the porcine teschovirus-1 2A sequence. Mshp53 coexpresses MYC and a shRNA against TP53; both vectors are flanked by loxP sites. HAUPins contains the UROS cDNA under the control of the erythroid chimeric HS-40 enhancer/ankyrin promoter. Arrows show the position of forward (F) and reverse (R) primers used for proviral integration analyses. The following abbreviations are used: HS40, enhancer of HBA1 (MIM 141800); Ank p, ankyrin promoter of ANKYRIN-1 (MIM 612641); UROS, cDNA of uroporphyrinogene III synthase; H1p, mammalian polymerase III H1 promoter; EF1α-p, elongation factor-1 alpha promoter; WPRE, woodchuck posttranscriptional regulatory element; PRE, a mutated WPRE sequence (WPREmut6); and cHS4, element of the chicken β-globin hypersensitivity site 4.
(C) Schematic strategy used for reprogramming human keratinocytes from a CEP-affected person and for HSC differentiation. The following abbreviations are used: HD, hematopoietic differentiation; and ED, erythroid differentiation.
The therapeutic lentiviral vector HAUPins has been previously described31 and is derived from the original HAUW vector containing UROS cDNA under the control of the chimeric HS40-Ankyrin promoter, a vector already used in gene therapy in the CEP mouse model.12 In brief, HAUPins is different from HAUW in that it contains (1) the insertion of a mutated WPRE sequence (PRE) without the potentially oncogenic WPRE-X32 and (2) the insertion of the 5′ cHS4 insulator in the ΔU3 sequence of the 3′ LTR.33 The HAEPins control vector contains exactly the same backbone but has EGFP instead of UROS cDNA (Figure 5C).
Figure 5.
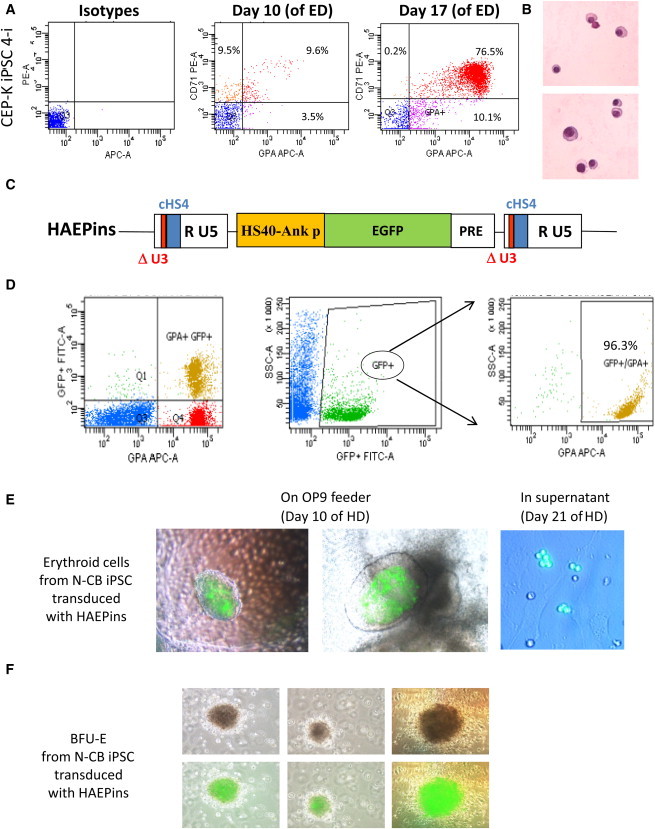
Efficient Erythroid Differentiation of HSCs Derived from iPSCs with Specific Transgene Expression
(A) Representative flow-cytometry data of in vitro erythroid-differentiation (ED) cultures from CEP-K iPSC 4-i. Cells were stained at day 10 and at day 17 of ED with anti-CD71-phycoerythrin (PE)- and anti-GPA-APC-conjugated antibodies (or they were stained with control isotype antibodies conjugated with PE and APC; left panels).
(B) Erythroblast maturation was evaluated at day 24 of ED by staining with May-Grünwald-Giemsa. Photographs show the late stage of erythroid maturation.
(C) Schema of the proviral form of the HAEPins control vector containing EGFP under the control of the HS40/ankyrin erythroid promoter.
(D) Representative FACS analysis of hematopoietic progenitors at day 21 of hematopoietic differentiation (HD) from the N-CB iPSCs previously transduced with the HAEPins lentivector.
(E) At day 10 of HD, formation of early erythroblast cells was revealed by GFP expression in sacks on OP9 stroma (middle and left panels) and by erythroblast cell suspension at day 21 of HD (right panel).
(F) Inverted microscopy of BFU-E colonies under a visible-light microscope (upper panels) and merged with GFP expression detected under UV light (lower panels).
VSV-G pseudotyped lentivectors were produced by triple transient transfection in 293T cells and were concentrated by ultrafiltration (Vivaspin 20, Sartorius Biotech SA, USA) as previously described.12 To determine viral production, we measured human immunodeficiency virus (HIV) p24 titles in the concentrated viral supernatant by ELISA assay (Innotest HIV, p24, Innogenetics, France). We estimated viral titers by comparing p24 antigen levels of each lentiviral supernatant with a green fluorescent protein (GFP) lentiviral supernatant produced simultaneously. Supernatants were found to be free of replication-competent virus.
Source of Somatic Cells for Human iPSC Generation and Transduction
Keratinocytes were isolated from a CEP-affected person and a healthy person undergoing plastic surgery after informed consent was obtained in accordance with the ethical standards of the responsible committee on human experimentation (Centre Hospitalier Universitaire de Bordeaux). Skin fragments were treated with trypsin-EDTA for 3 hr at 37°C for the separation of the epidermis from the dermis. Keratinocytes were seeded at a concentration of 105 cells per cm2 in MCDB153 medium, which included hydrocortisone (0.5 μg/ml), epidermal growth factor (10 ng/ml), insulin (5 μg/ml), and bovine pituitary extract (70 μg/ml) (all from Sigma). For reprogramming, keratinocytes were seeded at a concentration of 105 cells per well (in a 6-well plate) and were cotransduced 24 hr later with a combination of OSK 1 and Mshp53 lentivectors at a multiplicity of infection (MOI) of 10. After an additional 2 day culture in the same medium, cells were transferred onto mitomycined mouse embryonic fibroblasts (MEFs) and cultured in the embryonic stem (ES) medium described below.
CD34+ cells from a cord-blood sample were obtained after informed consent was obtained according to procedures approved by the Bagatelle Hospital review board. Mononuclear cells were isolated by Ficoll gradient. CD34+ cells were purified according to the manufacturer's (Miltenyi Biotech) instructions. Purity was analyzed by flow cytometry with a phycoerythrin-conjugated CD34 antibody (Becton Dickinson). CD34+ cells were cultured 2 days in expansion medium consisting of Stem Span SFEM (STEMCELL Technologies, Grenoble, France) supplemented with Flt3-L (50 ng/ml), SCF (50 ng/ml), and human TPO (50 ng/ml) (all from Peprotech, Rocky Hill, NJ, USA). For reprogramming, CD34+ cells were cotransduced with a combination of the two reprogramming lentivectors at a MOI of 100. After an additional 2 day culture in the same expansion medium, cells were transferred onto mitomycined MEFs and cultured in ES medium.
For keratinocytes and CD34+ cells, the individual iPSC-like colonies were picked up from days 14–21 for expansion. iPSC-like colonies from human skin fibroblasts were obtained with the same two lentivectors at a MOI of 30 and were later picked up from week 5 to week 6.
Human iPSC Culture
Human iPSC clones were maintained as undifferentiated cells in cocultures with mitomycined MEFs. The ES medium used was the following: KO-DMEM (Invitrogen, Villebon sur Yvette, France) containing 20% KOSR (Invitrogen) (vol/vol), 15 ng/ml human bFGF (Peprotech), 1 mM GlutaMAX (Invitrogen), 100 μM Non-Essential Amino Acids (Invitrogen), 100 μM 2-mercaptoethanol (Sigma-Aldrich, Saint Louis, MO, USA), 50 μg/ml ascorbic acid (Sigma-Aldrich), 0.5 mM sodium butyrate (Sigma-Aldrich), 50 U/ml penicillin, and 50 mg/ml streptomycin (Invitrogen). The ES medium was changed every day.
iPSC Characterization
Substrate staining for alkaline phosphatase (ALP) was carried out with a leukocyte ALP staining kit (Sigma-Aldrich).
For the detection of pluripotency markers, cells grown in 24-well plates were fixed by 4% paraformaldehyde and permeabilized with ice-cold 0.2% Triton X-100 in PBS. After saturation with 0.2% PBS-triton and 1% HSA, cells were stained with primary antibodies for 1 hr and were then incubated with a second fluorochrome-labeled antibody (Alexa Fluor, Invitrogen). The primary antibodies used were: OCT4 (clone C-10, Santa Cruz,CA, USA), SOX2 (Abcam, Cambridge, UK), KLF4 (Abcam), NANOG (Abcam), SSEA-4 (clone 813-70, STEMCELL Technologies), and TRA1-60 (STEMCELL Technologies).
For RT-PCR analysis of pluripotency markers, total RNA from iPSC clones and primary cells (CD34+ cells, keratinocytes, and fibroblasts) was isolated with TRIZOL reagent (Invitrogen). RT-PCR (27 cycles) was performed after RNase-free DNase pretreatment with a first-Strand cDNA synthesis kit for RT-PCR (Roche Applied Science, Meylan, France) and with oligonucleotides described by Papapetrou et al.27
For teratoma induction, iPSCs were plated in a 10 cm MEF feeder dish. At day 6, approximately 2 × 106 cells were harvested, resuspended in 100 μl of ES medium containing 10 μM of the Rho-associated kinase (Rock) inhibitor Y-27632 (Sigma), and injected into NOD Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NOD-SCID IL2Rg null:NSG) mice (subcutaneous space). The NSG mice were produced and housed in the University Bordeaux Segalen animal facility according to the rules and regulations governed and enforced by the Institutional Animal Care and Use Committee. The animal facility's institutional agreement number is A33063916. Animals were included in protocols between the ages of 6 and 8 weeks. Teratomas were harvested 8–12 weeks after injection. Paraffin-embedded tissue was sliced and stained with Alcian blue.
Karyotyping
After FrdU synchronization followed by a thymidine chase, standard R-banding analysis was performed on metaphase chromosomes obtained with all iPSC clones. At least 20 metaphase chromosomes were fully karyotyped.
Cre-Mediated Vector Excision
IPSC clones were transduced twice at an MOI of 100 with a Cre-expressing adenovirus (kindly provided by the Association Française contre les Myopathies, Généthon). At day 7, iPSCs were dissociated into single cells with accutase (STEMCELL Technologies) and cloned by a limiting dilution. Cre-Lox excision of proviral reprogramming cassettes was checked in each subclone by multiple PCR analyses. Primers used were designed by Primer3 software for both proviruses, and their positions in OSK 1 and Mshp53 are shown in Figure 1B. All oligonucleotide sequences are provided in Table S1, available online.
Quantitative PCR Detection of HAUPins Provirus
DNA was isolated with the NucleoSpin Tissue kit (Macherey-Nagel, Hoerdt, France) from 1 × 106 cells, and real-time PCR was performed on 100 ng of DNA with the use of Taqman technology (Applied Biosystems, Foster City, CA). DNA from nontransduced cells was isolated and amplified at the same time for demonstrating the absence of contamination. Samples were heated to 50°C for 2 min and to 95°C for 10 min, and then 40 cycles of PCR were performed for 15 s at 95°C and for 1 min at 60°C. UROS amplimers are described in Table S1. RNaseP was used for normalization (Applied Biosystems, Foster City, CA). DNA from two different cell clones (human 293T and K562 cells), containing a single integrated copy of the transgene, was used as a standard.
Characterization of Proviral Integration Site by LAM-PCR
For the identification of genomic proviral ISs, linear amplification-mediated PCR (LAM-PCR) was performed as previously described34 with 100 ng of DNA from each iPSC clone. Linear PCR was carried out with biotinylated primer LTRI∗. Selection of biotinylated extension products was performed with magnetic beads according to the manufacturer's (Invitrogen Dynal AS, Oslo, Norway) instructions. After double-stranded DNA synthesis, Tsp509I was used as the restriction enzyme, and a double-stranded asymmetric linker cassette was linked with LC1 and LC3 linkers. The first exponential PCR was carried out with linker-cassette primer LCI and vector-specific primer LTRII∗, whereas the second exponential PCR was carried out with internal primers LCII and LTRIII. PCR products were cloned into the TOPO TA cloning vector (Invitrogen, Carlsbad, CA) and sequenced. Sequences obtained were aligned by BLAST with the human genome.
ISs were confirmed by PCR with the forward primer specific to the cHS4 insulator sequence and the reverse primers specific to the genomic sequence adjacent to the integration (all oligonucleotides are provided in Table S1).
The proximity of ISs to proto-oncogenes was determined by comparison with the allOnco database.
Hematopoietic Differentiation of iPSCs
Hematopoietic differentiation of iPSCs was performed as described by Woods et al.35 with modifications. In brief, we first generated embryonic bodies (EBs). From day 0 to day 2, iPSCs were harvested and placed in EB medium (IMDM medium, 15% fetal bovine serum [FBS], 1% nonessential amino acid) overnight on nonadherent cell-culture dishes. Then, to induce mesodermal transition, we cultured newly generated EB colonies on mitomycined OP9 feeder cells (CRL-2749 from ATCC, Manassas, VA, USA) on Matrigel (BD Biosciences) for 12 additional days; we partially changed the medium every day in a mesodermal-specifying medium (DMEM/F12, 15% FBS with BMP4, low doses of VEGF, TPO, EPO, SCF, and Flt3L [all from Peprotech at the same concentrations described by Woods et al.35], holotransferrine and ascorbic acid [from Sigma-Aldrich], and PGE2 [Cayman Chemical]). From day 14 to day 21 of hematopoietic differentiation, medium was changed to serum-free expansion medium (STEMCELL Technologies) supplemented with TPO, EPO, SCF, Flt3L, and PGE2 for the promotion of hematopoietic differentiation and HSC expansion. Fluorescence-activated cell sorting (FACS) analysis of CD34+ and CD45+ cells was performed at day 21 for the evaluation of hematopoietic-differentiation efficiency.
Hematopoietic Colony Formation in Methylcellulose
Single-cell suspensions of hematopoietic cells were plated in a methylcellulose-based medium of MethoCult H4435 (STEMCELL Technologies) (approximately 105 cells) in 6-well plates. At day 14, colonies were scored by bright-field microscopy and fluorescence for the visualization of fluorocytes and GFP expression. Fluorescent images were acquired with a Nikon ECLIPSE Ti inverted microscope and captured with a digital sight camera and NIS-element imaging software.
Erythroid Differentiation
For erythroid differentiation, we performed a 3 week protocol after hematopoietic differentiation. In brief, cells were seeded in a 6-well low attachment plate with erythroid medium (Stem-alpha AE base [Stem Alpha, France] supplemented with 5% human plasma, 5 U/ml Epo, 50 ng/ml SCF from Peprotech, 1 mg/ml holotransferrine, 10−6 M dexamethasone, 20 ng/ml insulin, and 10−4 M β-mercapto-ethanol [Sigma Aldrich]). Hemoglobin analysis was performed by HPLC (Variant II, Bio-Rad). FACS analysis of CD71+ and GPA+ cells was performed at day 21 for the evaluation of erythroid-differentiation efficiency.
Flow Cytometry
Cells were individualized from the differentiation cultures, collected, and washed with 1% PBS-HSA. Cells were stained with phycoerythrin (PE)- or allophycocyanin (APC)-conjugated anti-CD34, PECy5- or PE-conjugated anti-CD45, PE-conjugated anti-CD14, APC-conjugated anti-CD19, PE-conjugated anti-CD33, PE-conjugated anti-CD71, and APC-conjugated anti-GpA (all from BD, Franklin Lakes, NJ, USA).
Fluorocytes were analyzed at 550 nm emission wavelength with a 405 nm excitation laser. Cells were analyzed on a FACS flow cytometer (Canto II, BD, San Jose, CA, USA).
UROS Enzymatic Activity and Metabolic Correction
UROS activity was determined by an enzyme-coupled assay as described previously.36 One unit was defined as the amount of enzyme that formed 1 nmol of uroporphyrinogen III per hour at 37°C.
Statistical Analysis
Results are expressed as mean ± standard deviation (SD). Statistical tests were performed with Student's t tests. p < 0.05 was considered statistically significant.
Results
Generation of iPSCs
Primary human epidermal keratinocytes were isolated from the skin of a healthy donor undergoing plastic surgery and from a skin biopsy from a CEP-affected individual after informed consent was obtained. Genetic testing revealed that the CEP-affected individual was compound-heterozygous for UROS (RefSeq NG_011557.1) mutations c.217T>C (p.Cys73Arg) and c.683C>T (p.Thr228Met), thus confirming the diagnosis of CEP (Figure 1A). The residual enzymatic activity measured by an enzyme coupled assay was below 2% of the normal level.
To increase the yield of iPSC generation, we designed the Mshp53 vector carrying the coexpression of MYC under the control of EF1-α promoter and a shRNA against TP53 under the control of the H1 promoter (Figure 1B). In addition to the expression of the four standard reprogramming factors, the repression of TP53 has been previously reported to improve the efficiency of the reprogramming process.37 iPSC generation was obtained by transduction of normal and deficient keratinocytes with the two SIN lentivectors (OSK 1 and Mshp53, Figure 1B). Colonies with ES-like morphology from normal keratinocytes (N-K iPSCs) and CEP keratinocytes (CEP-K iPSCs) were picked from days 14–21 (Figure 1C). Fibroblasts were also transduced for reprogramming as controls. In addition to transduction of these cells, transduction of neonatal CD34+ cells from umbilical-cord blood cells was performed for generating control iPSCs for further hematopoietic differentiation.
Generation of iPSC clones from keratinocytes was ten times more efficient than that from control skin fibroblasts (0.4% versus 0.04%, respectively) and was faster (picked from days 14–21 versus days 35–42 for fibroblasts). Although OSK 1 vector transduction alone led to efficient reprogramming with murine fibroblasts,30 the use of this single vector failed to generate stable iPSC clones from human cells. Indeed, the addition of the Mshp53 vector was mandatory for achieving the terminal reprogramming and generating high-quality iPSC clones. Efficiency and kinetics of keratinocyte reprogramming were quite similar to neonatal normal CD34+ cells (N-CB iPSCs). All the colonies harvested demonstrated the typical characteristics of pluripotent stem cells, i.e., similar morphology to human ES cells, strong alkaline-phosphatase activity (data not shown), and expression of pluripotent stem cell markers, such as OCT3/4, SOX2, KLF4, NANOG, SSEA-4, and TRA1-60 (Figure 2A), as evidenced by immunocytochemistry. We isolated a total of 23 iPSC clones from keratinocytes (N-K iPSCs and CEP-K iPSCs), fibroblasts, or normal CD34+ cells (N-CB iPSCs). Four putative iPSC clones (two CEP-K iPSCs [3 and 4] and two N-CB iPSCs [10 and 11]) were kept for further characterization. RT-PCR analysis revealed that the endogenous pluripotency-associated genes NANOG (MIM 607937), ESG1 (TLE1 [MIM 600189]), DNMT3B (MIM 602900), REX1 (ZFP42 [MIM 614572]), TERT (MIM 187270), DPPA4 (MIM 614125), and CRIPTO (TDGF1 [MIM 187395]) were robustly expressed in the isolated colonies (Figure 2B). iPSC xenografts into immunodeficient NSG mice resulted in the formation of teratomas, and all three embryonic germ layers demonstrated in vivo pluripotency of the iPSC clones (Figure 2C). Karyotypic analyses confirmed the absence of macroscopic genetic abnormality in all the tested iPSC clones (Figure 2D) except for CEP-K iPSC 3, in which there was a minor abnormality (some mitosis with one X chromosome deletion) (Figure 2D, left panel). As a precaution, this iPSC clone was kept as an uncorrected control for further experiments.
Figure 2.
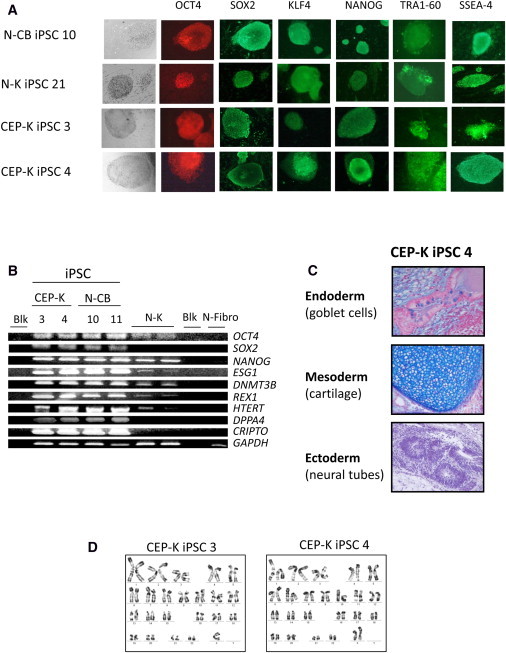
Characterization of iPSC Clones
(A) Representative immunofluorescence of pluripotency markers in human iPSC clones derived from normal CD34+ CB cells (N-CB iPSC 10) and in keratinocytes derived from a normal individual (N-K iPSC 21) and a CEP-affected individual (CEP-K iPSC 3 and CEP-K iPSC 4); staining is with anti-OCT4, anti-SOX2, anti-KLF4, anti-NANOG, anti-SSEA-4, and anti-TRA1-60. MEFs surrounding human iPSCs served as a negative control for immunofluorescence (magnification ×100 or ×200).
(B) Expression of pluripotency-associated genes (OCT4, SOX2, NANOG, ESG1, DNMT3B, REX1, HTERT, DPPA4, and CRIPTO) by RT-PCR cycles (28) from two independent CEP-K iPSC clones and two N-CB iPSC clones. Primary N-K cells and primary normal human fibroblast (N-Fibro) cells were used as controls. GAPDH served as an internal positive control. The following abbreviation is used: Blk, blank (PCR performed without cDNA).
(C) Alcian-blue staining of histological sections of a representative teratoma derived from human CEP-K iPSC 4 shows tissues of all three germ layers (magnification ×200).
(D) Representative karyotypic analysis of two human CEP-K iPSC clones (CEP-K iPSC 3 and CEP-K iPSC 4).
Generation of Safe and Corrected iPSC Clones without Exogenous Reprogramming Factors
To develop safe iPSCs without exogenous reprogramming factors, we excised the reprogramming cassettes, flanked by the loxP sites, by adenovirus-mediated CRE recombinase expression in the four iPSC clones tested (Figure 1C). After subcloning the four iPSCs (two CEP-K iPSCs [3 and 4] and two N-CB iPSCs [10 and 11]), we performed a DNA-PCR analysis to select clones with excision of both reprogramming cassettes (Figure 3A, upper panel). To check the absence of additional recombination with portions of the reprogramming vectors in the three excised clones (4-c, 4-e, and 4-i), we used an additional set of multiple primers all along both proviruses (Figure 3A, lower panel). Gene correction of CEP-K iPSC 4 was then obtained by lentiviral transduction with a HAUPins vector. This vector contains the UROS cDNA sequence under the control of the chimeric HS40 enhancer/ ankyrin-1 erythroid-specific promoter and is shielded by cHS4 insulators in the LTR (Figure 1B). Subsequently, we tested excised subclones from CEP-K iPSC 4 by quantitative-PCR analysis to identify those with only one copy of the HAUPins vector. The sequencing of the proviral IS after LAM-PCR allowed us to locate the IS of HAUPins (Figure 3C). For corrected subclone 4-c, the IS is located on chromosome 1 (position 148,683,731, orientation +) in the sixth intron of RPRD2. For corrected subclone 4-e, the IS is located on chromosome 11 (position 64,963,338, orientation +) in the tenth intron of CAPN1 (MIM 114220). For corrected subclone 4-i, the IS is located on chromosome 3 (position 17,418,684, orientation +) in the fifth intron of TBC1D5. Graphics depicting 600 kb of human genome of the IS region for each clone and all genes present are shown in Figure 3C. Sixteen genes, including three cancer-related genes (APH1A [MIM 607629], ECM1 [MIM 60220], and MCL1 [MIM 159552]; all labeled in red in Figure 3C, upper panel) were found in the 600 kb region around the IS of clone 4-c. In the same way, twenty-eight genes, including three cancer-related genes (SLC22A20 [MIM 611696], CDC42EP2 [MIM 606132], and FRMD8 [Figure 3C, middle panel]) were found near the IS of clone 4-e. In contrast, only two genes (neither of them related to cancer) were found in the 600 kb region of the 4-i IS (Figure 3C, lower panel). Then, we eliminated clone 4-e because of the density of genes in the neighbor of the IS. Characterization of the four excised iPSC clones (4-c, 4-e, 4-i, and 10-f) by RT-PCR (Figure 3B), immunocytochemistry (Figure 3D), and teratoma assays (Figure 3F) confirmed that the subclones retained their pluripotency. No abnormality was found by new karyotypic analysis (Figure 3E).
Figure 3.
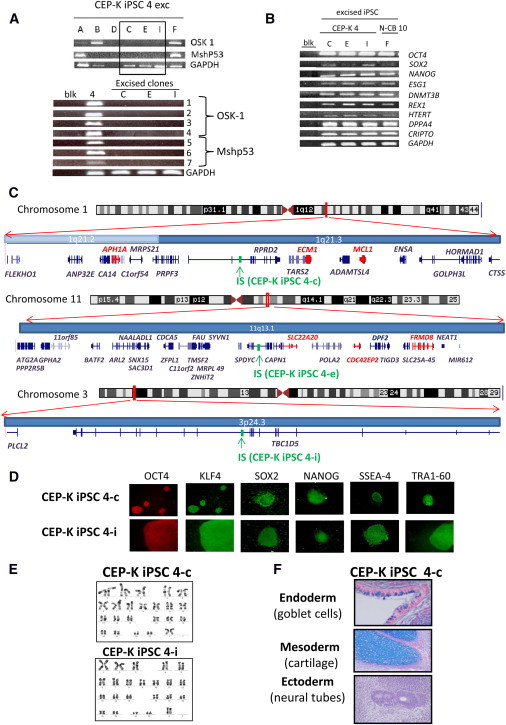
Characterization of iPSC Clones Free of Reprogramming Transgene
(A) Upper panel: PCRs for the integrated vectors OSK 1 and Mshp53 in seven CEP-K iPSC 4 subclones pretreated by CRE adenovirus (subclones are A, B, C, D, E, F, and I). Lower panel: Multiple PCRs performed on DNA from the three excised clones (C, E, and I) and the nonexcised iPSC 4 with seven couples of primers described in (B) for detecting the presence of portions of the recombinant provirus.
(B) Expression of pluripotency-associated genes by RT-PCR from three independent iPSC subclones from CEP-K iPSC 4 and one iPSC subclone from N-CB iPSC 10.
(C) Chromosome ideograms and graphics depicting 300 kb of the human genome on both sides of the proviral IS (in green) of HAUPins from CEP-K iPSC 4-c (upper panel), from CEP-K iPSC 4-e (middle panel), and from CEP-K iPSC 4-i (lower panel). Graphics were obtained with the UCSC Genome Graphs tool. All known genes present in the genomic region are shown (genes implicated in cancer according to the allOnco database are shown in red).
(D) Representative immunofluorescence of pluripotency markers in human iPSC clones that are free of reprogramming transgene and derived from CEP-K iPSC 4-c and 4-i (magnification ×100 or ×200).
(E) Karyotypic analysis of the two excised clones (4-c and 4-i).
(F) Alcian-blue staining of histological sections of a teratoma derived from human CEP-K iPSC 4-c, free of reprogramming transgene, shows tissues of all three germ layers.
Differentiation of iPSCs into HSCs
To generate hematopoietic cells, including hematopoietic stem and progenitors cells (HSPCs), we used the optimized 3 week protocol described by Woods et al.35 with some modifications (days 1–21, Figure 1C). At day 21, after hematopoietic differentiation, we obtained hematopoietic cells (CD45+) derived from all iPSC clones but with various yields of hematopoietic cells (CD45+) depending on the cell fractions (in adherent or nonadherent compartments) (Figures 4A and 4B).
Figure 4.
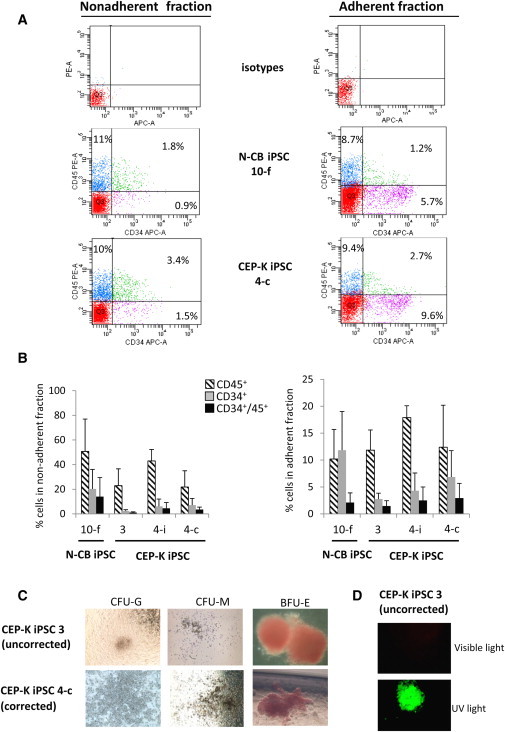
Efficient Hematopoietic Differentiation of iPSCs
(A) Representative FACS analysis of CD45+ and CD34+ cells obtained from N-CB iPSC 10-f (upper panels) and CEP-K iPSC 4-c (lower panels), both free of reprogramming transgenes, after hematopoietic differentiation (at day 21) in nonadherent fraction (left panels) and in adherent fraction (right panels) from the same experiment.
(B) Bar graphs show the average percentages of CD45+, CD34+, and CD34+ and CD45+ cells obtained from four iPSC clones (N-CB iPSC 10-f, uncorrected CEP-K iPSC 3, and corrected CEP-K iPSCs 4-i and 4-c) in nonadherent fractions (left panel) and in adherent fractions (right panel) at day 21 of hematopoietic differentiation (n = 5 independent experiments, mean ± SD). No statistical difference was observed between clones.
(C) Bright-field microscopy of CFUs (a granulocytic CFU [CFU-G], a monocytic CFU [CFU-M], and an erythroid burst-forming unit [BFU-E]) in methylcellulose medium by hematopoietic cells obtained from uncorrected (upper panel) or corrected (lower panel) CEP-K iPSCs (magnification ×100).
(D) Inverted microscopy of a BFU-E in methylcellulose derived from the uncorrected CEP-K iPSC 3 under visible (upper panel) or UV light (lower panel).
In the nonadherent fraction, high yields of CD45+ cells were obtained without a significant difference between clones: N-CB iPSC 10-f had a mean of 50.7%, CEP-K iPSC 3 had a mean of 22.8%, CEP-K iPSC 4i had a mean of 43%, and CEP-K iPSC 4c had a mean of 21.8% (Figures 4A and 4B, left panels). For the CD34+ and CD45+ population, we observed a higher mean of percentage for cells derived from N-CB iPSCs than for cells derived from K iPSCs, but there was no significant difference: N-CB iPSC 10-f had a mean of 14%, CEP-K-iPSC 3 had a mean of 1.1%, CEP-K iPSC 4i had a mean of 4.5%, and CEP-K-iPSC 4c had a mean of 3.5% (Figure 4B, left panel).
In the adherent fraction, no difference in hematopoietic-differentiation efficiency was observed for percentages of CD45+ cells and CD34+ and CD45+ cells between iPSCs derived from keratinocytes and those derived from cord-blood cells (Figure 4B, right panel). Interestingly, however, we found that the mean fluorescent intensity (MFI) of CD34-APC signals was ten times higher in the adherent fraction than in the nonadherent counterpart (Figure 4A).
A more complete cell-surface-marker signature was realized in nonadherent CD45+ cells compartment derived from the corrected CEP-K iPSC 4-i clone. FACS analysis showed the presence of myeloid cells (CD33+: 29.5%), monocytes (CD14+: 27.1%), erythroid cells (CD71+ and GPA+: 11.2%), and B lymphoid cells (CD19+: 2.6%).
In addition to performing cell-surface-marker analysis, we performed colony-forming unit (CFU) assays and observed erythroid burst-forming units (BFU-Es) and CFU granulocyte/monocyte (CFU-GM) colonies derived from all iPSC clones (Figure 4C). As a result of spontaneous porphyrin accumulation, BFU-Es from uncorrected CEP-K iPSC 3 fluoresced in red when viewed under UV light (excitation light 405 nm) (Figure 4D, lower panel), whereas BFU-Es from normal N-CB iPSCs, corrected CEP iPSC 4-c, and CEP iPSC 4-i did not fluoresce. A 3 week in vitro erythroid differentiation was then performed from nonadherent hematopoietic cells (Figure 1C). A very efficient erythroid differentiation was obtained (from 10% CD71+ and glycophorin-A+ at day 10 to 76% at day 17, for example, with CEP-K iPSC 4i) (Figure 5A). Cytological examination revealed abundant basophilic and polychromatic erythroblasts without enucleation (Figure 5B). HPLC analysis showed that these erythroblasts contained mainly fetal hemoglobin, but not adult hemoglobin (not shown).
Enzymatic and Metabolic Rescue of the Corrected Cells
After reprogramming, it is possible that epigenetic modifications induce silencing of the transgene promoter carried by a lentiviral vector. In our model, rescue of UROS activity was dependent on the activity of the HS40/Ank promoter during erythropoiesis. So, we first analyzed whether the chimeric HS40/Ank promoter was still functional and erythroid specific in our iPSC differentiation model. To carefully verify this main prerequisite, we transduced the N-CB iPSC 10-f clone with the HAEPins lentivector (the same as HAUPins but with EGFP cDNA instead of UROS cDNA [Figure 5C]) at a MOI of 10. Erythroid-specific GFP expression was confirmed by FACS analysis at day 21, when the hematopoietic differentiation was complete. Almost all GFP+ cells were Glycophorin A+ and had a high MFI, confirming the erythroid specificity of the vector in the iPSC model (Figure 5D). Surprisingly, inverted fluorescence microscopy at the early stage of hematopoietic differentiation (between days 10 and 14) revealed the presence of erythroid progenitor cells contained in sacs on the OP9 feeder cells; these sacs resembled the yolk sac described in early erythropoiesis during embryogenesis (Figure 5E, middle and left panels). These cells were found in the supernatant medium at day 21 (Figure 5E, right panel). GFP+ BFU-E colonies in methylcellulose assays were observed under an inverted microscope (Figure 5F).
The residual UROS activity in primary CEP keratinocytes used for iPSC generation was much lower than that in normal primary keratinocytes (<0.2 U/mg protein, Figure 6A). We performed quantification of UROS enzymatic activity of erythroid cells derived from iPSCs 21 days after the induction of erythroid differentiation. In erythroid cells derived from N-CB iPSC 10-f, a high level of enzymatic activity was observed in erythroblasts as a result of the strong activity of the endogenous erythroid-specific UROS promoter from the two alleles (mean = 46.3 ± 9.6 U/mg protein). By contrast, the erythroid cells from the uncorrected CEP iPSC 3 displayed very low levels of UROS activity (below <1 U/mg protein). We observed that the enzymatic activity in erythroid cells from the corrected clone (4-i) was six times higher than that in cells from the uncorrected clone 3 (mean = 5.2 ± 0.9 U/mg protein) (Figure 6 A).This activity represents 11% of the UROS activity of the erythroid cells derived from N-CB iPSCs and 52% of the UROS activity of normal keratinocytes (Figure 6A). To examine whether this partial enzymatic correction was translated into a metabolic rescue, we looked at the proportion of fluorocytes at the end of erythroid differentiation. Spontaneous accumulation of porphyrins in erythroid cells derived from the uncorrected CEP-K iPSC 3 clone resulted in a high percentage of fluorocytes (47.3% ± 1.8, n = 3), whereas fluorocytes were almost absent in the erythroblasts derived from N-CB iPSC 10-f (2.6% ± 0.4, n = 3, p < 0.001) (Figures 6B and 6C, upper panels). After gene therapy, metabolic correction was easily demonstrated by the complete disappearance of fluorocytes in the erythroid cells derived from the corrected CEP-K iPSC clones 4-c and 4-i (4.3% ± 1.8 and 6.3% ± 3.5, respectively; p < 0.01 for both clones versus the uncorrected CEP-K iPSC) (Figures 6B and 6C, lower panels). These data demonstrate that a single HAUPins proviral copy in CEP-K iPSC clones is sufficient to allow a metabolic correction of erythroid cells by the disappearance of 90% of porphyrins in erythroid cells.
Figure 6.
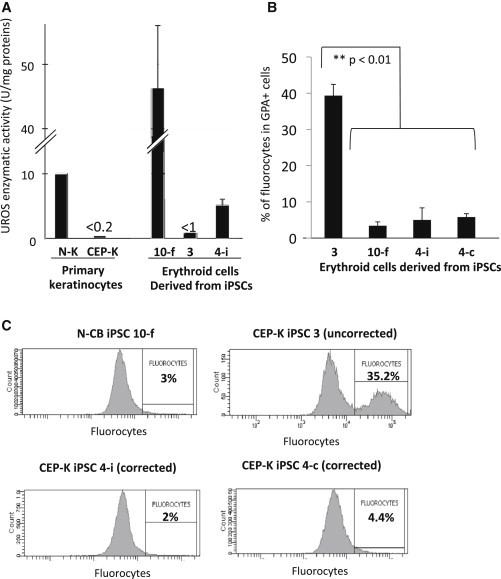
Enzymatic and Metabolic Correction
(A) UROS enzymatic activities in primary N-K and deficient CEP-K cells and in erythroid cells derived from N-CB iPSC 10-f, CEP-K iPSC 3 (uncorrected), and CEP-K iPSC 4-i (corrected) clones.
(B) Bar graphs show average percentages of fluorocytes gated on erythroid GPA+ cells from three independent experiments (mean ± SD).
(C) Representative FACS analysis of the fluorocytes gated on erythroid GPA+ cells derived from N-CB iPSC 10-f and uncorrected CEP-K iPSC 3 (upper panels) and from two corrected clones (4-i and 4-c; lower panels) at day 21 of erythroid differentiation.
Discussion
We generated iPSCs from keratinocytes at a high efficiency (up to 1.4% of input cells) with only two reprogramming vectors. On the basis of our preliminary results, we chose these cells because they had a reprogramming yield 10× higher and 2.5× faster than did human control fibroblasts. The low endogenous expression of pluripotency markers in keratinocytes is in agreement with the higher reprogramming yield in these cells compared with fibroblasts in our model. The efficiency of reprogramming human keratinocytes has also been observed by others studying normal and dystrophic-epidermolysis-bullosa-affected individuals.38,39 The low endogenous expression of KLF4 and MYC in keratinocytes could also explain the ease with which they are reprogramed.38
An efficient hematopoietic differentiation was obtained from two independent N-CB iPSC clones and three independent K iPSC clones. Interestingly, we found that the MFI of CD34-APC signals was ten times higher in the adherent fraction than in the nonadherent counterpart, suggesting the presence of more primitive HSCs in the adherent fraction, as previously observed by Woods et al.35 The type of donor cell can influence the epigenome and the differentiation potential of iPSCs.40,41 Hematopoietic differentiation was supposed to be more efficient with iPSCs derived from cord blood than with those derived from keratinocytes. Surprisingly, in our study, CEP-K iPSCs did not show a hematopoietic differentiation rate that was significantly different from that of N-CB iPSCs. We believe that both downregulation of TP53 and forced expression of MYC are key requisites for erasing the residual epigenetic memory of the primary source of cells. Importantly, our experimental strategy was set to excise the reprogramming vectors for the generation of “clean” iPSCs, i.e., those that are free of reprogramming cassettes. Lack of the remaining reprogramming gene in our iPSCs possibly prevents aberrant de novo methylations, which can be deleterious to efficient further differentiation.41 Methylcellulose assays showed evidence that the cells differentiated from both K iPSCs and N-CB iPSCs were functional hematopoietic progenitors. We can conclude that human primary keratinocytes are easily (1) available from human skin, (2) reprogrammed, and (3) differentiated in HSCs. Therefore, these cells are a very attractive somatic cell source for iPSC-based gene therapy for hematopoietic disorders.
We also report a highly efficient erythroid differentiation with HSCs derived from iPSCs. The erythroid specificity of our vector was demonstrated by a GFP-expressing control vector in which expression of the reporter gene was restricted to GPA+ cells. The UROS enzymatic activity generated in erythroid cells was similar to that previously observed in erythroblasts from normal primary CD34+ peripheral-blood cells.42 A single copy of this vector restored 11% of the UROS enzymatic activity, whereas one allele of the normal endogenous erythroid-specific UROS restored 50%, suggesting that the endogenous UROS enhancer and promoter is almost five times stronger than our enhancer and promoter construct.43 However, this partial activity (11% of the normal activity in erythroblasts) led to a disappearance of 90% of fluorocytes and was then enough to obtain a sufficient metabolic correction of the disease. This is in agreement with recently published results concerning a knockin CEP mouse model11 in which the substitutions p.Cys73Arg (c.217T>C) and p.Val99Leu (c.296G>C) described in this mouse model were associated with 11% of wild-type UROS activity; the animals were just very mildly anemic (hemoglobin: 14 g/dl). This is also in agreement with clinical observations of the absence of phenotypes or minor phenotypes in persons with genotypes inducing residual UROS enzymatic activity.44,45
In a 3 week protocol of in vitro erythroid differentiation, iPSC-derived erythroid cells expressed the majority of fetal hemoglobin (65%), as previously reported.27 This is probably due to an early stage of development, after only 21 days of culture. In vivo experiments in immunodeficient NSG mice could be helpful for modeling long-term erythroid differentiation with the use of iPSC clones derived from CEP cells. However, current humanized mice have not proved to be very useful in assessing human red blood cell function because macrophages limit reconstitution of human red blood cells in them.46
In current HSC-based gene therapy, the recommended transduction level is around 30%–40% so that multiple integration events and associated oncogenic risks can be avoided. However, in CEP, there is no metabolic cross-correction between nontransduced and transduced deficient cells, and the majority of cells have to be corrected.47 Fortunately, we previously demonstrated that gene therapy of CEP mice is facilitated by an in vivo survival advantage of corrected erythroid cells: only 40% of corrected cells were required for obtaining a complete phenotypic correction.12 Therefore, for current CEP gene therapy, a full conditioning regimen prior to grafting is likely to be necessary. By contrast, a major advantage of the iPSC clonal approach is that 100% of HSCs derived from a genetically corrected clone are metabolically corrected. Thus, reinjection of 100% of corrected cells with their survival advantage allows us to consider only a partial conditioning with the advantage of dramatically reducing the morbidity risks of full conditioning.
For clinical applications, safe iPSC-based gene therapy will require considerable efforts for establishing a sufficient number of iPSC clones with (1) a normal karyotype, (2) full pluripotential capability, (3) complete excision of reprogramming cassettes, (4) only one proviral copy number of therapeutic gene, and (5) a rare random GSH. The reprogramming vectors used in this study were flanked by two loxP sites in order to be excised by expression of CRE recombinase. Thus, iPSCs free of potentially oncogenic reprogramming genes could be obtained. We chose adenovirus-mediated CRE transient expression with no risk of genomic integration to obtain a fast and efficient proviral excision. However, with this approach, small DNA from RU5 and the two residual loxP sites (34 bp each) still remains and could potentially cause deleterious effects, such as alteration of regulatory sequences. For clinical application, nonintegrative reprogramming methods could be safer, even if the efficiency of these methods is still very low. Furthermore, one advantage of iPSC-based gene therapy is the clonal approach allowing the characterization of the proviral IS of the therapeutic gene. Here, the IS of the HAUPins vector was mapped by LAM-PCR from the three corrected iPSC clones with only one copy of the transgene. We characterized the exact position of the IS: in both clones, the HAUPins vector was located outside of the exons in the intronic regions of nononcogenic genes (according to the allOnco database). However, three proto-oncogenes were found in the 600 kb region surrounding the IS of both clones 4-c and 4-e. This method allows us to identify and eliminate clones with potential oncogenic risk for clinical translation. The HAUPins vector used is a new generation of lentiviral vector secured by an erythroid-specific promoter and by the insertion of the efficient chicken hypersensitive site 4 (cHS4) chromatin insulator in the ΔU3 sequence from the 3′ LTR. This insulator was designed to prevent deregulation of neighboring genes.48,49 Interestingly, no oncogene was found in the vicinity of the IS for the other clone (4-i). These would-be “GSHs” defined by Papapetrou et al.27 are relatively rare (fewer than 10% of ISs)29 and require a tedious characterization of each IS. In the same way, even the well-known safe AAVS1 locus does not respect all these strict rules because two oncogenes are in its vicinity and it even disrupts one gene.50 An interesting alternative to lentiviral additive therapy for clinical application could be to insert UROS cDNA by the zinc finger nuclease (ZFN) strategy into GSHs chosen in silico.50 Predicted high efficiency of this approach will certainly overpass the rarity of random retroviral ISs in GSHs. In the future, targeted repair of specific mutations will be the “ideal” correction approach but will require design of specific ZFNs for the most frequent UROS mutations.
In conclusion, this study reports iPSC-based gene therapy for porphyria. The next step of this project will be to evaluate the interest of iPSC-gene therapy in our CEP mouse model to study in vivo whether a long-term enzymatic and metabolic correction is possible. Although biosafety concerns still need to be addressed for clinical translation, regenerative therapy for CEP and other genetic red blood cell disorders is very promising.
Acknowledgments
We would like to thank Alice Biberan (from the vectorology platform, Bordeaux) for her technical assistance; Alban Giese for his technical assistance at the experimental histological platform, Canceropole Grand Sud-Ouest, Bordeaux; and Ollivier Milhavet (Institut de Génétique Humaine, Montpellier) for his helpful discussion in the field of iPSC culture. The authors also thank Maison de Santé Protestante de Bordeaux-Bagatelle (Bordeaux) for providing umbilical-cord blood. This work was supported by the Association Française contre les Myopathies, the Agence Nationale de la Recherche (program GENOPAT-2009; grant iPSGENTHER), the Biomedicine Agence, and the Conseil Régional d'Aquitaine.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
allOnco database, http://microb230.med.upenn.edu/protocols/cancergenes.html
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Anderson K.E., Sassa S., Bishop D.F., Desnick R.J. Disorders of heme biosynthesis: X-linked sideroblastic anemia and the porphyrias. In: Scriver R., Beaudet A.L., Sly W.S., Valle E., editors. The Metabolic and Molecular Bases of Inherited Disease. C McGraw-Hill; New York: 2001. pp. 2961–3062. [Google Scholar]
- 2.Richard E., Robert-Richard E., Ged C., Moreau-Gaudry F., de Verneuil H. Erythropoietic porphyrias: animal models and update in gene-based therapies. Curr. Gene Ther. 2008;8:176–186. doi: 10.2174/156652308784746477. [DOI] [PubMed] [Google Scholar]
- 3.Shaw P.H., Mancini A.J., McConnell J.P., Brown D., Kletzel M. Treatment of congenital erythropoietic porphyria in children by allogeneic stem cell transplantation: A case report and review of the literature. Bone Marrow Transplant. 2001;27:101–105. doi: 10.1038/sj.bmt.1702738. [DOI] [PubMed] [Google Scholar]
- 4.Kauffman L., Evans D.I.K., Stevens R.F., Weinkove C. Bone-marrow transplantation for congenital erythropoietic porphyria. Lancet. 1991;337:1510–1511. doi: 10.1016/0140-6736(91)93198-i. [DOI] [PubMed] [Google Scholar]
- 5.Lagarde C., Hamel-Teillac D., De Prost Y., Blanche S., Thomas C., Fischer A., Nordmann Y., Ged C., De Verneuil H. [Allogeneic bone marrow transplantation in congenital erythropoietic porphyria. Gunther's disease] Ann. Dermatol. Venereol. 1998;125:114–117. [PubMed] [Google Scholar]
- 6.Tezcan I., Xu W., Gurgey A., Tuncer M., Cetin M., Oner C., Yetgin S., Ersoy F., Aizencang G., Astrin K.H., Desnick R.J. Congenital erythropoietic porphyria successfully treated by allogeneic bone marrow transplantation. Blood. 1998;92:4053–4058. [PubMed] [Google Scholar]
- 7.Harada F.A., Shwayder T.A., Desnick R.J., Lim H.W. Treatment of severe congenital erythropoietic porphyria by bone marrow transplantation. J. Am. Acad. Dermatol. 2001;45:279–282. doi: 10.1067/mjd.2001.114730. [DOI] [PubMed] [Google Scholar]
- 8.Dupuis-Girod S., Akkari V., Ged C., Galambrun C., Kebaïli K., Deybach J.C., Claudy A., Geburher L., Philippe N., de Verneuil H., Bertrand Y. Successful match-unrelated donor bone marrow transplantation for congenital erythropoietic porphyria (Günther disease) Eur. J. Pediatr. 2005;164:104–107. doi: 10.1007/s00431-004-1575-x. [DOI] [PubMed] [Google Scholar]
- 9.Ged C., Mendez M., Robert E., Lalanne M., Lamrissi-Garcia I., Costet P., Daniel J.Y., Dubus P., Mazurier F., Moreau-Gaudry F., de Verneuil H. A knock-in mouse model of congenital erythropoietic porphyria. Genomics. 2006;87:84–92. doi: 10.1016/j.ygeno.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 10.Bishop D.F., Johansson A., Phelps R., Shady A.A., Ramirez M.C., Yasuda M., Caro A., Desnick R.J. Uroporphyrinogen III synthase knock-in mice have the human congenital erythropoietic porphyria phenotype, including the characteristic light-induced cutaneous lesions. Am. J. Hum. Genet. 2006;78:645–658. doi: 10.1086/502667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bishop D.F., Clavero S., Mohandas N., Desnick R.J. Congenital erythropoietic porphyria: characterization of murine models of the severe common (C73R/C73R) and later-onset genotypes. Mol. Med. 2011;17:748–756. doi: 10.2119/molmed.2010.00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robert-Richard E., Moreau-Gaudry F., Lalanne M., Lamrissi-Garcia I., Cario-André M., Guyonnet-Dupérat V., Taine L., Ged C., de Verneuil H. Effective gene therapy of mice with congenital erythropoietic porphyria is facilitated by a survival advantage of corrected erythroid cells. Am. J. Hum. Genet. 2008;82:113–124. doi: 10.1016/j.ajhg.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kohn D.B., Candotti F. Gene therapy fulfilling its promise. N. Engl. J. Med. 2009;360:518–521. doi: 10.1056/NEJMe0809614. [DOI] [PubMed] [Google Scholar]
- 14.Hacein-Bey-Abina S., Hauer J., Lim A., Picard C., Wang G.P., Berry C.C., Martinache C., Rieux-Laucat F., Latour S., Belohradsky B.H. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2010;363:355–364. doi: 10.1056/NEJMoa1000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aiuti A., Cattaneo F., Galimberti S., Benninghoff U., Cassani B., Callegaro L., Scaramuzza S., Andolfi G., Mirolo M., Brigida I. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N. Engl. J. Med. 2009;360:447–458. doi: 10.1056/NEJMoa0805817. [DOI] [PubMed] [Google Scholar]
- 16.Gaspar H.B., Cooray S., Gilmour K.C., Parsley K.L., Zhang F., Adams S., Bjorkegren E., Bayford J., Brown L., Davies E.G. Hematopoietic stem cell gene therapy for adenosine deaminase-deficient severe combined immunodeficiency leads to long-term immunological recovery and metabolic correction. Sci. Transl. Med. 2011;3:97ra80. doi: 10.1126/scitranslmed.3002716. [DOI] [PubMed] [Google Scholar]
- 17.Biffi A., Aubourg P., Cartier N. Gene therapy for leukodystrophies. Hum. Mol. Genet. 2011;20(R1):R42–R53. doi: 10.1093/hmg/ddr142. [DOI] [PubMed] [Google Scholar]
- 18.Cavazzana-Calvo M., Payen E., Negre O., Wang G., Hehir K., Fusil F., Down J., Denaro M., Brady T., Westerman K. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature. 2010;467:318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nathwani A.C., Tuddenham E.G., Rangarajan S., Rosales C., McIntosh J., Linch D.C., Chowdary P., Riddell A., Pie A.J., Harrington C. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hacein-Bey-Abina S., Garrigue A., Wang G.P., Soulier J., Lim A., Morillon E., Clappier E., Caccavelli L., Delabesse E., Beldjord K. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest. 2008;118:3132–3142. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 22.Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 23.Yu J., Vodyanik M.A., Smuga-Otto K., Antosiewicz-Bourget J., Frane J.L., Tian S., Nie J., Jonsdottir G.A., Ruotti V., Stewart R. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 24.Park I.H., Zhao R., West J.A., Yabuuchi A., Huo H., Ince T.A., Lerou P.H., Lensch M.W., Daley G.Q. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–146. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- 25.Raya A., Rodríguez-Pizà I., Guenechea G., Vassena R., Navarro S., Barrero M.J., Consiglio A., Castellà M., Río P., Sleep E. Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature. 2009;460:53–59. doi: 10.1038/nature08129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ye L., Chang J.C., Lin C., Sun X., Yu J., Kan Y.W. Induced pluripotent stem cells offer new approach to therapy in thalassemia and sickle cell anemia and option in prenatal diagnosis in genetic diseases. Proc. Natl. Acad. Sci. USA. 2009;106:9826–9830. doi: 10.1073/pnas.0904689106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Papapetrou E.P., Lee G., Malani N., Setty M., Riviere I., Tirunagari L.M., Kadota K., Roth S.L., Giardina P., Viale A. Genomic safe harbors permit high β-globin transgene expression in thalassemia induced pluripotent stem cells. Nat. Biotechnol. 2011;29:73–78. doi: 10.1038/nbt.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu D., Alipio Z., Fink L.M., Adcock D.M., Yang J., Ward D.C., Ma Y. Phenotypic correction of murine hemophilia A using an iPS cell-based therapy. Proc. Natl. Acad. Sci. USA. 2009;106:808–813. doi: 10.1073/pnas.0812090106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Papapetrou E.P., Sadelain M. Derivation of genetically modified human pluripotent stem cells with integrated transgenes at unique mapped genomic sites. Nat. Protoc. 2011;6:1274–1289. doi: 10.1038/nprot.2011.362. [DOI] [PubMed] [Google Scholar]
- 30.Chang C.W., Lai Y.S., Pawlik K.M., Liu K., Sun C.W., Li C., Schoeb T.R., Townes T.M. Polycistronic lentiviral vector for “hit and run” reprogramming of adult skin fibroblasts to induced pluripotent stem cells. Stem Cells. 2009;27:1042–1049. doi: 10.1002/stem.39. [DOI] [PubMed] [Google Scholar]
- 31.Robert-Richard E., Lalanne M., Lamrissi-Garcia I., Guyonnet-Duperat V., Richard E., Pitard V., Mazurier F., Moreau-Gaudry F., Ged C., de Verneuil H. Modeling of congenital erythropoietic porphyria by RNA interference: A new tool for preclinical gene therapy evaluation. J. Gene Med. 2010;12:637–646. doi: 10.1002/jgm.1478. [DOI] [PubMed] [Google Scholar]
- 32.Kingsman S.M., Mitrophanous K., Olsen J.C. Potential oncogene activity of the woodchuck hepatitis post-transcriptional regulatory element (WPRE) Gene Ther. 2005;12:3–4. doi: 10.1038/sj.gt.3302417. [DOI] [PubMed] [Google Scholar]
- 33.Chung J.H., Bell A.C., Felsenfeld G. Characterization of the chicken beta-globin insulator. Proc. Natl. Acad. Sci. USA. 1997;94:575–580. doi: 10.1073/pnas.94.2.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmidt M., Schwarzwaelder K., Bartholomae C., Zaoui K., Ball C., Pilz I., Braun S., Glimm H., von Kalle C. High-resolution insertion-site analysis by linear amplification-mediated PCR (LAM-PCR) Nat. Methods. 2007;4:1051–1057. doi: 10.1038/nmeth1103. [DOI] [PubMed] [Google Scholar]
- 35.Woods N.B., Parker A.S., Moraghebi R., Lutz M.K., Firth A.L., Brennand K.J., Berggren W.T., Raya A., Izpisúa Belmonte J.C., Gage F.H., Verma I.M. Brief report: Efficient generation of hematopoietic precursors and progenitors from human pluripotent stem cell lines. Stem Cells. 2011;29:1158–1164. doi: 10.1002/stem.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsai S.F., Bishop D.F., Desnick R.J. Coupled-enzyme and direct assays for uroporphyrinogen III synthase activity in human erythrocytes and cultured lymphoblasts. Enzymatic diagnosis of heterozygotes and homozygotes with congenital erythropoietic porphyria. Anal. Biochem. 1987;166:120–133. doi: 10.1016/0003-2697(87)90554-9. [DOI] [PubMed] [Google Scholar]
- 37.Li H., Collado M., Villasante A., Strati K., Ortega S., Cañamero M., Blasco M.A., Serrano M. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature. 2009;460:1136–1139. doi: 10.1038/nature08290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aasen T., Raya A., Barrero M.J., Garreta E., Consiglio A., Gonzalez F., Vassena R., Bilić J., Pekarik V., Tiscornia G. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat. Biotechnol. 2008;26:1276–1284. doi: 10.1038/nbt.1503. [DOI] [PubMed] [Google Scholar]
- 39.Itoh M., Kiuru M., Cairo M.S., Christiano A.M. Generation of keratinocytes from normal and recessive dystrophic epidermolysis bullosa-induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA. 2011;108:8797–8802. doi: 10.1073/pnas.1100332108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim K., Doi A., Wen B., Ng K., Zhao R., Cahan P., Kim J., Aryee M.J., Ji H., Ehrlich L.I. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467:285–290. doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim K., Zhao R., Doi A., Ng K., Unternaehrer J., Cahan P., Huo H., Loh Y.H., Aryee M.J., Lensch M.W. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat. Biotechnol. 2011;29:1117–1119. doi: 10.1038/nbt.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Géronimi F., Richard E., Lamrissi-Garcia I., Lalanne M., Ged C., Redonnet-Vernhet I., Moreau-Gaudry F., de Verneuil H. Lentivirus-mediated gene transfer of uroporphyrinogen III synthase fully corrects the porphyric phenotype in human cells. J. Mol. Med. 2003;81:310–320. doi: 10.1007/s00109-003-0438-7. [DOI] [PubMed] [Google Scholar]
- 43.Aizencang G., Solis C., Bishop D.F., Warner C., Desnick R.J. Human uroporphyrinogen-III synthase: Genomic organization, alternative promoters, and erythroid-specific expression. Genomics. 2000;70:223–231. doi: 10.1006/geno.2000.6373. [DOI] [PubMed] [Google Scholar]
- 44.Shady A.A., Colby B.R., Cunha L.F., Astrin K.H., Bishop D.F., Desnick R.J. Congenital erythropoietic porphyria: Identification and expression of eight novel mutations in the uroporphyrinogen III synthase gene. Br. J. Haematol. 2002;117:980–987. doi: 10.1046/j.1365-2141.2002.03558.x. [DOI] [PubMed] [Google Scholar]
- 45.Ged C., Moreau-Gaudry F., Richard E., Robert-Richard E., de Verneuil H. Congenital erythropoietic porphyria: Mutation update and correlations between genotype and phenotype. Cell. Mol. Biol. (Noisy-le-grand) 2009;55:53–60. [PubMed] [Google Scholar]
- 46.Hu Z., Van Rooijen N., Yang Y.G. Macrophages prevent human red blood cell reconstitution in immunodeficient mice. Blood. 2011;118:5938–5946. doi: 10.1182/blood-2010-11-321414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moreau-Gaudry F., Mazurier F., Bensidhoum M., Ged C., de Verneuil H. Metabolic correction of congenital erythropoietic porphyria by retrovirus-mediated gene transfer into Epstein-Barr virus-transformed B-cell lines. Blood. 1995;85:1449–1453. [PubMed] [Google Scholar]
- 48.Arumugam P.I., Scholes J., Perelman N., Xia P., Yee J.K., Malik P. Improved human beta-globin expression from self-inactivating lentiviral vectors carrying the chicken hypersensitive site-4 (cHS4) insulator element. Mol. Ther. 2007;15:1863–1871. doi: 10.1038/sj.mt.6300259. [DOI] [PubMed] [Google Scholar]
- 49.Robert-Richard E., Richard E., Malik P., Ged C., de Verneuil H., Moreau-Gaudry F. Murine retroviral but not human cellular promoters induce in vivo erythroid-specific deregulation that can be partially prevented by insulators. Mol. Ther. 2007;15:173–182. doi: 10.1038/sj.mt.6300030. [DOI] [PubMed] [Google Scholar]
- 50.Sadelain M., Papapetrou E.P., Bushman F.D. Safe harbours for the integration of new DNA in the human genome. Nat. Rev. Cancer. 2012;12:51–58. doi: 10.1038/nrc3179. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.