

Abstract
The distal hereditary motor neuropathies (dHMNs) are a heterogeneous group of neurodegenerative disorders affecting the lower motoneuron. In a family with both autosomal-dominant dHMN and dHMN type V (dHMN/dHMN-V) present in three generations, we excluded mutations in all genes known to be associated with a dHMN phenotype through Sanger sequencing and defined three potential loci through linkage analysis. Whole-exome sequencing of two affected individuals revealed a single candidate variant within the linking regions, i.e., a splice-site alteration in REEP1 (c.304-2A>G). A minigene assay confirmed complete loss of splice-acceptor functionality and skipping of the in-frame exon 5. The resulting mRNA is predicted to be expressed at normal levels and to encode an internally shortened protein (p.102_139del). Loss-of-function REEP1 mutations have previously been identified in dominant hereditary spastic paraplegia (HSP), a disease associated with upper-motoneuron pathology. Consistent with our clinical-genetic data, we show that REEP1 is strongly expressed in the lower motoneurons as well. Upon exogeneous overexpression in cell lines we observe a subcellular localization defect for p.102_139del that differs from that observed for the known HSP-associated missense mutation c.59C>A (p.Ala20Glu). Moreover, we show that p.102_139del, but not p.Ala20Glu, recruits atlastin-1, i.e., one of the REEP1 binding partners, to the altered sites of localization. These data corroborate the loss-of-function nature of REEP1 mutations in HSP and suggest that a different mechanism applies in REEP1-associated dHMN.
Main Text
The distal hereditary motor neuropathies (dHMNs), also known as distal spinal muscular atrophies or spinal Charcot-Marie-Tooth (spinal CMT) diseases, are a group of clinically and genetically heterogeneous disorders characterized mainly by slowly progressive distal-limb-muscle weakness and wasting, i.e., a lower-motoneuron phenotype. Minor sensory disturbances or pyramidal signs may also be present. The dHMNs have been subdivided into several categories on the basis of phenotype and inheritance.1 There has been considerable recent progress on the genetic side; 11 genes have been identified to date. Still, for a large fraction of individuals affected with dHMN, a mutation in an as-yet-unidentified gene has to be assumed.2 dHMN type V (dHMN-V [MIM 600794]) is an autosomal-dominant form in which muscle weakness and wasting are predominantly confined to the hands and often exclusively involve thenar and/or interosseus dorsalis I eminences. Mutations in two genes have been described so far. Missense mutations in GARS (MIM 600287) were found in classical dHMN and dHMN-V but also in the clinically overlapping CMT2D syndrome (MIM 601472).3 In BSCL2 (MIM 606158), two missense mutations (c.263A>G [p.Asn88Ser] and c.269C>T [p.Ser90Leu]) affecting glycosylation were identified.4 They are both associated with a broad, often intrafamilial spectrum of phenotypes, which includes classical dHMN-V but also Silver syndrome and spastic paraplegia.5,6 Our recent finding that GARS and BSCL2 explain < 20% of cases suggests further genetic heterogeneity for dHMN-V also.7
The initial aim of the present study was to elucidate the genetic defect in an Austrian family with classical dHMN/dHMN-V, inherited in an autosomal-dominant fashion. After informed consent and approval of the study were obtained from the local ethical committee, nine members (four affected individuals, three at-risk individuals, and two spouses) were enrolled in the study (for pedigree see Figure 1A). Within the first or second decade, all affected individuals had presented with symptoms suggestive of a predominant peripheral motor neuropathy that started in the hands of two individuals and was exclusively confined to the thenar and dorsalis interosseus I muscles (Figure 1B). Family members II-1, III-5 and IV-5 showed lower-limb involvement (Figures 1C and 1D) and walked with a mild steppage gait. None of the affected individuals showed elevated muscle tone or a spastic gait. Affected individuals were diagnosed as either dHMN-V (II-1, III-3, and III-5) or dHMN (IV-5) on the basis of clinical and electrophysiological findings. Detailed clinical and electrophysiological data are provided in Tables S1 and S2, respectively, available online.
Figure 1.
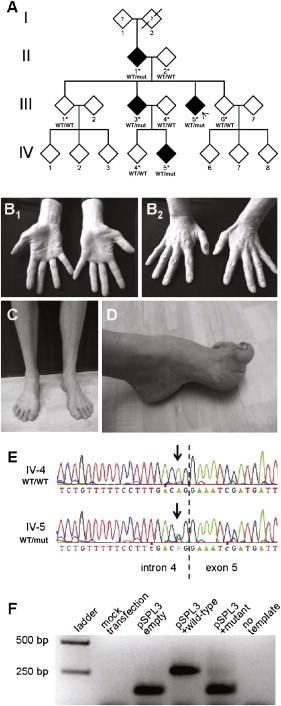
Pedigree Structure, Clinical Images, REEP1 Sanger Sequencing, and Minigene Assay
(A) Partial pedigree. Gender information was removed to preserve anonymity. White and black symbols indicate unaffected and affected individuals, respectively. Asterisks mark family members whose DNA was available and was applied in linkage analysis. Indicated below the identifiers are the results of REEP1 Sanger sequencing (WT: wild-type allele, mut: mutant allele). The arrow denotes the index case.
(B) Wasting of the thenar (B1) and interosseus dorsalis I (B2) muscles in individual III-3.
(C) Distal-lower-limb atrophy in individual IV-5.
(D) Pes cavus foot deformity in individual III-5.
(E) Exemplary results of Sanger sequencing of REEP1 exon 5. The stippled line indicates the intron/exon boundary. The arrow marks nucleotide c.304-2 (NM_022912).
(F) Minigene assay. RT-PCR on HeLa cells transfected with the indicated constructs shows that mutation c.304-2A>G results in skipping of REEP1 exon 5.
All dominant genes known to be mutated in dHMN (BSCL2, GARS, HSPB1 [MIM 602195], HSPB8 [MIM 608014], SETX [MIM 608465], DCTN1 [MIM 601143], and VAPB [MIM 605704]) were excluded previously through the use of Sanger sequencing in the index proband III-5.8 We therefore performed multipoint linkage analysis by applying Affymetrix GeneChip Human Mapping 10K arrays (Affymetrix, Santa Clara, CA, USA) to the DNA of all nine family members available. This revealed three genomic intervals for which the affected individuals, but not the unaffected individuals, share a common haplotype. The corresponding LOD scores of 1.51 represented the maximum score, given the family structure (Figure S1). The three regions reside in chromosomes 2p, 4q, and 8q. They do not contain any of the known genes mutated in dHMN (see Table S3 for numbers of genes contained in each region).2 As these findings argued strongly for a mutation in a hitherto unknown gene for dHMN, we turned to whole-exome sequencing. We selected individuals II-1 and IV-5 (grandparent and grandchild) in order to minimize the expected number of shared variants. Sequencing was performed on a Genome Analyzer HiSeq 2000 system (Illumina, San Diego, CA, USA) after in-solution enrichment of exonic and adjacent intronic sequences (SureSelect Human All Exon 50Mb kit, Agilent, Santa Clara, CA, USA) and indexing of samples for multiplex sequencing (Multiplexing Sample Preparation Oligonucleotide Kit, Illumina). We performed 100 bp paired-end runs, yielding 8.4 and 8.7 Gb of sequence for individuals II-1 and IV-5, respectively. The average read depths were 106 and 111, respectively, with 89% of the targeted regions covered at least 20-fold. Read alignment was performed with Burrows-Wheeler Aligner (version 0.5.8) with application to the human genome assembly hg19. Single-nucleotide variants and small insertions and deletions were called with SAMtools (version 0.1.7). We filtered variants to exclude HapMap SNPs present in dbSNP132 that had an average heterozygosity greater than 0.02, as well as those present in more than two of approximately 800 in-house exomes from individuals with unrelated diseases. Variant annotation was performed with custom scripts. Only rare variants present in both individuals were considered further. This approach left 97 shared candidate variants. However, considering those variants that were neither presented as rare variants in the HapMap SNP database nor present in our in-house controls resulted in a reduction to 53 nonsynonymous sequence variations. Two of these mapped to a region of linkage as defined above, but one of them (rs141088571) was updated in the dbSNP135 database. The second variant at position g.86,479,195 on chromosome 2 localized to a highly conserved nucleotide in intron 4 of REEP1 (MIM 609139) (Figure S2A) and altered one of the two critical bases in the splice acceptor of exon 5 (c.304-2A>G, NM_022912; Genome Reference Consortium h37.p5). As expected, complete segregation was subsequently confirmed with Sanger sequencing (Figure 1E). In silico splice algorithms predicted complete loss of splice-acceptor function (Table S4), thereby suggesting skipping of exon 5 (r.304_417del). Unfortunately, we could not test for an effect on splicing using proband-derived material; consistent with previous reports,9,10 all our attempts to amplify REEP1 cDNA from peripheral blood failed (data not shown). We therefore employed a minigene assay based on a strategy originally developed for exon trapping.11 An ∼800 bp fragment, consisting of REEP1 exon 5 and ∼350 bp of the neighboring introns, was amplified from DNA of the index proband and cloned into the multiple cloning site (MCS) of the pSPL3 vector (Invitrogen, now Life Technologies, Carlsbad, CA, USA). Clones derived from the wild-type allele or from the mutant allele were identified through sequencing. HeLa cells (105 cells in 3.5 cm wells) were transfected with 3 μg of vector. Transfections of empty pSPL3 and mock transfections served as positive and negative controls, respectively. After 16 hr, total RNA was prepared (RNeasy Mini Kit, QIAGEN, Hilden, Germany) and reverse transcribed with the use of random hexamers (RevertAID First Strand cDNA Synthesis Kit, Fermentas, now Thermo Fisher Scientific, Waltham, MA). PCR amplification with vector-specific primers revealed no product from mock-transfected cells and a 153 bp product from cells transfected with empty vector. Transfection of pSPL3 containing the wild-type genomic fragment resulted in an enlarged product (size = 267 bp), consistent with the splicing-mediated inclusion of the exon 5 sequence. With the mutant fragment, in contrast, a product of this size was virtually absent (Figure 1F). The minigene assay thus confirmed that c.304-2A>G results in skipping of exon 5 due to abolition of splice-acceptor function. Though this exon, together with the preceding exon 4, is also missing in one of four Reference Sequence entries for the gene, the existence of significant amounts of a corresponding mRNA species is not supported by the > 100 REEP1-derived expressed sequence tag (EST) sequences currently available at the UCSC genome browser. We therefore conclude that c.304-2A>G, by removing exon 5 from the major isoform, results in an aberrant internal shortening of the encoded 201-residue protein (p.102_139del). This removal of a significant portion of the primary structure (almost 20% of the proteins' residues) that is highly conserved in evolution (Figure S2B), together with perfect segregation and the lack of any other candidate variant, strongly suggested that REEP1 c.304-2A>G was the mutation underlying dHMN in the family studied. Moreover, the variant was neither found in > 10,000 publicly available control chromosomes (∼4,000 exomes in the National Heart, Lung, and Blood Institute [NHLBI] Exome Variant Server; 1,094 genomes in the 1000 Genomes database) nor in our previous analysis of the gene in 366 control individuals,10 in our 800 in-house exomes, or in 88 specifically screened local control individuals (data not shown). Although we cannot definitively exclude the presence of a copy-number aberration or of a deep intronic mutation in the linking regions, it appears highly likely that REEP1 can be mutated in individuals with dominant dHMN.
Heterozygous REEP1 mutations have previously been shown to underlie SPG31 (MIM 610250), a relatively frequent, autosomal-dominant form of hereditary spastic paraplegia (HSP).9 In the HSPs, degeneration of the upper motoneuron leads to a progressively spastic gait.12 The vast majority of individuals suffering from SPG31 show clinically pure HSP; i.e., pyramidal signs predominate, and there is no evidence for lower-motoneuron involvement. Consistent with these clinical and genetic observations, endogenous REEP1 has been detected in the brain13 and in cultured cortical neurons.14 Our initially surprising finding that a REEP1 mutation underlies a lower-motoneuron phenotype prompted us to address the expression of the gene in the spinal cord. The exon 2–6 cDNA sequence of murine Reep1 was amplified from whole-embryo cDNA and cloned into pCRII-TOPO (Invitrogen). Following linearization by appropriate restriction digests, sense and antisense riboprobes were transcribed by T7 and SP6 RNA polymerases, respectively (New England Biolabs, Ipswich, MA). Riboprobes were labeled with digoxigenin (DIG RNA Labeling Kit, Roche Diagnostics, Mannheim, Germany) according to the manufacturer's instructions. Spinal cord was obtained from 4-month-old, fresh-frozen C57BL/6 mice, and series of frontal 20 μm cryotome sections were thawed onto glass slides (SuperFrost Plus, Menzel, Braunschweig, Germany). Representative slides from the cervical, thoracal, and lumbar regions were hybridized with the labeled riboprobes as described previously.15 For neuroanatomical orientation, adjacent sections were stained with thionine.16 This staining, by highlighting all neuronal perikarya, revealed the typical butterfly shape of the gray matter (Figure 2, left panel). No signal was obtained upon hybridization of the Reep1 sense probe (not shown), whereas the signal from the antisense probe specifically labeled neurons with large somata in the ventral horns. This pattern, which was observed at all rostrocaudal levels analyzed (Figure 2, right panel), indicates predominant expression of Reep1 in the spinal cord motoneurons and, therefore, is consistent with a causative association of the REEP1 alteration with the lower-motoneuron phenotype in our family.
Figure 2.
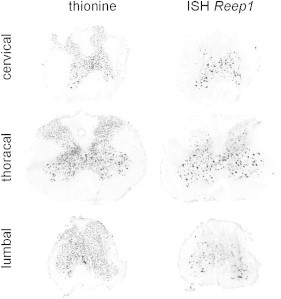
Expression of Reep1 in the Spinal Cord at Different Rostrocaudal Levels
Alternating spinal cord sections (10 μm) from 4-month-old adult mice were stained with thionine or hybridized with a Reep1-specific RNA probe. Thionine labels all neuronal cell bodies, thereby defining the gray matter, whereas the signal from Reep1 in situ hybridization is restricted to large somata in the ventral horns.
A total of 40 HSP-associated REEP1 mutations have been reported thus far.9,10,17–22 As most of these are truncating (i.e., create a preterminal stop codon), nonsense-mediated decay (NMD) of the mutant mRNA is likely.23 Consequently, HSP-associated REEP1 mutations have been proposed to act by a (haplo)insufficiency loss-of-function mechanism.10 The c.304-2A>G mutation identified in the present study, in a family with dHMN rather than HSP, is predicted to result in an mRNA that lacks exon 5 (r.304_417del). Interestingly, this is the only exon of REEP1 whose absence does not result in a frameshift and, thus, would not trigger NMD of the mRNA. Instead, mRNA from the mutant allele should be present at normal levels and result in translation of a normal amount of mutant protein (p.102_139del). We therefore decided to investigate the consequences of removing residues 102-139 from the protein. The coding sequence of human REEP1, with inclusion of the stop codon, was amplified from brain-derived cDNA (Human Multiple Tissue cDNA Panel I, Clontech, Palo Alto, CA, USA) and cloned into the MCS of pCS2+; thus, expression was enabled under the cytomegalovirus promotor of an mRNA coding for the untagged protein. Mutation c.304_417del was introduced through the application of centrally overlapping, seminested PCRs. Using a similar strategy, we also created c.59C>A, i.e., a recurrent HSP-associated alteration,9,10 which, due to its representation of a missense change (p.Ala20Glu), is also predicted to not trigger NMD. The identity of all three constructs was verified through sequencing. After seeding of HeLa cells into 8-well chamber slides (Lab-Tek, Thermo Fisher Scientific) and growth for 16 hr, constructs were transfected with lipofectamine (Invitrogen) and cells were grown for another 20 hr to allow for expression of the vector-derived mRNAs and translation of the corresponding proteins. Cells were then fixed in 4% paraformaldehyde, permeabilized in 0.1% Triton X-100/PBS for 15 min, and incubated in blocking buffer containing goat serum for 30 min. Overexpressed REEP1 was detected with the use of a rabbit polyclonal antibody (SAB2101976, Sigma-Aldrich, St. Louis) and visualized with the use of appropriately labeled secondary antibodies. After counterstaining of the cell nuclei with the use of DAPI, chambers were removed and slides were mounted in ProLong antifade reagent (Invitrogen). Images were obtained via confocal laser-scanning microscopy (LSM510 Meta, Zeiss, Oberkochen, Germany). Wild-type REEP1 localized to a cytoplasmic network (Figure 3A, upper panel). Similar to findings on COS7 cells in a previous report,14 co-overexpression with green fluorescent protein (GFP)-tagged Sec61b revealed a high degree of overlap (Figure S3), thereby confirming a targeting to the tubular portion of the peripheral endoplasmic reticulum (ER).14 The p.Ala20Glu construct showed a severely altered localization. Whereas a reticular pattern was never observed, numerous punctate structures, some of which had a hollow, doughnut-like appearance, were present throughout the cytoplasm (Figure 3A, middle panel). The apparent failure of p.Ala20Glu to localize to the ER is consistent with the proposed involvement of the hydrophobic N terminus in subcellular targeting.10,14 It also corroborates the haploinsufficiency mechanism generally considered to give rise to HSP-causing REEP1 mutations:10 unavailability of the protein at the site of physiological function would effectively resemble an absence of the protein. Interestingly, REEP1 p.102_139del showed a completely different (mis)localization. Similar to the wild-type, some of the signal labeled a reticular network that colocalized with GFP-tagged Sec61b (Figure S3). In addition, however, we observed 5–20 irregularly shaped compact structures per cell, which were confined to the cytoplasm. They were of varying size, with the largest ones measuring several μm and occupying perinuclear positions (Figure 3A, lower panel). Taken together, these observations represented an initial hint that the pathomechanism that applies to p.102_139del and leads to dHMN may be fundamentally different from the mechanism relevant for HSP-associated REEP1 mutations.
Figure 3.
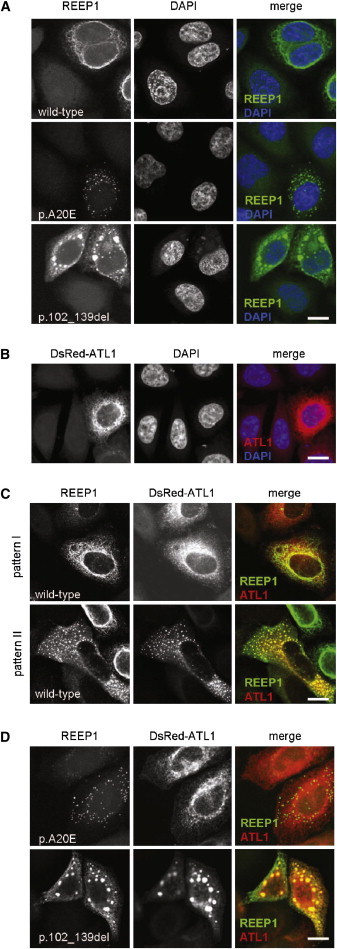
Subcellular Distribution of Overexpressed REEP1 and Atlastin-1
HeLa cells were transfected with the indicated constructs. Untagged REEP1 was detected by immunofluorescence, atlastin-1 by fluorescence of the DsRed tag. Nuclei were stained with DAPI. The scale bar represents 20 μm.
(A) Subcellular targeting of wild-type REEP1 is differentially altered by the indicated pathogenic mutations. Wild-type REEP1 localizes to a cytoplasmic network representing ER14 (upper panel, see also Figure S3A). REEP1 p.Ala20Glu is exclusively found in numerous small, sometimes hollow structures (middle panel). REEP1 p.102_139del, in addition to being found in the ER network (see also Figure S3B), accumulates in 5–20 compact structures, the largest of which occupy a perinuclear position (lower panel).
(B) Atlastin-1, in single transfections, is targeted to a highly branched ER network.27
(C) Cotransfections of wild-type REEP1 and atlastin-1 result in a spectrum of distribution patterns. Shown are two extremes, i.e., a cell in which REEP1 distribution appears unaltered whereas some atlastin-1 is redistributed to small peripheral spots (upper panel) and a cell in which atlastin-1 and much of REEP1 coaccumulate in cytoplasmic punctate structures, which are evenly distributed throughout the cytoplasm (lower panel).
(D) Mutation p.102_139del, but not p.Ala20Glu, impacts on the subcellular distribution of atlastin-1. Whereas cotransfection of REEP1 p.Ala20Glu leaves the atlastin-1 pattern unaltered (upper panel), REEP1 p.102_139del recruits atlastin-1 to the large perinuclear structures also observed in single transfections of this mutant protein (lower panel, compare lower panel in A).
Recent in vitro studies have provided some ideas regarding the probable cellular roles of the family of REEP proteins. REEP5, also known as DP1, has been shown to act in concert with the reticulons in generation and/or stabilization of membrane curvature in the tubular ER.24 Such “shaping” activity depends on the presence of a central, unusually long hydrophobic domain,25 which is found in all REEPs and, thereby, defines the protein family.14 In REEP1, it occupies residues 37 to 76 and, therefore, remains unaffected by both the p.Ala20Glu missense change and the p.102_139 deletion. As it also represents an intramembrane site of interaction with numerous other ER-resident proteins,14,25,26 we hypothesized that REEP1 mislocalization may have an influence on the localization of its binding partners. We chose human atlastin-1, a well-characterized REEP1 interactor,14 and cloned it behind a DsRed tag (pDsRed-C1, Clontech). Overexpression in HeLa cells revealed the reticular pattern (Figure 3B), which has been reported previously and was shown to represent tubular ER.27 Upon co-overexpression of atlastin-1 with wild-type REEP1, a wide range of localization patterns was observed for both proteins. In a fraction of cells, targeting of REEP1 was virtually unaltered whereas some atlastin-1 was redistributed to small punctuate structures in the cell's periphery (Figure 3C, upper panel). This pattern resembled the one previously observed in COS7 cells.14 In other cells, there were many more (up to several hundred) punctuate structures, and these also represented a major site of REEP1 localization (Figure 3C, lower panel). Co-overexpression of atlastin-1 with REEP1 p.Ala20Glu, in contrast, did not have any effect on atlastin-1 distribution. Moreover, there was hardly any overlap with the structures labeled by REEP1 p.Ala20Glu (Figure 3D, upper panel). Finally, atlastin-1 and REEP1 p.102_139del completely colocalized upon co-overexpression. Notably, this included a recruitment of atlastin-1 to the 5–20 large perinuclear “clumps” characteristic of REEP1 p.102_139del (Figure 3D, lower panel; compare Figure 3A, lower panel). Our coexpression data, therefore, strongly argue for the association of an interaction-mediated, possibly toxic gain-of-function effect with p.102_139del (with atlastin-1 not necessarily representing the pathologically relevant interactor), but not with p.Ala20Glu. We suggest that, in general, heterozygous loss-of-function mutations in REEP1 entail upper-motoneuron pathology, whereas mutations possibly gaining a toxic function lead to a lower-motoneuron phenotype. The mechanistic basis of this difference remains enigmatic. Its elucidation, however, can be expected to significantly enhance our understanding of the diverse group of motor neuron diseases.
Our REEP1 mutation associated with classical dHMN differs from the HSP-associated alterations in that it (1) leaves the N-terminal half intact, (2) removes exon 5 residues, and (3) is unlikely to trigger NMD. Only a small fraction of changes in REEP1 are expected to have similar consequences (other alterations which affect the exon 5 splice acceptor, genomic deletions of exon 5, and small in-frame deletions in exon 5). REEP1-associated dHMN may therefore be a rare condition. Consistent with this notion, our screening of a well-characterized cohort of nine additional dHMN-V index cases, for whom GARS and BSCL2 had already been excluded,7 failed to identify a second family. One may, however, imagine that certain mutations confer both a loss- and a gain-of-function effect. Pertinent alterations would be predicted to (1) leave the N-terminal half intact, (2) remove or alter exon 5 residues, and (3) partially trigger NMD and partially escape it. A total of five such mutations have been described (nomenclature refers to NM_022912): c.320T>C (p.Leu107Pro and defective splicing predicted),18 c.337C>T (p.Arg113X),19 c.340_347del (p.Ser114fsX70),18 c.345C>A (p.Tyr115X),18 and c.366G>T (p.Gly122GlyfsX46).22 In the one case analyzed, presence of at least some of the mutant mRNA was shown.22 Clinical details are available for this (c.366G>T) and for one of the other mutations (c.337C>T).19 Strikingly, in both cases, the diagnosis was complicated HSP, based on clear signs of an additional motor neuropathy and the wasting of the small hand muscles in one affected individual.19,22 It thus appears that the combination of too little REEP1 in general and the presence of possibly toxic gain-of-function REEP1 can result in a mixed HSP-dHMN phenotype.
Our identification of REEP1 to represent a gene mutated in classical dHMN considerably broadens the phenotypic spectrum associated with REEP1 mutations. This significant qualitative extension resembles the case of ATL1 (MIM 606439), mutations of which are classically associated with HSP (SPG3A [MIM 182600]) and have recently been found by us to be associated with hereditary sensory neuropathy (HSN1D [MIM 613708]) as well.28 The REEP1 clinical spectrum now strongly matches that of BSCL2 missense mutations p.Asn88Ser and p.Ser90Leu.4 Both protein products are also tightly associated with ER-related functions.14,29 Given the two BSCL2 mutations found all along the phenotypic spectrum5 versus a clear genotype-phenotype correlation for REEP1 (in the present study), the mechanisms underlying differential damage of lower and upper motoneurons appear to differ. From the clinical genetic perspective, however, our findings indicate that both BSCL2 and REEP1 should be considered for a spectrum of disorders ranging all the way from HSP to dHMN. More generally, they strengthen the recent notion that similar molecular themes may underlie clinically differing axonopathies.30
Acknowledgments
We would like to thank the family members for their participation in this study. This work was supported by the Austrian Science Fund (FWF, P23223-B19), the German Research Foundation (DFG) (BE 4069-1/1, DE 307/8-1), and the Tom Wahlig Foundation.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org/
NHLBI Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Harding A.E. Inherited neuronal atrophy and degeneration predominantly of lower motoneurons. In: Dyck P.J., Thomas P.K., editors. Peripheral Neuropathy. W. B. Saunders Company; Philadelphia: 1993. pp. 1051–1064. [Google Scholar]
- 2.Rossor A.M., Kalmar B., Greensmith L., Reilly M.M. The distal hereditary motor neuropathies. J. Neurol. Neurosurg. Psychiatry. 2012;83:6–14. doi: 10.1136/jnnp-2011-300952. [DOI] [PubMed] [Google Scholar]
- 3.Antonellis A., Ellsworth R.E., Sambuughin N., Puls I., Abel A., Lee-Lin S.Q., Jordanova A., Kremensky I., Christodoulou K., Middleton L.T. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am. J. Hum. Genet. 2003;72:1293–1299. doi: 10.1086/375039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Windpassinger C., Auer-Grumbach M., Irobi J., Patel H., Petek E., Hörl G., Malli R., Reed J.A., Dierick I., Verpoorten N. Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome. Nat. Genet. 2004;36:271–276. doi: 10.1038/ng1313. [DOI] [PubMed] [Google Scholar]
- 5.Auer-Grumbach M., Schlotter-Weigel B., Lochmüller H., Strobl-Wildemann G., Auer-Grumbach P., Fischer R., Offenbacher H., Zwick E.B., Robl T., Hartl G., Austrian Peripheral Neuropathy Study Group Phenotypes of the N88S Berardinelli-Seip congenital lipodystrophy 2 mutation. Ann. Neurol. 2005;57:415–424. doi: 10.1002/ana.20410. [DOI] [PubMed] [Google Scholar]
- 6.Irobi J., Van den Bergh P., Merlini L., Verellen C., Van Maldergem L., Dierick I., Verpoorten N., Jordanova A., Windpassinger C., De Vriendt E. The phenotype of motor neuropathies associated with BSCL2 mutations is broader than Silver syndrome and distal HMN type V. Brain. 2004;127:2124–2130. doi: 10.1093/brain/awh232. [DOI] [PubMed] [Google Scholar]
- 7.Rohkamm B., Reilly M.M., Lochmüller H., Schlotter-Weigel B., Barisic N., Schöls L., Nicholson G., Pareyson D., Laurà M., Janecke A.R. Further evidence for genetic heterogeneity of distal HMN type V, CMT2 with predominant hand involvement and Silver syndrome. J. Neurol. Sci. 2007;263:100–106. doi: 10.1016/j.jns.2007.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dierick I., Baets J., Irobi J., Jacobs A., De Vriendt E., Deconinck T., Merlini L., Van den Bergh P., Rasic V.M., Robberecht W. Relative contribution of mutations in genes for autosomal dominant distal hereditary motor neuropathies: a genotype-phenotype correlation study. Brain. 2008;131:1217–1227. doi: 10.1093/brain/awn029. [DOI] [PubMed] [Google Scholar]
- 9.Züchner S., Wang G., Tran-Viet K.N., Nance M.A., Gaskell P.C., Vance J.M., Ashley-Koch A.E., Pericak-Vance M.A. Mutations in the novel mitochondrial protein REEP1 cause hereditary spastic paraplegia type 31. Am. J. Hum. Genet. 2006;79:365–369. doi: 10.1086/505361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beetz C., Schüle R., Deconinck T., Tran-Viet K.N., Zhu H., Kremer B.P., Frints S.G., van Zelst-Stams W.A., Byrne P., Otto S. REEP1 mutation spectrum and genotype/phenotype correlation in hereditary spastic paraplegia type 31. Brain. 2008;131:1078–1086. doi: 10.1093/brain/awn026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buckler A.J., Chang D.D., Graw S.L., Brook J.D., Haber D.A., Sharp P.A., Housman D.E. Exon amplification: a strategy to isolate mammalian genes based on RNA splicing. Proc. Natl. Acad. Sci. USA. 1991;88:4005–4009. doi: 10.1073/pnas.88.9.4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reid E. Many pathways lead to hereditary spastic paraplegia. Lancet Neurol. 2003;2:210. doi: 10.1016/s1474-4422(03)00345-4. [DOI] [PubMed] [Google Scholar]
- 13.Saito H., Kubota M., Roberts R.W., Chi Q., Matsunami H. RTP family members induce functional expression of mammalian odorant receptors. Cell. 2004;119:679–691. doi: 10.1016/j.cell.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 14.Park S.H., Zhu P.P., Parker R.L., Blackstone C. Hereditary spastic paraplegia proteins REEP1, spastin, and atlastin-1 coordinate microtubule interactions with the tubular ER network. J. Clin. Invest. 2010;120:1097–1110. doi: 10.1172/JCI40979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hertel N., Redies C. Absence of layer-specific cadherin expression profiles in the neocortex of the reeler mutant mouse. Cereb. Cortex. 2011;21:1105–1117. doi: 10.1093/cercor/bhq183. [DOI] [PubMed] [Google Scholar]
- 16.Redies C., Takeichi M. Expression of N-cadherin mRNA during development of the mouse brain. Dev. Dyn. 1993;197:26–39. doi: 10.1002/aja.1001970104. [DOI] [PubMed] [Google Scholar]
- 17.Battini R., Fogli A., Borghetti D., Michelucci A., Perazza S., Baldinotti F., Conidi M.E., Ferreri M.I., Simi P., Cioni G. Clinical and genetic findings in a series of Italian children with pure hereditary spastic paraplegia. Eur. J. Neurol. 2011;18:150–157. doi: 10.1111/j.1468-1331.2010.03102.x. [DOI] [PubMed] [Google Scholar]
- 18.Schlang K.J., Arning L., Epplen J.T., Stemmler S. Autosomal dominant hereditary spastic paraplegia: novel mutations in the REEP1 gene (SPG31) BMC Med. Genet. 2008;9:71. doi: 10.1186/1471-2350-9-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hewamadduma C., McDermott C., Kirby J., Grierson A., Panayi M., Dalton A., Rajabally Y., Shaw P. New pedigrees and novel mutation expand the phenotype of REEP1-associated hereditary spastic paraplegia (HSP) Neurogenetics. 2009;10:105–110. doi: 10.1007/s10048-008-0163-z. [DOI] [PubMed] [Google Scholar]
- 20.Liu S.G., Che F.Y., Heng X.Y., Li F.F., Huang S.Z., Lu de G., Hou S.J., Liu S.E., Wang Q., Wang H.P., Ma X. Clinical and genetic study of a novel mutation in the REEP1 gene. Synapse. 2009;63:201–205. doi: 10.1002/syn.20602. [DOI] [PubMed] [Google Scholar]
- 21.de Bot S.T., van de Warrenburg B.P., Kremer H.P., Willemsen M.A. Child neurology: hereditary spastic paraplegia in children. Neurology. 2010;75:e75–e79. doi: 10.1212/WNL.0b013e3181fc2776. [DOI] [PubMed] [Google Scholar]
- 22.Goizet C., Depienne C., Benard G., Boukhris A., Mundwiller E., Solé G., Coupry I., Pilliod J., Martin-Négrier M.L., Fedirko E. REEP1 mutations in SPG31: frequency, mutational spectrum, and potential association with mitochondrial morpho-functional dysfunction. Hum. Mutat. 2011;32:1118–1127. doi: 10.1002/humu.21542. [DOI] [PubMed] [Google Scholar]
- 23.Lykke-Andersen J. mRNA quality control: Marking the message for life or death. Curr. Biol. 2001;11:R88–R91. doi: 10.1016/s0960-9822(01)00036-7. [DOI] [PubMed] [Google Scholar]
- 24.Voeltz G.K., Prinz W.A., Shibata Y., Rist J.M., Rapoport T.A. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell. 2006;124:573–586. doi: 10.1016/j.cell.2005.11.047. [DOI] [PubMed] [Google Scholar]
- 25.Shibata Y., Voss C., Rist J.M., Hu J., Rapoport T.A., Prinz W.A., Voeltz G.K. The reticulon and DP1/Yop1p proteins form immobile oligomers in the tubular endoplasmic reticulum. J. Biol. Chem. 2008;283:18892–18904. doi: 10.1074/jbc.M800986200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu J., Shibata Y., Zhu P.P., Voss C., Rismanchi N., Prinz W.A., Rapoport T.A., Blackstone C. A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell. 2009;138:549–561. doi: 10.1016/j.cell.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanderson C.M., Connell J.W., Edwards T.L., Bright N.A., Duley S., Thompson A., Luzio J.P., Reid E. Spastin and atlastin, two proteins mutated in autosomal-dominant hereditary spastic paraplegia, are binding partners. Hum. Mol. Genet. 2006;15:307–318. doi: 10.1093/hmg/ddi447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guelly C., Zhu P.P., Leonardis L., Papić L., Zidar J., Schabhüttl M., Strohmaier H., Weis J., Strom T.M., Baets J. Targeted high-throughput sequencing identifies mutations in atlastin-1 as a cause of hereditary sensory neuropathy type I. Am. J. Hum. Genet. 2011;88:99–105. doi: 10.1016/j.ajhg.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ito D., Suzuki N. Seipinopathy: a novel endoplasmic reticulum stress-associated disease. Brain. 2009;132:8–15. doi: 10.1093/brain/awn216. [DOI] [PubMed] [Google Scholar]
- 30.Timmerman, V., Clowes, V.E., and Reid, E. (2012). Overlapping molecular pathological themes link Charcot-Marie-Tooth neuropathies and hereditary spastic paraplegias. Exp Neurol., in press. http://dx.doi.org/10.1016/j.expneurol.2012.01.010. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.