

Abstract
Hyperphosphatasia with mental retardation syndrome (HPMRS), an autosomal-recessive form of intellectual disability characterized by facial dysmorphism, seizures, brachytelephalangy, and persistent elevated serum alkaline phosphatase (hyperphosphatasia), was recently shown to be caused by mutations in PIGV, a member of the glycosylphosphatidylinositol (GPI)-anchor-synthesis pathway. However, not all individuals with HPMRS harbor mutations in this gene. By exome sequencing, we detected compound-heterozygous mutations in PIGO, a gene coding for a membrane protein of the same molecular pathway, in two siblings with HPMRS, and we then found by Sanger sequencing further mutations in another affected individual; these mutations cosegregated in the investigated families. The mutant transcripts are aberrantly spliced, decrease the membrane stability of the protein, or impair enzyme function such that GPI-anchor synthesis is affected and the level of GPI-anchored substrates localized at the cell surface is reduced. Our data identify PIGO as the second gene associated with HPMRS and suggest that a deficiency in GPI-anchor synthesis is the underlying molecular pathomechanism of HPMRS.
Main Text
More than 100 cell-surface proteins are attached to the plasma membrane by covalent attachment to a glycosylphosphatidylinositol (GPI) anchor that is assembled in the endoplasmic reticulum (ER) and added to the C terminus of the proteins. Biosynthesis of GPI anchors involves more than 30 different genes.1 Genetic defects in various components of the GPI-anchor-synthesis pathway have been identified in a number of phenotypically diverse diseases that are now also referred to as deficiencies of the GPI-anchor-glycosylation pathway; these diseases belong to a subclass of congenital disorders of glycosylation.2 Somatic mutations in the X-linked gene phosphatidylinositol glycan class A (PIGA, MIM 311770) in hematopoietic stem cells cause paroxysmal nocturnal hemoglobinuria, which manifests as bone-marrow failure, hemolytic anemia, smooth-muscle dystonias, and thrombosis (MIM 300818);3 germline mutations in this gene result in a severe neurological phenotype (MIM 300868).4 Germline mutations in PIGL (MIM 605947), a gene of the early GPI-anchor glycosylation, cause CHIME syndrome (MIM 280000).5 Germline promoter mutations in phosphatidylinositol glycan class M (PIGM [MIM 610273]; Figure 1) result in a severe deficiency of GPI-anchored proteins (GPI-AP) and were found in individuals with portal- and hepatic-vein thrombosis and intractable absence seizures (MIM 610293).6 An autosomal-recessive syndrome caused by mutations in phosphatidylinositol glycan class N (PIGN [MIM 606097]; Figure 1) and characterized by dysmorphic features and multiple congenital anomalies, severe neurological impairment, chorea, and seizures leading to early death was described (MIM 614080).7 We have recently identified mutations in phosphatidylinositol glycan class V (PIGV [MIM 610274]; Figure 1) in individuals with HPMRS (MIM 239300).8–10 However, mutations in this gene are only found in approximately half of the individuals with HPMRS. The purpose of the current study was therefore to investigate the molecular etiology of HPMRS in PIGV-negative individuals.
Figure 1.
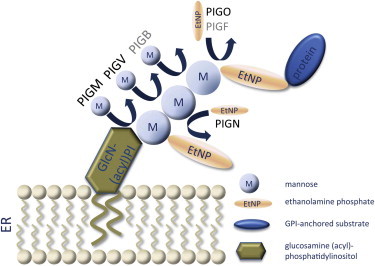
Schematic Illustration of Biochemical Reactions of Late GPI-Anchor Synthesis
The first, second, and third mannose residues are sequentially transferred to GlcN-(acyl)PI by PIGM, PIGV, and PIGB. EtNP is transferred to the first mannose by PIGN and to the third mannose by PIGO and PIGF. Proteins for which the corresponding mutated genes are known to cause congenital disorders of GPI-anchor glycosylation are colored black.
This study was approved by the Charité University Medicine ethics board, and informed consent was obtained from responsible persons (parents) on behalf of all study participants.
We performed whole-exome sequencing of all subjects in family A (see Figure 2 for photos, Figure 4A for a pedigree, and Table S1, available online, for clinical details). The affected sisters, who are 12 and 15 years old, are offspring of nonconsanguineous healthy parents of white British origin.
Figure 2.
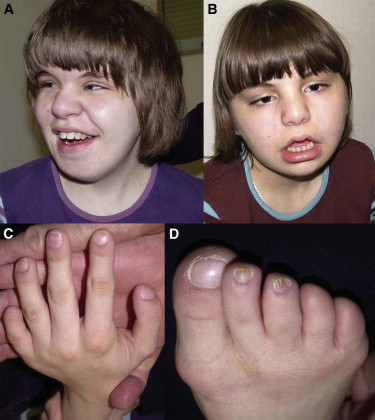
Affected Individuals from Family A
(A) Facial appearance of individual II-1 at the age of 15 years.
(B) Individual II-2 at the age of 12 years.
(C) Nail hypoplasia of the second and fourth digits and absent nail of the fifth digit in individual II-1.
(D) Broad hallux, small nails of the second and third toes, and aplasia of the nails of the fourth and fifth digits in individual II-1.
Figure 4.
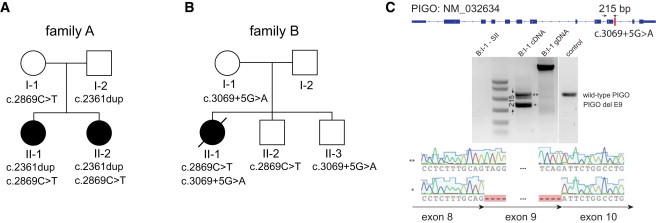
Mutations in PIGO
(A) PIGO mutations in family A as demonstrated by whole-exome sequencing. Both affected daughters were found to be compound heterozygous for the PIGO mutations c.2869C>T and c.2361dup. The father is heterozygous for c.2361dup, and the mother is heterozygous for c.2869C>T.
(B) The affected individual from family B is compound heterozygous for c.2869C>T and c.3069+5G>A. Her mother and healthy brother II-3 are heterozygous only for c.3069+5G>A, and her healthy brother II-2 is heterozygous only for c.2869C>T.
(C) The intronic mutation results in an aberrant splicing product of transcript NM_032634, which is missing 215-bp-long exon 9.
Genomic DNA of all family members was enriched with the target region of all human consensus coding sequence (CCDS) exons with Agilent's SureSelect Human All Exon Kit according to the manufacturer's protocol. Single-read clusters were then generated on the Cluster Station (Illumina). The captured, purified, and clonally amplified library targeting the exome was then sequenced on an Illumina Genome Analyzer II. Whole-exome sequencing (with two lanes of 120 bp unpaired reads) was performed according to the manufacturer's protocol and resulted in more than 5 Gb of high-quality short-read sequence data.
Novoalign was used for the alignment of the sequence reads to the human genome (GRCh37). The percentage alignment of the reads to the targeted exome was calculated with Perl scripts and Bedtools.11 In each individual from family A, around 90% of the target region was covered by more than ten unique sequence reads (Figure S1). Samtools and Perl scripts were used for the detection of single-nucleotide variants as well as small indels (<20 bp) on the short-read alignments.12–14 After common polymorphisms that are also listed in dbSNP (build 132) were filtered out, all detected variants were reduced to family-specific rare variants and annotated with ANNOVAR.15 Assuming an autosomal-recessive pattern of inheritance with 100% penetrance of the phenotype, we filtered for genes with rare homozygous or compound-heterozygous nonsynonymous variants in the affected siblings and identified phosphatidylinositol glycan class O (PIGO) as the single candidate gene (Table S2). The two detected variants in PIGO were c.2869C>T (p.Leu957Phe) (NM_032634.3) and c.2361dup (i.e., the mutation inserts an additional cytosine residue into a homopolymer tract consisting of seven cytosine residues), which led to a frameshift (p.Thr788Hisfs∗5) (Table S3). These variants were compound heterozygous in the affected sisters and were thus compatible with an autosomal-recessive mode of inheritance. The mother was found to be heterozygous for c.2869C>T, and the father was heterozygous for c.2361dup (Figure 4 A and Figure S2).
After validating these variants by Applied Biosystems Sanger sequencing, we subsequently screened 11 unrelated individuals without PIGV mutations for mutations in PIGO. We identified the compound-heterozygous candidate mutations c.2869C>T and c.3069+5G>A in individual II-1 from family B (see Figure 3 for photos, Figure 4B for a pedigree, Table S1 for clinical details, and Table S3). Individual II-1 is the second child of nonconsanguineous parents of European descent. The mother is heterozygous for c.3069+5G>A, and two unaffected siblings are also heterozygous for one of the two detected mutations.
Figure 3.
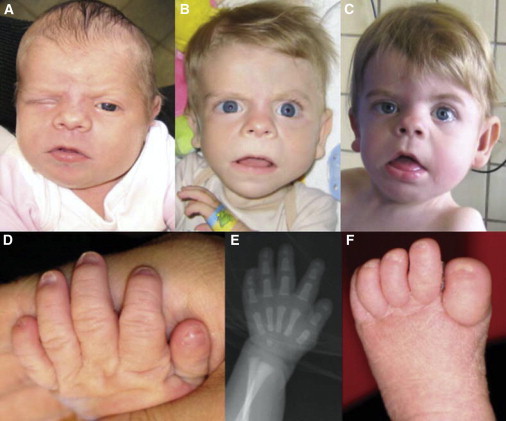
Individual II-1 from Family B at Different Ages
(A) When individual II-1 was 6 weeks of age, facial dysmorphism included wide and downward-slanting palpebral fissures, a broad nasal bridge and tip, ptosis of the right eye, a tented upper lip, large ears with fleshy and uplifted ear lobules, and facial asymmetry.
(B) Facial appearance when individual II-1 was 9 months old.
(C) At the age of 18 months.
(D) Nail hypoplasia of the second and fifth digits and clinodactyly V.
(E) Hand radiograph when individual II-1 was 1 week old. Note brachytelephalangy II to V, mostly affecting fingers II and V, and a broad distal phalanx of the thumb.
(F) Nail hypoplasia of all toes.
We hypothesized that the intronic mutation, c.3069+5G>A, identified in this family would interfere with splicing of the transcript, and we analyzed the effect of this variant on the RNA level. Approximately 3 μg of RNA was isolated from a blood sample of the mother carrying this variant and was used for the first-strand cDNA synthesis. The quality of cDNA was verified by amplification of β-actin cDNA. PIGO transcripts were amplified and sequenced from this cDNA pool. The intronic mutation c.3069+5G>A results in an aberrant splicing product with a skipped exon 9 (Figure 4C); this product was not observed in 13 cDNA controls (Figure S3). The deletion of this 215 bp exon causes a frameshift followed by a premature stop codon. According to data from the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project, there is one heterozygous individual for this intronic mutation out of 5,379 tested individuals, which is consistent with the expected incidence of the disease.
In mammals, PIGO encodes a 1,089 amino acid protein, GPI ethanolamine phosphate transferase 3 (also known as phosphatidylinositol-glycan biosynthesis class O), that is involved in GPI biosynthesis.16,17 The substitution p.Leu957Phe affects the second of four leucine residues in a polyleucine stretch within a hydrophobic transmembrane domain of PIGO. The residue is evolutionarily highly conserved in most species, including mammals, frogs, and zebrafish (Figure S4), and the effect of the detected substitution was classified as disease causing by MutationTaster18 and Polyphen.19 The heterozygote frequency of all three alleles in the European population is below 0.0005, which is expected for rare recessive disorders.20
We first investigated the influence of two PIGO mutations on PIGO function. To test the variants p.Leu957Phe and p.Thr788Hisfs∗5 for effects on PIGO function, we cloned a human PIGO cDNA from a cDNA library derived from Hep3B (a hepatoma cell line) cells, tagged it with FLAG at the N-terminus, and subcloned it into pME.21 PIGO mutants were generated by site-directed mutagenesis. Mutant and wild-type PIGO plasmids were transfected by electroporation into human CD59-expressing PIGO-deficient CHO cells that were derived from aerolysin-resistant clones from chemically mutagenized Chinese hamster ovary (CHO) cells as previously described.22 We determined the levels of GPI-APs CD59 (at the cell surface) and endogenous urokinase plasminogen activator receptor (uPAR) by staining cells with anti-CD59 (5H8) and anti-hamster uPAR antibodies, and we used a flow cytometer (Cant II; BD Biosciences, Franklin Lakes, NJ) with Flowjo software (Tommy Digital, Tokyo, Japan) to analyze them. Wild-type PIGO efficiently restored the levels of CD59 at the cell surface and of uPAR, whereas p.Thr788Hisfs∗5 PIGO did not rescue protein levels at all and the Leu957Phe PIGO induced only very low levels of CD59 and uPAR (Figure 5A). Levels of PIGO in cells were determined by immunoblot analysis. Band intensities of endogenous glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were used as loading controls. Compared with that of wild-type PIGO, the level of Leu957Phe was slightly reduced, whereas the c.2361dup mutation resulted in an increased level of the truncated Thr788Hisfs∗ protein (Figure 5B).
Figure 5.
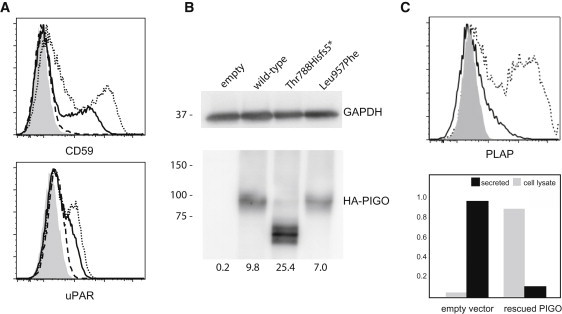
PIGO Activity Is Required for Linking GPI-Anchored Substrates to the Cell Membrane
(A) PIGO-deficient CHO cells were transiently transfected with human wild-type (dotted lines), p.Thr788Hisfs∗5 (dashed line), or p.L957F (solid lines) PIGO cDNA expression constructs. Restoration of the levels of CD59 at the cell surface and of uPAR was assessed 2 days later. Wild-type PIGO efficiently restored levels of CD59 at the cell surface and of uPAR, whereas Thr788Hisfs∗5 PIGO did not restore the level of CD59 at all and the Leu957Phe PIGO induced only very low levels of CD59 and uPAR. The shadowed area indicates an empty-vector transfectant (control).
(B) PIGO levels. The level of the truncated Thr788Hisfs∗5 PIGO (lane 3) was about 2.5× higher than that of wild-type PIGO (lane 2), and the level of Leu957Phe PIGO (lane 4) was slightly lower than that of wild-type PIGO (lane 2).
(C) The level of PLAP at the cell surface after cotransfection with PIGO into PIGO-deficient CHO cells. PIGO-deficient CHO cells were transiently transfected with pME HA-PLAP together with pME PIGO (dotted line) or an empty vector (solid line). The level of PLAP at the cell surface was analyzed by fluorescence-activated cell sorting. PLAP activity was measured in culture medium and cell lysates after cotransfection of PLAP and PIGO cDNAs into PIGO-deficient CHO cells. Relative ALP activity was measured in culture medium (black bars) and in cell lysates (dark gray bar) against the total ALP activity in PIGO-restored CHO cells. Restoration of PIGO activity reduces ALP activity in the medium and increases activity at the cell membrane.
The three individuals carrying PIGO mutations were born with normal measurements, but they had anal stenosis (individuals II-1 and II-2 from family A) or anal atresia with perineal fistula (individual II-1 from family B) (Figures 2 and 3 and Table S1). In individual II-1 from family B, additional malformations included an atrial septal defect, peripheral pulmonary stenosis, left coronal synostosis resulting in plagiocephaly, and an enlarged supratentorial ventricular system. Growth development was delayed in individuals II-1 from family A and II-1 from family B, who also showed marked microcephaly of −5.5 standard deviations (SDs) at the age of 20 months. Psychomotor development was severely retarded in individuals II-1 from family A and II-1 from family B and was moderately delayed in individual II-2 of family A. Individual II-1 from family B developed tonic-clonic seizures at the age of 21 months and died at the age of 22 months as a result of a convulsive crisis. Their common facial signs included wide-set eyes that appeared large because of long palpebral fissures, a short nose with a broad nasal bridge and nasal tip, and a tented mouth. Their fingers showed nail hypoplasia, especially of the second, fourth, and fifth digits, and absent nails of the fifth digits (individuals II-1 and II-2 from family A). Their halluces were broad, but the toes showed small nails or aplasia of nails, especially of the fourth and fifths digits. Serum alkaline phosphatase (ALP) activity was elevated in repeated tests (1,872 U/l in individual II-1 and 1,381 U/l in individual II-2 from family A [the normal range is 200–700 U/l] and 1,436 U/l in individual II-1 from family B [the normal range is 124–341 U/l]).
We next hypothesized that the hyperphosphatasia observed in our patients was due to the PIGO defect. We performed measurements of relative placental ALP (PLAP) activity in cell lysates and medium of PIGO-deficient CHO cells, and we compared them with those of wild-type cells. The mutant CHO cells were cotransfected with pME HA-PLAP (a previously described construct23), pME Luc, and either a PIGO-expressing plasmid (pME F-PIGO) or an empty vector (pME). Media were changed 6 hr later, and the ALP activity in cell lysates and culture media was measured on the following day with the SEAP assay kit (Clontec). Luciferase activities of cell lysates were measured with the Luciferase assay kit (Promega) and were used for the normalization of gene expression. Relative activity in the culture medium was the ratio of normalized ALP activity in the culture medium to the total normalized ALP activity of PIGO-rescued cells. PLAP failed to express on the cell surface (Figure 5C, upper panel), but 90% of the activity was found to reside in the medium from the PIGO-deficient cells (Figure 5C, lower panel). In contrast, transfection with the PIGO-expressing vector restored the surface expression (Figure 5C, upper panel) and prevented oversecretion of PLAP; the majority of PLAP activity remained in the cell (Figure 5C, lower panel). These results indicate that hyperphosphatasia is a result of the release of ALP into serum, which itself is due to a GPI deficiency caused by the PIGO mutation.
A combination of hyperphosphatasia, intellectual disability, and various neurological problems, mainly seizures, has been described by different authors, and the condition has been designated as “hyperphosphatasia with mental retardation.”24,25 In a subset of individuals affected by HPMRS in addition to specific facial features and brachytelephalangy, missense mutations of PIGV have been identified.8–10 Anorectal malformations, Hirschsprung disease, and other organ malformations broaden the clinical spectrum associated with PIGV mutations.8–10 Because PIGV mutations are only found in some of the affected individuals with the core manifestations, genetic heterogeneity seems to be likely. Our study identifies compound-heterozygous mutations in PIGO as a further genetic cause of HPMRS. The characteristic facial appearance, moderate-to-severe developmental delay, hypoplastic or even absent terminal phalanges (including nails), and hyperphosphatasia were present in all affected individuals studied here. In individual II-1 from family B, a significant deceleration of head growth was seen during infancy (the occipital frontal circumference fell to more than 5 SDs below the mean). Whereas individuals carrying PIGV mutations often show growth parameters at or above the mean, individuals with PIGO mutations seem to show more pronounced growth delay. Malformations of the urinary system and heart should be considered as part of this condition.
Our results suggest that PIGO is essential for GPI anchoring of GPI-APs alkaline phosphatase, CD59, and uPAR, and we show that the two tested PIGO mutations have a deleterious effect on PIGO function in a cell-based assay. We have recently shown that GPI transamidase plays a critical role in the secretion of proproteins in the absence of mature GPI, as observed in PIGV-deficient HPMRS.23 The presence of mannose residue of immature GPI is critical for cleavage of the proproteins by GPI transamidase, which provides an explanation for the observation that PIGV deficiency, which affects a relatively late step in the GPI-biosynthesis pathway, leads to hyperphosphatasia, whereas PIGM deficiency, which leads to a defect in the first GPI mannosyltransferase, does not.23 This observation provides a plausible explanation for the fact that mutations in both PIGV, which encodes a protein that transfers the second mannose residue to the GPI anchor,26 and PIGO, which encodes a protein that transfers phosphoethanolamine to the third mannose residue of the GPI anchor,16 are associated with HPMRS. Determining whether there are any clinical differences between these groups will require phenotypic characterization of more individuals with PIGO and PIGV mutations. Given the fact that neither PIGV nor PIGO mutations were identified in ten individuals with a similar phenotype, it seems likely that mutations in other GPI-pathway genes might represent further etiologies of HPMRS.
Acknowledgments
This work was supported by grant 0313911 from the Bundesministerium für Forschung und Technologie and by grants from the Ministry of Education, Culture, Sports, Science, and Technology and the Ministry of Health, Labour, and Welfare of Japan. We wish to thank all patients and their families involved in this study for their generous help.
Contributor Information
Peter N. Robinson, Email: peter.robinson@charite.de.
Denise Horn, Email: denise.horn@charite.de.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org
Agilent eArray, https://earray.chem.agilent.com/earray/
Online Mendelian Ineritance in Man (OMIM), http://www.omim.org
NHLBI Exome Sequencing Project (ESP), http://evs.gs.washington.edu/EVS/
References
- 1.Kinoshita T., Fujita M., Maeda Y. Biosynthesis, remodelling and functions of mammalian GPI-anchored proteins: Recent progress. J. Biochem. 2008;144:287–294. doi: 10.1093/jb/mvn090. [DOI] [PubMed] [Google Scholar]
- 2.Jaeken J. Congenital disorders of glycosylation (CDG): It's (nearly) all in it! J. Inherit. Metab. Dis. 2011;34:853–858. doi: 10.1007/s10545-011-9299-3. [DOI] [PubMed] [Google Scholar]
- 3.Takeda J., Miyata T., Kawagoe K., Iida Y., Endo Y., Fujita T., Takahashi M., Kitani T., Kinoshita T. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell. 1993;73:703–711. doi: 10.1016/0092-8674(93)90250-t. [DOI] [PubMed] [Google Scholar]
- 4.Johnston J.J., Gropman A.L., Sapp J.C., Teer J.K., Martin J.M., Liu C.F., Yuan X., Ye Z., Cheng L., Brodsky R.A., Biesecker L.G. The phenotype of a germline mutation in PIGA: The gene somatically mutated in paroxysmal nocturnal hemoglobinuria. Am. J. Hum. Genet. 2012;90:295–300. doi: 10.1016/j.ajhg.2011.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ng B.G., Hackmann K., Jones M.A., Eroshkin A.M., He P., Wiliams R., Bhide S., Cantagrel V., Gleeson J.G., Paller A.S. Mutations in the Glycosylphosphatidylinositol Gene PIGL Cause CHIME Syndrome. Am. J. Hum. Genet. 2012;90:685–688. doi: 10.1016/j.ajhg.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Almeida A.M., Murakami Y., Layton D.M., Hillmen P., Sellick G.S., Maeda Y., Richards S., Patterson S., Kotsianidis I., Mollica L. Hypomorphic promoter mutation in PIGM causes inherited glycosylphosphatidylinositol deficiency. Nat. Med. 2006;12:846–851. doi: 10.1038/nm1410. [DOI] [PubMed] [Google Scholar]
- 7.Maydan G., Noyman I., Har-Zahav A., Neriah Z.B., Pasmanik-Chor M., Yeheskel A., Albin-Kaplanski A., Maya I., Magal N., Birk E. Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J. Med. Genet. 2011;48:383–389. doi: 10.1136/jmg.2010.087114. [DOI] [PubMed] [Google Scholar]
- 8.Horn D., Krawitz P., Mannhardt A., Korenke G.C., Meinecke P. Hyperphosphatasia-mental retardation syndrome due to PIGV mutations: Expanded clinical spectrum. Am. J. Med. Genet. A. 2011;155A:1917–1922. doi: 10.1002/ajmg.a.34102. [DOI] [PubMed] [Google Scholar]
- 9.Krawitz P.M., Schweiger M.R., Rödelsperger C., Marcelis C., Kölsch U., Meisel C., Stephani F., Kinoshita T., Murakami Y., Bauer S. Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat. Genet. 2010;42:827–829. doi: 10.1038/ng.653. [DOI] [PubMed] [Google Scholar]
- 10.Thompson M.D., Roscioli T., Marcelis C., Nezarati M.M., Stolte-Dijkstra I., Sharom F.J., Lu P., Phillips J.A., Sweeney E., Robinson P.N. Phenotypic variability in hyperphosphatasia with seizures and neurologic deficit (Mabry syndrome) Am. J. Med. Genet. A. 2012;158A:553–558. doi: 10.1002/ajmg.a.35202. [DOI] [PubMed] [Google Scholar]
- 11.Quinlan A.R., Hall I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krawitz P., Rödelsperger C., Jäger M., Jostins L., Bauer S., Robinson P.N. Microindel detection in short-read sequence data. Bioinformatics. 2010;26:722–729. doi: 10.1093/bioinformatics/btq027. [DOI] [PubMed] [Google Scholar]
- 15.Wang K., Li M., Hakonarson H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong Y., Maeda Y., Watanabe R., Inoue N., Ohishi K., Kinoshita T. Requirement of PIG-F and PIG-O for transferring phosphoethanolamine to the third mannose in glycosylphosphatidylinositol. J. Biol. Chem. 2000;275:20911–20919. doi: 10.1074/jbc.M001913200. [DOI] [PubMed] [Google Scholar]
- 17.Flury I., Benachour A., Conzelmann A. YLL031c belongs to a novel family of membrane proteins involved in the transfer of ethanolaminephosphate onto the core structure of glycosylphosphatidylinositol anchors in yeast. J. Biol. Chem. 2000;275:24458–24465. doi: 10.1074/jbc.M003844200. [DOI] [PubMed] [Google Scholar]
- 18.Schwarz J.M., Rödelsperger C., Schuelke M., Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 19.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bell C.J., Dinwiddie D.L., Miller N.A., Hateley S.L., Ganusova E.E., Mudge J., Langley R.J., Zhang L., Lee C.C., Schilkey F.D. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci. Transl. Med. 2011;3:ra4. doi: 10.1126/scitranslmed.3001756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takebe Y., Seiki M., Fujisawa J., Hoy P., Yokota K., Arai K., Yoshida M., Arai N. SR alpha promoter: An efficient and versatile mammalian cDNA expression system composed of the simian virus 40 early promoter and the R-U5 segment of human T-cell leukemia virus type 1 long terminal repeat. Mol. Cell. Biol. 1988;8:466–472. doi: 10.1128/mcb.8.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hong Y., Ohishi K., Inoue N., Kang J.Y., Shime H., Horiguchi Y., van der Goot F.G., Sugimoto N., Kinoshita T. Requirement of N-glycan on GPI-anchored proteins for efficient binding of aerolysin but not Clostridium septicum alpha-toxin. EMBO J. 2002;21:5047–5056. doi: 10.1093/emboj/cdf508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murakami Y., Kanzawa N., Saito K., Krawitz P.M., Mundlos S., Robinson P.N., Karadimitris A., Maeda Y., Kinoshita T. Mechanism for release of alkaline phosphatase caused by glycosylphosphatidylinositol deficiency in patients with hyperphosphatasia mental retardation syndrome. J. Biol. Chem. 2012;287:6318–6325. doi: 10.1074/jbc.M111.331090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mabry C.C., Bautista A., Kirk R.F., Dubilier L.D., Braunstein H., Koepke J.A. Familial hyperphosphatase with mental retardation, seizures, and neurologic deficits. J. Pediatr. 1970;77:74–85. doi: 10.1016/s0022-3476(70)80047-6. [DOI] [PubMed] [Google Scholar]
- 25.Kruse K., Hanefeld F., Kohlschütter A., Rosskamp R., Gross-Selbeck G. Hyperphosphatasia with mental retardation. J. Pediatr. 1988;112:436–439. doi: 10.1016/s0022-3476(88)80331-7. [DOI] [PubMed] [Google Scholar]
- 26.Kang J.Y., Hong Y., Ashida H., Shishioh N., Murakami Y., Morita Y.S., Maeda Y., Kinoshita T. PIG-V involved in transferring the second mannose in glycosylphosphatidylinositol. J. Biol. Chem. 2005;280:9489–9497. doi: 10.1074/jbc.M413867200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.