

Abstract
Copy-number variants (CNVs) are a major contributor to the pathophysiology of autism spectrum disorders (ASDs), but the functional impact of CNVs remains largely unexplored. Because brain tissue is not available from most samples, we interrogated gene expression in lymphoblasts from 244 families with discordant siblings in the Simons Simplex Collection in order to identify potentially pathogenic variation. Our results reveal that the overall frequency of significantly misexpressed genes (which we refer to here as outliers) identified in probands and unaffected siblings does not differ. However, in probands, but not their unaffected siblings, the group of outlier genes is significantly enriched in neural-related pathways, including neuropeptide signaling, synaptogenesis, and cell adhesion. We demonstrate that outlier genes cluster within the most pathogenic CNVs (rare de novo CNVs) and can be used for the prioritization of rare CNVs of potentially unknown significance. Several nonrecurrent CNVs with significant gene-expression alterations are identified (these include deletions in chromosomal regions 3q27, 3p13, and 3p26 and duplications at 2p15), suggesting that these are potential candidate ASD loci. In addition, we identify distinct expression changes in 16p11.2 microdeletions, 16p11.2 microduplications, and 7q11.23 duplications, and we show that specific genes within the 16p CNV interval correlate with differences in head circumference, an ASD-relevant phenotype. This study provides evidence that pathogenic structural variants have a functional impact via transcriptome alterations in ASDs at a genome-wide level and demonstrates the utility of integrating gene expression with mutation data for the prioritization of genes disrupted by potentially pathogenic mutations.
Introduction
Autism, also known as autism spectrum disorders (ASDs [MIM 209850]), is a heterogeneous syndrome defined by impairments in three core domains: social interaction, language, and range of interests.1,2 Autism is not viewed in isolation, but rather as one of several entities collectively referred to as ASDs.1 Both family3,4 and twin studies5 indicate that ASDs are highly heritable neuropsychiatric disorders. A growing body of literature reveals that rare mutations or structural variations dramatically increase disease risk.6–11 This evidence suggests that rare genetic variation plays a larger role in ASDs than was previously suspected.2,12–14
The discovery of rare and recurrent copy-number variants (CNVs) as important pathogenic mutations in ASDs was a watershed in ASD genetics.7,8 Recurrent CNVs such as those at 16p11.2, 22q11.2, 1q21.1, 7q11.23, and 15q11-q13 show statistically significant association with ASDs.15–19 However, the functional impact of these CNVs on downstream RNA expression at both a collective and individual level remains largely unknown. Because CNVs alter copy number and must presumably act via changes in downstream gene expression, an initial study that explored the transcriptome-wide effects of CNVs in human lymphoblast cell lines (LCLs) reported that changes in gene copy number explained roughly 20% of detected transcriptional alterations.20 Although widely assumed, it remains unknown whether rare CNVs identified in autistic individuals have similar effects on transcription levels and subsequent pathophysiology. Evidence certainly exists for the association between rare CNVs as a group and ASDs, but the paucity of cases prohibits proof of genetic association for most individual rare CNVs. Alternative lines of evidence, such as gene-expression data, might confirm the presence of functional alterations related to a particular CNV and would thus be of significant utility.
No risk locus has been identified with a frequency exceeding ∼1% in affected samples, which is consistent with heterogeneity.18,21 Our experimental strategy is predicated on the assumption that analyzing individuals at the resolution of the single gene, rather than as a single group, would yield valuable insight. First, we analyzed gene-expression variance in families with discordant siblings (i.e., one affected and one unaffected sibling) from the Simons Simplex Collection (SSC). Because brain or neuronal tissue is not available from large numbers of individuals with ASDs, we used lymphoblasts, and although they do not express all relevant CNS genes, they do provide useful data for a significantly overlapping set of genes expressed in the CNS.22–24 To assess which dysregulated genes could direct us to pathogenic mutations, we investigated expression variance in each subject and identified genes with significant deviations in expression in individuals' lymphoblasts. To explore the functional impact of CNVs in ASDs at a genome-wide scale, our interrogation utilized the overlap between structural-variation data in a recently published manuscript18 and transcriptional data in a subset of the same population. Our data support the notion that the intersection of gene expression with mutation data, such as CNV calls or single-nucleotide variants (SNVs) derived from exome sequence data, represents an efficacious approach to identifying new mutations and prioritizing autism-susceptibility genes associated with chromosomal structural variation.
Material and Methods
Individuals and LCLs Analyzed in This Study
We analyzed individuals from the SSC in two stages. In the first stage, we collected 386 individuals from 196 families (190 matched sibling pairs plus 5 siblings and 1 proband). In the second stage, we prioritized 53 samples: 42 probands and 8 siblings with de novo CNVs and 3 mothers who carry 16p11.2 events.18 Phenotype information can be found at the Simons Foundation Autism Research Initiative (SFARI) database, and inclusion information is shown in Table S1, available online. This study was approved by the institutional review boards at all participating institutions. The LCLs of the subjects were grown in RPMI 1640 medium with 2 mM L-glutamine and 25 mM HEPES (Invitrogen, Carlsbad, CA, USA), 10% fetal bovine serum, and 1X Antibiotic-Antimycotic solution (Invitrogen) at 37°C in a humidified 5% CO2 chamber. Cells were grown to a density of 6 × 105/ml. Cell lines were maintained in the same conditions so that environmental variation could be kept to a minimum.
Microarray Experiments
A total of 9 × 106 lymphoblasts were seeded in a T-75 flask in 30 ml of fresh medium. After 24 hr, total RNA was extracted from the cells with an RNeasy Mini Kit with DNase treatment (QIAGEN, Valencia, CA, USA) according to the manufacturer's protocol. RNA quantity and quality were measured by ND-100 (Nanodrop, Wilmington, DE, USA) and 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA), respectively. mRNA was hybridized on the Illumina Whole Human Genome Array Human REF-8 version 3.0 according to the manufacturer's protocol.
Sample Quality Control
We used GenomeStudio to convert image data to numerical data as per our typical protocols.25–27 In all, 439 samples (chips) were cross correlated with the use of expression levels for all probe sets. These interarray correlations (IACs)28 were averaged for each array and compared to the resulting distribution of IACs for the dataset.25 Samples with an average IAC < 2 standard deviations (SDs) below the mean IAC for the dataset were removed. We clustered the remaining samples by using average linkage and one IAC as a distance metric to identify the 27 samples (6%) with poor quality. After sample removal, quantile normalization28 was performed in R. To eliminate batch effects, we performed additional normalization by using the R package ComBat29 with the default parameters. ComBat successfully eliminated batch effects, as evidenced by hierarchical clustering and significant improvement of the mean IAC (Figure S1). After data preprocessing, 412 microarrays remained for follow-up analysis; 333 of these had genomic array data and expression data. Three samples (of the 333) are mothers of probands. We used the remaining 330 samples for all of the analyses except the 16p11.2-event analysis. Among the 412 samples, we have 168 pairs of individuals (each pair is from the same family). Ninety-eight out of 168 pairs are matched for sex. To control for potential confounding factors, we used linear regression to remove sex and age effects. We checked the average CNV number per individual, and with the exception of African Americans (60 CNVs per individual), there was no effect of ancestry on CNV frequency (35 CNVs per individual). Because African American samples only compose 3% of our cohort, we retained them to have more statistical power and a better overlap between microarray and genetic data.
Probe Quality Control
We only used probes with evidence of robust expression (detection p value ≤ 0.05 in at least 50% of the samples). By filtering out unexpressed probes, we were left with 11,150 probes (corresponding to 9,524 genes) for analysis. To study the functional impact of CNVs on expression, we filtered the 9,524 genes by restriction to genes that had 30 or more markers (SNPs and monomorphic probes) covering them. For any of these “high-quality” genes that had multiple gene-expression reads, we took the average expression for each gene. This resulted in a set of 8,006 unique genes with gene-expression values.
Outlier-Gene Analysis
For outlier-gene analysis, we calculated the Z statistic for each gene by using the “scale” function in R. We calculated the mean and SD for each expressed gene in cases and controls separately. We selected a cutoff (2 SDs or 3 SDs) to define whether a gene was an outlier in probands or siblings. For outlier analysis not performed in conjunction with CNV data, we used a more stringent cutoff (3 SDs). Subsequently, for the comparison of the overlap between CNVs and transcriptional alterations, we used 2 SDs as a cutoff. These different thresholds were used for two major reasons. When analyzing expression changes in isolation, we used the more conservative 3 SD cutoff to increase stringency. When we integrated genotyping and expression data, we relaxed the statistical threshold to 2 SDs so as to increase power by increasing the number of potentially dysregulated, outlier genes. We use the term “outlier genes” unless we have evidence that the gene is also affected at the genetic level by a CNV. In that case, we call the gene dysregulated to reflect the concept that it is contained within a structural variation and shows significant alteration in gene expression.
Odds-Ratio Analysis
We calculated an odds ratio (OR) with the epitools library in R by using the Wald method, an unconditional likelihood-estimation method. For calculating the OR of dysregulated genes within CNVs (or near CNVs) versus within the genome background, we used the genes not within CNVs in a certain individual as the control group. The two-by-two contingency matrix was made for the calculation of the OR. The two columns are (1) sum of the gene number within CNVs for all probands or all siblings and (2) sum of the gene number not within CNVs for all probands or all siblings; the two rows are (1) sum of the dysregulated genes and (2) sum of the normally expressed genes. We used a Bonferroni correction30 to correct for multiple testing of the OR analysis.
Integrating Expression Data with CNV Data
The CNV list was taken from Table S4 and Table S8 in Sanders et al. (2011). The criteria for subgrouping CNVs were as described,18 and de novo CNVs were determined by the CNV-calling algorithm described therein. Rare CNVs are defined as CNVs with less than 50% overlap with those in the Database of Genomic Variants (DGV).31
Multivariate Linear-Regression Analysis of Expression and Copy Number
For analysis of the relationship between gene expression (genes within CNVs and genes nearby [500 kb]) and copy number, we applied a generalized estimating equation (GEE)32 model by using the “geeglm” function in R. We regressed out the effects of age and sex from the standardized gene-expression data by using a linear model. We then used the residuals obtained from the linear model as the continuous, predicted variable for our expression-value analysis. Next, we (1) obtained a biased sample of 100 gene-expression residuals in which the CNVs were equally represented (50 were duplications and 50 were deletions); (2) matched (by gene) each subject with CNVs to a subject with no CNVs; (3) fit a GEE linear model (which, in all instances that follow, we use to account for any unknown, within-individual correlation among gene expressions) between the gene-expression residuals and the two predictor variables, proband status and copy number; and (4) repeated steps 1–3 for 500 runs to obtain a distribution of coefficients and p values for each predictor. To measure the effects of rareness and CNV size on outlier status in gene expression, we defined as outliers those standardized gene-expression scores with absolute value ≥ 2, which we then encoded as a binary variable. We then used a GEE model with a binomial link for a logistic regression to accommodate the binary nature of the outlier-status variable. Rareness was defined as it was in Sanders et al. (2011). The contrast is with genes falling within CNVs that do not meet these criteria. The estimated size of the CNV was entered as a continuous variable. To study the cis-regulation of CNVs, we performed a similar analysis by using genes 500 kb upstream and downstream of CNVs. The predictors are the same as for genes within CNVs.
Detecting Outlier Genes within CNVs
For 330 individuals, 12,068 CNVs were identified by Sanders et al. (2011). A total of 2,215 out of 12,068 CNVs contain at least one gene expressed in LCLs. We used this list of 2,215 CNVs to study the functional impact of copy number on transcription. For expressed genes within these CNVs, we identified outliers as genes that are ± 2 SDs from the mean expression in all samples. According to this method, 10.7% (238 out of 2,215) of CNVs contain at least one outlier gene.
Enrichment Analysis of Outlier Genes in Rare De Novo CNVs
To compare the dysregulated genes residing in rare de novo CNVs with rare transmitted CNVs and common CNVs, we analyzed all CNVs containing as least one gene expressed in the LCLs; this led to 38 rare de novo CNVs from 37 probands, 419 rare transmitted CNVs from 170 probands, and 353 common CNVs from 184 probands. We used two methods to control for the gene number in each type of CNV. (1) We compared the ratio of dysregulated genes (the number of dysregulated genes divided by the number of genes expressed) between these three groups. The Kruskal-Wallis test, a general form of the multigroup nonparametric test, was used. (2) We matched CNVs for gene-number content: 16 rare de novo CNVs, 18 rare transmitted CNVs, and 31 common CNVs matched for gene number. We used the Bonferroni correction to correct for multiple testing.
To compare the dysregulated genes residing in rare de novo CNVs between probands and siblings, we compared 38 rare de novo CNVs found in 37 probands and three rare de novo CNVs found in three siblings. We calculated the ratio of dysregulated genes within each CNV and compared the rank difference of the ratio by using the Mann Whitney U test.
Permutation Test of Outlier Genes in the Whole Genome
To compute the empirical p value of the significance of the number of dysregulated genes within each rare de novo CNV, we applied a permutation test. We randomly picked one individual and one chromosomal region and selected the adjacent genes to match the number of expressed genes in each rare de novo CNV. We then calculated the number of dysregulated genes in this randomly picked region. We performed 100,000 permutations for each rare de novo CNV.
Multivariate Linear-Regression Analysis of Expression and Copy Number at 16p11.2 and 7q11.23
We used the “geeglm” function in R to fit a linear-regression model between the copy number and expressed genes in 16p11.2 and 7q11.23: expression value ∼ copy number + age + sex. We used general estimating equations to correct for family structure. For 16p11.2 events, we fitted the model by treating the copy number as both a quantitative variable and a factor variable; both methods provided similar results. The p value from the quantitative variable approach is reported in Figure 5. For 7q11.23 duplications, we fitted the model by treating the copy number as a quantitative variable.
Figure 5.
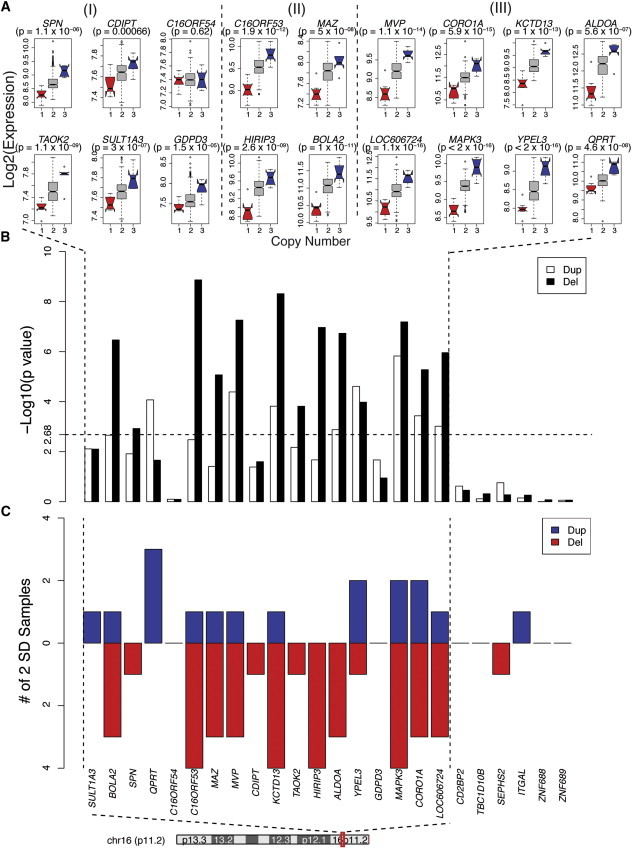
Gene Expression in the 16p11.2 Duplication and Deletion Interval
(A) For each of the expressed genes within the 16p11.2 interval, the log2 expression level is shown for deletions (red), duplications (blue), and controls (gray). The p value was calculated with a multivariable linear-regression model with 16p11.2 cases and 398 controls without a known 16p11.2 event (Material and Methods). Twelve out of 19 expressed genes in deletions have at least a 1.3-fold change measured by microarray, whereas 8 out of 19 genes in duplications show a 1.3-fold or greater change. Group I represents genes that don't reach 1.3-fold change in either duplications or deletions; group II represents genes that have greater than 1.3-fold change in deletions only; and group III represents genes that have greater than 1.3-fold change in both duplications and deletions (dash lines separate the three groups). Error bars are defined as 1.5× the interquartile range.
(B) The –log10 of the p value (t test) for duplications and deletions is shown on the y axis for each gene within the 16p11.2 region and within 500 kb upstream and downstream. The dashed vertical lines show the p value threshold after Bonferroni correction (corrected for 24 genes, p value < 2.1 × 10−3).
(C) Genes showing expression deviating by at least 2 SDs from the mean across 13 samples (seven deletions and six duplications) with 16p11.2 CNVs.
Genome-wide Differential-Expression Analysis
The Limma33 package in R was applied for standard differential expression (DE) analysis in the cases of 16p11.2 deletions, 16p11.2 duplications, and 7q11.23 duplications. Controls were chosen from the pool of all controls with a matched sex ratio to specific cases. In total, seven 16p11.2 deletions (six males and one female) and 120 controls (100 males and 20 females) were used for DE analysis, whereas six 16p11.2 duplications (five males and one female) and 117 controls (100 males and 17 females) were used. In total, three 7q11.23 duplications (two females and one male) and 142 controls (46 males and 96 females) were used.
Multivariate Linear-Regression Analysis of Phenotype
We used the “lm” function in R to fit a linear-regression model between the expressed genes in 16p11.2 and head circumference, which was adjusted for age and sex.34 Age, sex, and expression value were used together as predictors, and the expression value of each gene was normalized by the “scale” function in R program before the linear model was fit.
Principle-Component Analysis
We used the “prcomp” function in R to calculate the first two principle components. Seven 16p11.2 deletions, six 16p11.2 duplications, and three 7q11.23 cases were used. The 20 sporadic cases and 20 controls were selected randomly in our samples. Samples were clustered by the differentially expressed genes (p < 0.01) identified in 16p11.2 duplications, 16p11.2 deletions, and 7q11.23 duplications.
Pathway Analysis
We used DAVID (Database for Annotation, Visualization, and Integrated Discovery) gene ontology (GO) and MetaCore by GeneGo (Thompson Reuters) for pathway analyses. For both analyses, the background was set to the total list of genes expressed in our dataset. The statistical-significance threshold level for all GO enrichment analyses was p < 0.05.
qPCR Validation of CNVs
We used quantitative PCR (qPCR) to confirm the presence or absence of predicted CNVs in lymphoblast DNA. Two control primers were designed within “house-keeping genes” RPP21 (MIM 612524) and ZNF80 (MIM 194553), genes in which no CNVs were reported in the DGV. One microliter of DNA with the concentration of 0.2 μg/μl was used for the qPCR reaction by 2X MyTaq Red Mix (Bioline). A pooled sample from 96 normal SSC siblings was used as the control sample. qPCR was performed on the ABI Prism 7900 (Applied Biosystems, Foster City, CA, USA) with Platinum SYBR Green qPCR SuperMix UDG with ROX (Invitrogen). Thermal cycling consisted of an initial step at 50°C for 2 min, another step of 95°C for 2 min, and 45 cycles each of 95°C for 15 s and 60°C for 30 s. The primers used for qPCR are listed in Table S6. We used the following formula to estimate copy number:18
Estimated copy number = 2(-ΔΔCt [cycle threshold]), for which
-
•
ΔΔCt = (Ct Region:Sample – Ct Ref:Sample) – (Ct Region:Control – Ct Ref:Control)
-
•
Ct Region:Sample = mean Ct values for the region of interest and sample of interest (e.g. ExpPrimer1 and ExpSample1)
-
•
Ct Ref:Sample = mean Ct values for the reference region and sample of interest (e.g. RNase P Primer and ExpSample1)
-
•
Ct Region:Control = mean Ct values for the region of interest and the control sample (e.g. ExpPrimer1 and Ct_pooled_control)
-
•
Ct Ref:Control = mean Ct values for the reference region and the control sample (e.g. RNase P Primer and Ct_pooled_control)
qPCR Validation for Expression Alteration
We used 500 ng of total RNA to make cDNA with SuperScript III First-Strand Synthesis SuperMix (Invitrogen) and random hexamers (Invitrogen). We performed qPCR on an ABI Prism 7900 (Applied Biosystems, Foster City, CA, USA) by using Platinum SYBR Green qPCR SuperMix UDG with ROX (Invitrogen). Thermal cycling consisted of an initial step at 50°C for 2 min, another step of 95°C for 2 min, and 45 cycles each of 95°C for 15 s and 60°C for 30 s. Data were normalized by the quantity of glyceraldehyde-3-phosphate dehydrogenase (GAPDH [MIM 138400]). The gene Ct value of targeted probands was compared to the average Ct values from five unaffected siblings matched for sex and age. The primers used are listed in Table S6.
The ΔCt, ΔΔCt, and fold change of the tested gene were calculated by the following formulas:
-
•
ΔCt for each sample: ΔCt = Ct (tested gene) – Ct (GAPDH)
-
•
ΔΔCt for each sample: ΔΔCt = ΔCt of tested gene in the targeted proband – average ΔCt of test gene in siblings
-
•
Fold change for upregulated genes: fold change = 2−ΔΔCt
-
•
Fold change for downregulated genes: fold change = −2ΔΔCt
Results
Neural-Related Pathways Are Altered in the LCLs of Probands, but Not Siblings
We performed gene-expression profiling by using LCLs from 439 individuals in 244 SSC families consisting of one proband and their unaffected sibling. Data collection occurred in two stages: first, we analyzed 386 individuals from 196 families, and second, we prioritized 53 individuals with de novo CNVs from Sanders et al., 2011 (42 probands, 8 siblings, and 3 mothers who carry 16p11.2 events) (Material and Methods). We cleaned the data to control for confounding factors, such as batch, race, and sex effects (Material and Methods, Figure S1). Four hundred twelve microarrays, accounting for 221 probands, 188 siblings, and 3 mothers and containing a total of 11,150 expressed probes, remained for analysis (360 from stage 1 and 52 from stage 2) (Figure 1). Because the genetic contribution to ASDs includes rare mutations of intermediate to large effect size, differential gene expression is more likely to occur as a consequence within the CNV region in those specific cases relative to other cases and controls. On the basis of this, we applied a simple statistical framework to identify “outlier genes” in individuals, defined as those whose expression is either 2 or 3 SDs from the overall mean expression for that gene across the cohort (Material and Methods).23 We initially took a strict, conservative approach by defining an outlier gene as being ± 3 SDs (99.7% confidence interval) from the mean expression of that gene across all samples (Material and Methods). Probands and siblings had a similar number of outlier genes per individual (8.1 and 10.2, respectively, [p = 0.60] for downregulated genes; 16.6 and 17.6, respectively, [p = 0.76] for upregulated genes; unpaired t test), similar to what is observed when all CNVs are treated as a homogeneous class of events.18 Restricting analysis to brain-expressed genes35 demonstrated that 77% and 73% of outlier genes were expressed in the human fetal brain35 in probands and siblings, respectively (Chi-square p = 1.5 × 10−3). However, no such enrichment was observed for genes expressed in the adult human brain26 (76% of outlier genes in both probands and siblings were expressed in the adult cerebral cortex26; p = 0.95). Similar results were obtained from a comparison with the Allen Brain Atlas data on the human adult brain (see Web Resources) (81% of outlier genes in both probands and siblings were expressed in the human adult brain; p = 0.93). This agrees with most ASD-origin models that posit a fetal or prenatal origin in most cases.1,36–39
Figure 1.
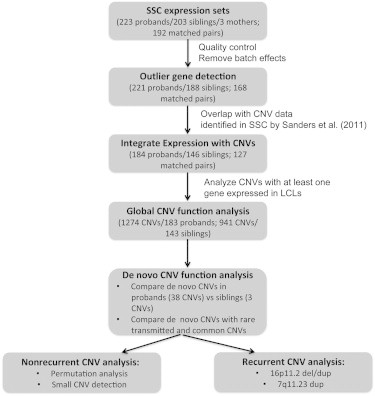
Flow Chart of Expression-Data Analysis and Integration with CNV Data in the SSC
Quality control was done before any data analysis (Figure S1, Material and Methods). The numbers of individuals and CNVs used for downstream analysis are shown in the flow chart.
We next used MetaCore by GeneGo and DAVID GO to explore whether the outlier genes had divergent biological functions or were related to specific pathways (Material and Methods). To control for effects related to transformation, we removed differentially expressed (DEX) genes known to be caused by Epstein-Barr virus (EBV) transformation.40 Remarkably, in addition to several non-neural pathways, a significant enrichment of neural-related pathways in probands was observed. GeneGo (Figure 2) captured enrichment of pathways representing signal transduction, neuropeptide signaling (p = 1.3 × 10−6), development, neurogenesis, and synaptogenesis (p = 3.8 × 10−3). DAVID GO (Table S2) also identified enrichment of similar CNS-related pathways, none of which were enriched in siblings (Table S2). This is not solely due to CNVs (see the overlap analysis below) because >90% of the dysregulated genes in GeneGo neural pathways are outside CNVs. Analyses of the stage-one samples in isolation revealed the same enrichment phenomena, a clear indication that sample selection bias had no impact on the results and a confirmation of the robustness of the GO observations. Thus, despite profiling a peripheral non-neural tissue, we identified significant neural pathways, including some identified in a recent pathway analysis of SCC CNVs,41 previously related to ASDs.26 Our investigation also identified several previously known ASD-susceptibility genes as being outliers; these included OXTR (MIM 167055), PCDH9 (MIM 603581), CNTN4 (MIM 607280), and UBE3A (MIM 601623) (Table S3).
Figure 2.
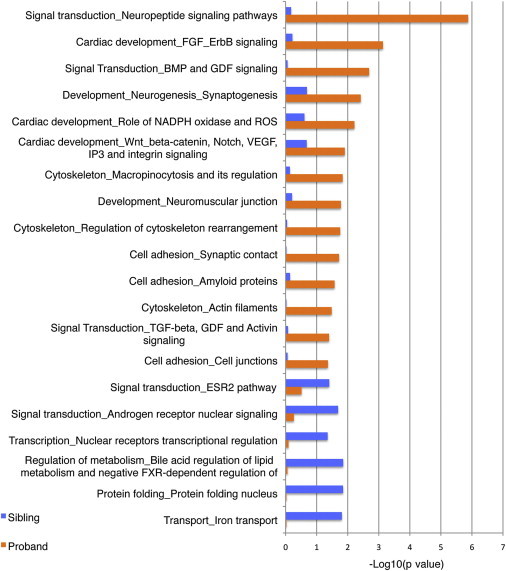
Neural-Related Pathways Are Enriched in Probands versus Siblings
GeneGo was used for the ontology analysis for outlier genes identified in probands and siblings. The –log10 p value is shown with the pathways that were significant (with uncorrected p value < 0.05) in either probands or siblings.
CNVs Affect Transcript Levels in Both Probands and Siblings
We next asked whether CNVs result in transcriptional changes and, conversely, whether dysregulated genes can aid in characterizing structural chromosomal variation. We compared CNVs identified in the SSC, which represents the most extensively validated cohort of CNV calls in ASDs.18 In this study, we used three independent algorithms to identify a robust set of CNVs. Over 500 qPCRs were done in randomly selected individuals representing 403 de novo and 120 transmitted events, providing a high confidence group of CNVs. These CNV data were integrated with microarray gene-expression data, resulting in 330 samples characterized by both genotyping data and expression data (Figure 1).
To analyze the functional impact of CNVs on expression, we employed linear regression to interpret the relationship between copy number and the standard expression value (Z score) by taking a random sample conditional on copy-number status (Material and Methods). We found a significant correlation between copy number and extreme expression (β = 0.524, p value = 1.30 × 10−5); that is, genes in regions of duplication or deletion were far more likely to show extreme expression values than were genes in the genome background. We increased statistical power with a larger sample of outliers by assessing the percentage of CNVs bearing dysregulated genes in 330 samples by using a cutoff of ± 2 SDs (95% confidence interval) (Material and Methods). By calculating the percentage of CNVs with dysregulated genes, we found that 238 out of 2,215 (10.7%) CNVs contained at least one dysregulated gene, and there was a similar ratio between probands (11.5%) and siblings (9.7%) (Material and Methods). Next, we calculated an OR by comparing the average ratio of outlier genes among all expressed genes in the genome to the average ratio of outlier genes from expressed genes within CNVs of the 330 cases and siblings (Material and Methods). We observed that outlier genes were more likely to be present in CNVs than anywhere else in the genome (in probands, OR = 4.3 and Bonferroni p = 2.97 × 10−102; in siblings, OR = 2.6 and Bonferroni p = 2.16 × 10−21). Moreover, in both probands and siblings, the direction of differential expression strongly correlated with the direction of copy-number change. This presents further evidence that outliers are not random. The expected direction of dysregulation was observed in 92% of events (downregulation in deletions and upregulation in duplications) (Table S5).
Previous studies have suggested that CNVs can affect not only the transcriptional level of genes within them but also genes in nearby regions up to 500 kb on either side.20,42 We observed that there were dysregulated genes within 500 kb upstream or downstream of 18.3% of CNVs in both probands and siblings; compared to that in the rest of the genome, this is a significant enrichment (Bonferroni corrected p = 1.4 × 10−7 for probands; Bonferroni corrected p = 1.5 × 10−6 for siblings; Fisher's exact test) (Material and Methods). Interestingly, these changes were less likely to show the expected directionality shift than were those inside the CNVs. Only 43% changed in the direction of CNV dosage, indicating a more complex mechanism of regulation (Table S5). Furthermore, our linear-regression model did not capture a significant relationship between copy number and the expression value of these nearby genes (β = 0.029, p value = 0.234) (Material and Methods), indicating that the relationship between cis gene expression and copy number is not linear.
Outlier Genes Are Enriched in Large Rare De Novo CNVs
Previous studies have shown that rare CNVs, especially rare de novo CNVs, are associated with autism.7,18,43 Here, in general, the rarer the CNV, the higher the chance that it harbors an expression outlier (p = 4.9 × 10−19; Material and Methods). On the basis of the degree of CNV pathogenicity suggested by previous studies (rare de novo > rare transmitted > common), we next investigated whether there was an observable gradient in transcriptional change. Because rare de novo CNVs might be larger or contain more genes than rare transmitted CNVs and common CNVs (Sanders et al., 2011; Levy et al., 2011), we used two methods to control for the potential confounding effect of CNV size (Material and Methods). We calculated the proportion of dysregulated genes within a given CNV by dividing the number of dysregulated genes by the number of expressed genes within CNVs. This yielded a significantly higher proportion of dysregulated genes in rare de novo CNVs than in rare transmitted CNVs and common CNVs in probands (p < 2.0 × 10−16, Kruskal-Wallis test) (Figure 3A). We then compared an arbitrary cohort of CNVs matched for gene number in probands (16 rare de novo CNVs, 18 rare transmitted CNVs, and 31 common CNVs). This comparison detected significantly more dysregulated genes in probands' rare de novo CNVs than in the other two CNV classes (p = 1.5 × 10−5, Kruskal-Wallis test) (Figure 3B). The results signify that, not only are genic segments enriched in rare de novo CNVs in probands, but these rare de novo CNVs are enriched with dysregulated genes even after correction for gene number within the CNV.
Figure 3.
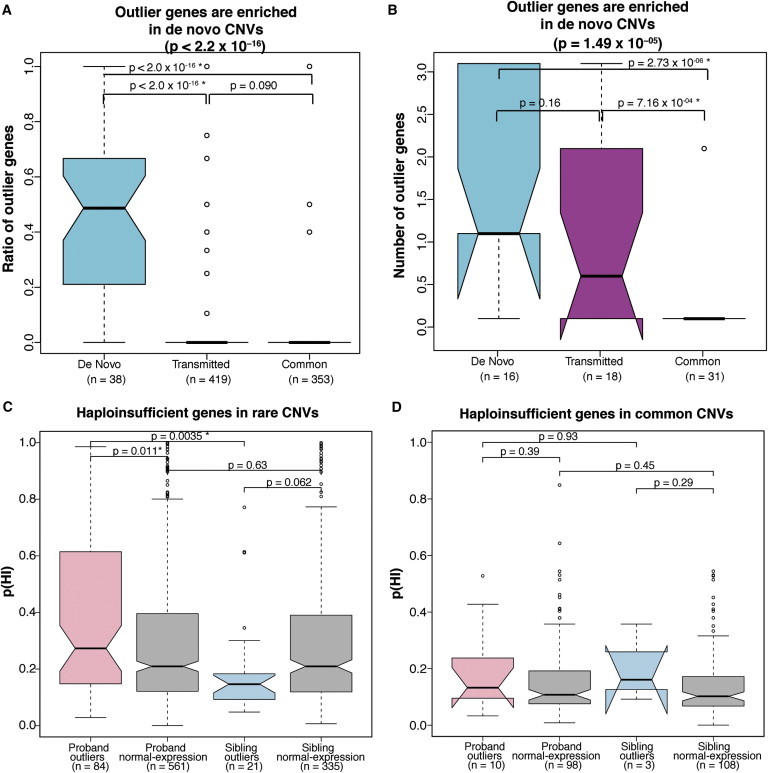
Outlier Genes Are Enriched in Rare De Novo CNVs in Probands
(A) The box plot depicts the ratio of dysregulated genes (the number of dysregulated genes within a CNV versus the total number of genes within that CNV) in each of the three types of CNVs (rare de novo CNVs, rare transmitted CNVs, and common CNVs). The Krusakal-Wallis test p value is shown.
(B) The box plot shows the number of dysregulated genes in three types of CNVs with matched gene number.
(C) The box plot compares haploinsufficiency (HI) scores of downregulated genes (2 SDs) in rare deletions in probands and siblings with those of normally expressed genes within CNVs. The HI score of dysregulated genes in rare deletions in probands is significantly higher than that of the normally expressed genes, whereas the HI score of dysregulated genes in rare deletions in siblings is significantly lower than that of the normally expressed genes (Mann Whitney U test).
(D) The box plot compares HI scores of downregulated genes (2 SDs) in common deletions in probands and siblings with those of normally expressed genes within CNVs. The Mann Whitney U test p value is shown for each pairwise comparison.
A star indicates a statistically significant p value after Bonferroni correction (p < 0.017 in A and B; p < 0.0125 in C and D). Error bars for these four panels are defined as 1.5× the interquartile range.
We next performed an independent assessment of predictions of CNV pathogenicity on the basis of the gene-expression data by employing a recently developed bioinformatics method for the assessment of haploinsufficiency (HI).44 To assess haploinsufficiency on a gene-by-gene level and correct for the potential confound of CNV size, we calculated HI probabilities (pHI), which estimate the likelihood of being haploinsufficient for each dysregulated gene involved in rare deletions in probands versus siblings.44 We combined rare de novo CNVs with rare transmitted CNVs to increase statistical power and focused our analysis on deletions because deletions, not duplications, are associated with HI. A significantly higher pHI probability was observed in probands than in siblings, consistent with increased pathogenicity of CNVs in probands (Figure 3C). We also compared dysregulated genes with nondysregulated genes within the same CNV. Importantly, the pHI of genes that are downregulated in probands is significantly greater than in those genes that do not change expression within rare deletions, showing a relationship between expression dysregulation and predicted pathogenicity (Bonferroni p = 4.4 × 10−2, Mann Whitney U test) (Figure 3C). In contrast, downregulated genes in siblings actually have a lower HI than non-differentially-expressed genes within rare deletions, as would be predicted on the basis of the presumed relative nonpathogenicity of these expression changes (Bonferroni p = 0.25, Mann Whitney U test; Figure 3C). We tested the gene pHI in common deletions in probands versus siblings as a control. No difference was observed, which is expected on the basis of the presumed lack of pathogenicity of these events (Figure 3D).
Transcriptional Data Aids Prioritization of Small and Nonrecurrent CNVs
We next reasoned that gene expression could help prioritize the potential pathogenicity of rare nonrecurrent CNVs, an important step, given that even large de novo CNVs occur in 1%–2% of controls. To identify whether genes within a defined genomic region were significantly dyregulated, we compared the percentage of dysregulated genes within each CNV with random expectations on the genome background (Material and Methods). Twenty-seven out of 40 rare de novo CNVs identified in probands had significantly more dysregulated genes than did the genome background (p < 0.05, permutation test) (Table 1). Our analysis highlights a number of nonrecurrent CNVs that have not previously been shown to be associated with ASDs; these include deletions at 3q27, 3p13, and 3p26 and duplications at 2p15 and 13q14. To verify the altered expression detected by microarrays, we selected 12 genes in 8 corresponding nonrecurrent CNVs to validate by qPCR (Material and Methods). Nine of 12 (75%) genes were confirmed by qPCR, supporting the robustness of these analyses (Figures S5A and S5B).
Table 1.
Gene Dysregulation in De Novo CNVs
| Individual | Loci | Type | Size (kb) | % Outlier Genes | Empirical p Valuea | Outlier Genes |
|---|---|---|---|---|---|---|
| 12184.p1 | 12p11.22 | deletion | 13,000 | 63% | 1.00 × 10−5 | >10 genes |
| 11233.p1 | 15q23 | deletion | 5,000 | 53% | 1.00 × 10−5 | ADPGK,BBS4,KIF23,MYO9A,NPTN,PARP6,PKM2,RPLP1 |
| 11090.p1 | 16p11.2 | deletion | 600 | 47% | 1.00 × 10−5 | ALDOA,BOLA2,C16ORF53,CORO1A,HIRIP3,KCTD13,LOC606724,MAPK3,MAZ |
| 11540.p1 | 16p11.2 | deletion | 600 | 58% | 1.00 × 10−5 | >10 genes |
| 12451.p1 | 16p11.2 | deletion | 600 | 62% | 1.00 × 10−5 | ALDOA,C16ORF53,CDIPT,CORO1A,HIRIP3,KCTD13,MAPK3,MAZ,MVP,YPEL3 |
| 11435.p1 | 16p13.3 | deletion | 1,200 | 76% | 1.00 × 10−5 | >10 genes |
| 11080.p1 | 1p34.3 | duplication | 5,000 | 64% | 1.00 × 10−5 | >10 genes |
| 12239.p1 | 22q11.21 | deletion | 1,400 | 93% | 1.00 × 10−5 | >10 genes |
| 11129.p1 | 7q11.23 | duplication | 1,400 | 57% | 1.00 × 10−5 | BAZ1B,BCL7B,EIF4H,LAT2,NSUN5,STX1A,TBL2,WBSCR22 |
| 12420.p1 | 1q21.1 | duplication | 1,000 | 71% | 3.00 × 10−5 | ACP6,BCL9,CHD1L,GPR89A,PRKAB2 |
| 12032.p1 | 3p13 | deletion | 5,000 | 67% | 5.00 × 10−5 | ARL6IP5,C3ORF64,SUCLG2,TMF1,FOXP1,LMOD3 |
| 11154.p1 | 7q11.23 | duplication | 1,000 | 43% | 0.00011 | BAZ1B,BCL7B,CLIP2,EIF4H,LAT2,WBSCR22 |
| 11046.p1 | 3p26.2 | deletion | 700 | 100% | 0.00012 | ITPR1,SETMAR,SUMF1 |
| 12343.p1 | 13q14.11 | duplication | 500 | 75% | 0.00039 | ELF1,MRPS31,WBP4 |
| 11551.p1 | 16p13.2 | duplication | 500 | 75% | 0.00039 | CARHSP1,PMM2,USP7 |
| 12594.p1 | 7q11.23 | duplication | 300 | 75% | 0.00039 | BCL7B,NSUN5,TBL2 |
| 12647.p1 | 16p11.2 | duplication | 500 | 32% | 0.00046 | BOLA2,CORO1A,KCTD13,MAPK3,MVP,SULT1A3 |
| 11353.p1 | 17q12 | deletion | 1,600 | 50% | 0.00106 | AATF,ACACA,TADA2L |
| 12235.p1 | 9q34.11 | duplication | 600 | 36% | 0.00108 | ODF2,PTGES2,SET,SLC27A4 |
| 12435.p1 | 16p11.2 | duplication | 600 | 25% | 0.00365 | CORO1A,IMAA,MAZ,SPN |
| 11433.p1 | 16p11.2 | deletion | 500 | 21% | 0.006 | ALDOA,KCTD13,MVP,SPN |
| 11555.p1 | 16p11.2 | duplication | 700 | 21% | 0.006 | C16ORF53,LOC606724,MAPK3,QPRT |
| 11435.p1 | 9p24.2 | duplication | 3,000 | 33% | 0.01022 | DOCK8,KIAA0020 |
| 11962.p1 | 10q11.23 | duplication | 1,700 | 100% | 0.02 | CSTF2T |
| 12339.p1 | 3q27.2 | deletion | 100 | 100% | 0.02 | SFRS10 |
| 12224.p1 | 22q13.1 | deletion | 200 | 50% | 0.035 | ADSL |
| 11343.p1 | 2p15 | duplication | 1,700 | 50% | 0.035 | XPO1 |
| 12007.p1 | 15q11.2 | duplication | 2,200 | 33% | 0.05 | UBE3A |
| 11680.p1 | 16p11.2 | deletion | 500 | 12% | 0.05 | MAPK3,MVP |
| 12100.p1 | 16p11.2 | deletion | 600 | 12% | 0.05 | C16ORF53,HIRIP3 |
| 11532.p1 | 17p13.1 | duplication | 800 | 33% | 0.05 | FAM64A |
| 12295.s1 | 19p13.3 | duplication | 300 | 50% | 0.00038 | C19ORF22,POLRMT,PTBP1,RNF126 |
| 12117.s1 | 17q23.1 | duplication | 2,000 | 67% | 0.0026 | APPBP2,PPM1D |
The p values were calculated by a permutation test (Material and Methods).
We next examined whether expression data could inform our analysis of small, potentially pathogenic CNVs. Figure 4 shows four examples of small rare CNVs observed in probands with a relatively high ratio of outlier genes. Both the CNVs and expression alterations in these four examples were validated by PCR (Material and Methods). One example involves a case with both a 16p11.2 deletion (Figure 4D) and a small rare Xq28 deletion affecting the expression level of TMLHE (MIM 300777).45 A recent study46 has shown that a deletion of TMLHE exon 2 was 2.82× more frequent in probands from male-male multiplex autism families than in controls,46 suggesting that TMLHE is a putative autism candidate gene. Because transcription levels are affected within these CNVs, the data presented in Figure 4 clearly warrant follow up in additional cohorts.
Figure 4.
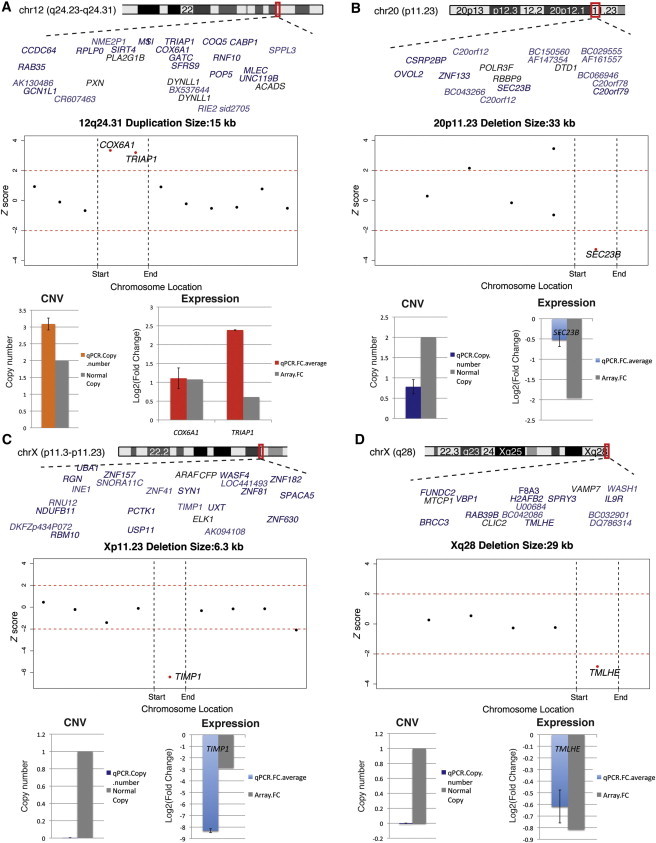
Outlier Genes Highlight Small but Likely Functional CNVs
(A) A small duplication with a high ratio of dysregulated genes.
(B, C, and D) Small deletions with high ratios of dysregulated genes. The Z scores of all expressed genes within the CNV interval and within 500 kb upstream and downstream are shown. Outlier genes (2 SDs; red) within the CNVs are shown. A bar plot shows the qPCR validation for both copy-number change and the expression alteration. Error bars represent the SD of three replicates of qPCR experiments.
Transcriptional Alterations in Recurrent CNVs: 16p11.2 Duplications and Deletions and 7q11.23 Duplications
To determine whether gene-expression analysis could help differentiate 16p11.2 deletions and duplications (MIM 611913) and identify dysregulated candidate genes, we conducted an examination of the effects of the 16p11.2 CNV on gene expression within the interval (Figure 5). First, we validated the dysregulation of three genes of interest, ALDOA (MIM 103850), MAPK3 (MIM 601795), and CORO1A (MIM 605000), from across the interval in five cases of 16p11.2 deletion by using qPCR to provide technical validation of a cross section of the microarray results (Figure S5C).
This examination generated several notable observations. Using a multivariate linear-regression model, we observed a positive correlation between transcription level and 16p11.2 copy number; this highlighted the group of genes most correlated with 16p11.2 dosage: MAPK3 (p < 2 × 10−16), YPEL3 (MIM 609724) (p < 2 × 10−16), CORO1A (p = 6 × 10−15), and KCTD13 (MIM 608947) (p = 1 × 10−13) (Figure 5A) (Material and Methods). Second, deletions had a larger effect on transcriptional level and, compared with duplications, contained more genes with altered expression (Figure 5), which agrees with a recently published 16p11.2 mouse model.47 We also studied the expression pattern in the three mothers carrying 16p11.2 events (two duplications and one mosaic deletion) (Figure S3). Consistent with their lack of clinical ASD diagnosis, carriers looked similar to controls and, relative to cases, had few changes in gene expression (p = 8.5 × 10−5, Kruskal-Wallis test) (Figure S3). This suggests that changes in expression levels might at least partially explain the molecular mechanism of incomplete penetrance of 16p11.2 events observed in parents and some offspring.
To determine trans-regulation of 16p11.2 events and explore whether 16p11.2 duplications and deletions affect similar or divergent biological pathways, we performed genome-wide DEX analysis and focused on changes outside of the CNV region (Material and Methods). We observed 70 DEX genes in 16p11.2-deletion cases and 135 DEX genes in 16p11.2-duplication cases (p < 0.01). Strikingly, no overlap was evident in DEX genes between the two conditions. GO enrichment analysis revealed that in deletions, pathways containing DEX genes were enriched with neural-related ontologies, whereas no such enrichment was observed in duplications (Figures 7A and 7B) (Material and Methods). This suggests that 16p11.2 deletions and duplications interrupt distinct molecular pathways, providing a functional basis for the different phenotypes observed in these two conditions.
Figure 7.
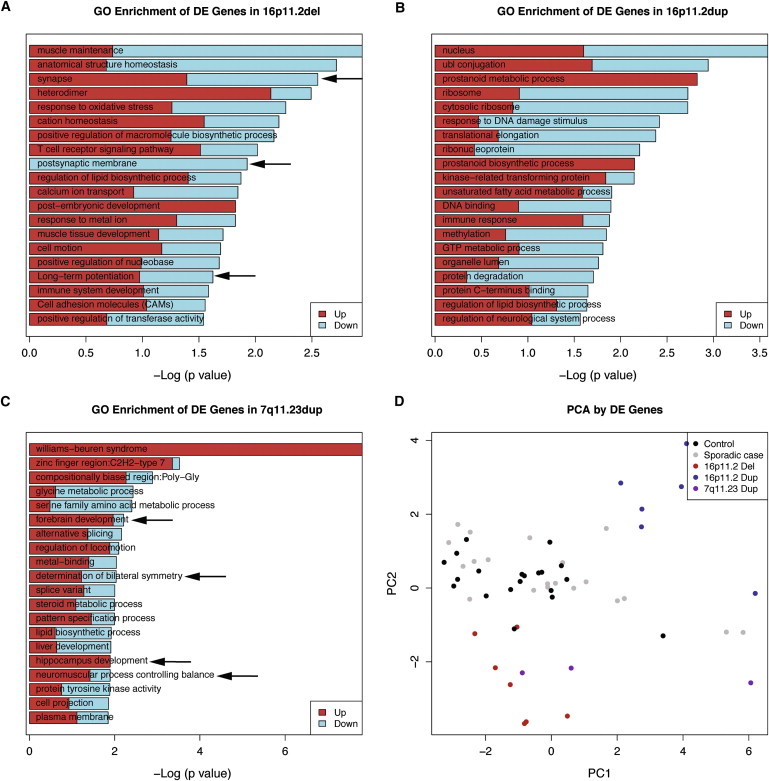
GO Enrichment Analysis and PCA Highlight Distinct Molecular Pathways in 16p11.2 Duplications and Deletions
(A) GO enrichment analysis of the 307 genes (p < 0.05) showing altered expression in deletions (DAVID). The –log10 of the uncorrected p value is shown in (A)–(C).
(B) GO enrichment analysis of the 698 genes (p < 0.05) showing altered expression in duplications (DAVID).
(C) GO enrichment of the 439 genes (p < 0.05) showing altered expression in 7q11.23 duplications (DAVID).
(D) Scatter plot of the first two components of 16p11.2 cases, 7q11.23 cases, sporadic-autism cases, and controls. Samples are clustered on the basis of PCA. Seven 16p11.2-deletion probands (red), six 16p11.2-duplication probands (green), and three 7q11.23-duplication (purple) probands were included. As a comparison group, 20 randomly selected sporadic-autism probands (blue) and 20 randomly selected controls (black) were included. The first two principle components were used for the formation of a two-dimensional space. The merged list of DEX genes (p < 0.01) in 16p11.2 duplications, 16p11.2 deletions, and 7q11.23 duplications was utilized for PCA.
Previous studies indicate that 16p11.2-deletion cases have significant macrocephaly, whereas cases with duplications have microcephaly.48–54 To explore whether variance in gene expression in the 16p11.2 region can be related to variance in head circumference, we applied a multivariate linear-regression model (Material and Methods). The most significantly associated genes within the CNVs are TAOK2 (MIM 613199), CORO1A, KCTD13, and QPRT (MIM 606248) (Figure S4). This is not a circular association reflecting the confounding of DE with gene dosage and head circumference because several of the genes, including TAOK2, most associated with HC are not among the most DEX genes in the region. Remarkably, the changes in these genes' expression accounted for more than 50% of the variance in head circumference. Given the sample size, this should be treated as a preliminary observation that warrants follow up. However, it suggests that alterations in gene expression in peripheral blood can be related to disease-relevant CNS phenotypes.
Another recurrent event associated with autism is the Williams-Beuren syndrome (MIM 194050) 7q11.23 deletion.18,19 We observed that, similar to the 16p11.2 events, this region contains multiple dysregulated genes, including BCL7B (MIM 605846), EIF4H (MIM 603431), and LAT2 (MIM 605719), consistently changing in all three cases (Figure 6). Outside of the region, we observed 85 DEX genes in individuals with 7q11.23 duplications (p < 0.01). In this gene list, GO analysis identified several enriched developmental pathways, including forebrain development, determination of bilateral symmetry, and hippocampus development, providing another demonstration that CNS-relevant pathways can be recovered from peripheral blood.
Figure 6.
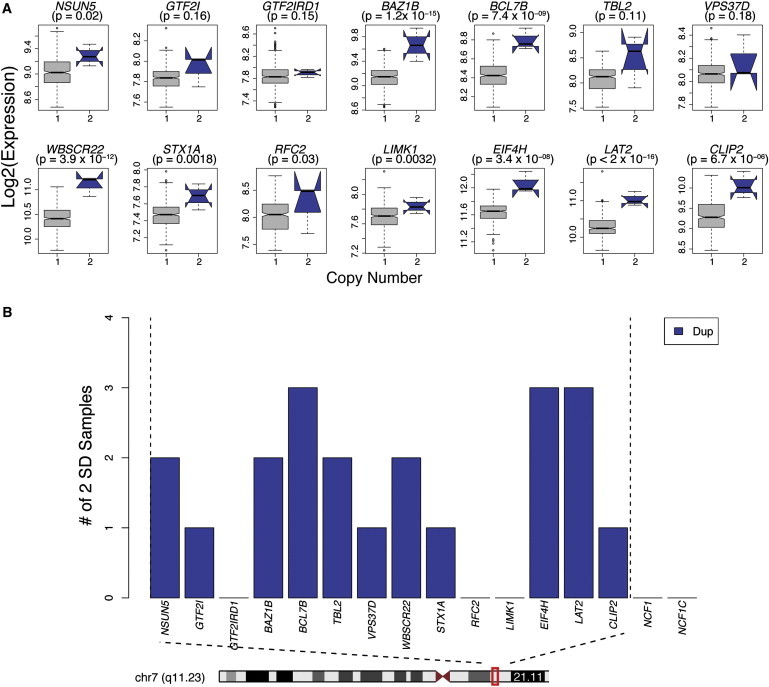
Gene Expression in the 7q11.23 Interval
(A) For each of the expressed genes within the 7q11.23 interval, the log2 expression level is shown for duplications (blue) and controls (gray). The p value was calculated with a multivariate linear regression with 7q11.23-duplication cases and 411 controls without a known 7q11.23 event (Material and Methods). Error bars are defined as 1.5× the interquartile range.
(B) Genes showing expression deviating by at least 2 SDs from the mean across three samples with 7q11.23 duplications.
To explore whether genome-wide expression changes were sufficient to separate the different genotypes from each other and controls, we performed principle-component analysis (PCA) for 16p11.2 and 7q11.23 cases and compared them with 20 controls and 20 sporadic cases in different families (Material and Methods). This analysis (Figure 7D) suggests that 16p11.2 deletions and duplications might be distinct from each other, consistent with the analysis of gene expression within the CNV and the GO analysis of trans effects of each mutation on genome-wide expression. Furthermore, the 7q11.23 cases appear to cluster more with the 16p11.2-deletion cases even though the number is small, consistent with the observation that both disrupt CNS-related GO categories; however, the 16p11.2-duplication cases do not cluster with the 16p11.2-deletion cases. Interestingly, the fact that sporadic-autism cases clustered with the controls (Figure 7D) is consistent with the absence of significant shared genome-wide gene-expression changes differentiating between randomly selected cases versus controls (Figure S6). To further study the relationship between recurrent variants that are associated with autism, we compared the DEX genes from 16p11.2 deletions, 16p11.2 duplications, and 7q11.23 duplications with the DEX genes identified previously in 15q11-13 duplications (15qdup) and fragile X mutation carriers with autism (FMR1-FM).22 Interestingly, RIMS3 (MIM 611600)55 is DEX in 16p11.2dup, 7q11.23dup, 15qdup, and FMR1-FM, indicating convergent dysregulation of this gene in multiple ASDs.
Discussion
These results demonstrate the utility of gene-expression analysis in evaluating the functional consequences of rare functional structural variations in a human neuropsychiatric disease (ASD). Given the difficulty in interpreting whole-genome-level data in the context of rare variation, our data demonstrate a wealth of transcriptional alterations that are associated with structural variation. By integrating expression and genomic data, we show that the more pathogenic classes of CNVs are associated with increased odds of harboring transcriptional alterations either within or nearby the CNV; this is consistent with previous studies that demonstrate the impact of CNVs on genome-wide expression.20,42 We also found that the CNVs only explain a portion of outlier genes; further studies are needed for the identification of potential mutations or epigenetic modifications that might contribute to the expression alterations observed in these outlier genes. Additionally, for recurrent CNVs known to be associated with ASDs, cis- and trans-expression analyses suggest distinct molecular mechanisms for 16p11.2 deletions and duplications.
It is well recognized that any method based on expression profiling would be optimal in the tissue most involved in the disorder (i.e., CNS tissue), preferably during early brain development, when ASDs unfold. There is no doubt that this analysis has missed some disease-relevant genes that are not expressed in lymphoblasts.56 Unfortunately, postmortem brain tissue is only available from a very small number of individuals, and tissue from early developmental stages is not available. Thus, the use of lymphoblasts has the advantage that these cells are widely available and permit a high-throughput, genome-wide analysis. Advances in induced pluripotent stem cell (IPSc) technology might eventually permit analyses of neuronal development in vitro.57 Our successful use of expression data in lymphoblasts supports the use of such an approach in the future for determining the functional consequences of rare SNVs and CNVs. This is especially germane given the recent results of exome sequencing in ASDs.58–61 These studies reveal an excess of rare de novo nonsense SNVs and, to a lesser extent, missense SNVs, in ASDs. Except for in a few cases, the extent to which a given variant is functional is hard to predict. Thus, integration of gene expression with SNV data would most likely be helpful.
Analysis of outliers was performed independently from analysis of CNVs, and there were equivalent numbers of outliers in probands and siblings. However, GO analysis demonstrates that there is specific enrichment of CNS pathways in outliers detected in probands, supporting the hypothesis that ASD risk in simplex families is associated with the position and size of the CNV and not necessarily its overall burden. The GO pathways dysregulated specifically in probands also include known autism candidate genes, for example, the oxytocin receptor (OXTR)62 and ubiquitin protein ligase E3A (UBE3A).17 We also observed enrichment of non-neural pathways in probands. Although some of these are not annotated as neural in GeneGo, they include signaling pathways, such as BMP, TGF-β or FGF signaling, that play crucial roles in neural development. Few pathways are enriched in siblings, and all are non-neural, consistent with the interpretation that these probably represent noise, such as that introduced during the EBV transformation process63 or based on the effect of variability in genetic background.
The pathogenic role of de novo CNVs in ASDs has been previously established.7,17,43,64,65 Although it has been assumed that underlying changes in gene expression contribute to pathogenicity, this has not been demonstrated previously. If a CNV encompasses a region where biologically critical genes are more likely to be haploinsufficient, then it has a higher chance of having a functional impact on transcription.44 The fact that we observed a higher pHI of genes in rare CNVs only in probands and not in sibling CNVs provides independent validation of the outlier analysis by showing clear differences between the functional impact of these CNVs on expression. Previous studies have shown that many factors can contribute to the pathogenicity of CNVs; these factors can include size, gene density, segmental duplication density, enrichment of certain functional pathways, and a higher-than-average expression correlation compared with that of the genome background.66,67 Here, we show that analysis of peripheral-blood gene expression can provide a useful and direct assessment of the functional consequences of chromosomal structural variation in a neuropsychiatric condition.
Assessment of the functional and potentially pathogenic impact of individual rare nonrecurrent CNVs in disease remains an important challenge. Here, we use the outlier approach to identify candidate ASD loci at 12p11.22, 15q23, 1p34.3, 3q27, and 3p26.2. For example, the 3p26.2 deletion in one proband contains three expressed genes: inositol 1,4,5-triphosphate receptor, type 1 (ITPR1 [MIM 147265]), SET domain and mariner transposase fusion gene (SETMAR [MIM 609834]), and sulfatase modifying factor 1 (SUMF1 [MIM 607939]), all of which are downregulated. Although none of these genes has been previously associated with autism, they are all functionally linked to the nervous system.68–70 Another example is a 100 kb deletion at 3q27.2, which includes only one gene, the SR-like splicing factor SFRS10/TRA2b (Htra2-beta1; also known as TRA2b [MIM 602719]), which was downregulated in the probands. TRA2b has recently been implicated in activity-dependent regulation of RNA splicing via interaction with DARPP-32 (MIM 604399).71 This is particularly interesting given the involvement of another neuronal splicing factor, Fox1/A2BP1 (MIM 605104) in ASDs,73 regulated by neuronal activity72 and previous data implicating activity-dependent regulation of gene expression in ASDs.74
Here, we also explore the functional impact of 16p11.2 microdeletions and microduplications on gene expression. Previously, it was unclear which genes are dysregulated in or near the 16p11.2 region or whether there is a common expression signature shared by 16p11.2 cases. Our analysis shows a significant positive correlation between expression level and copy number, as recently observed in mouse models,47 and highlights genes with the most consistent alterations across all 16p11.2 cases; these genes include potassium channel tetramerisation domain containing 13 (KCTD13), aldolase A, fructose-bisphosphate (ALDOA), and MYC-associated zinc finger protein (MAZ [MIM 600999]). Genes encoding potassium-channel proteins, such as KCNJ3 (MIM 601534) and KCNMA1 (MIM 600150), have been associated with neurodevelopmental abnormalities.75,76 ALDOA is involved in glycolysis and energy balance, which is important for synaptic metabolism and neurotransmitter release.77 MAZ enhances the NMDA receptor subunit type 1 activity during neuronal differentiation.78 This study provides a source for candidate-gene prioritization for future functional and mutational analyses. Although our analysis of differential expression highlights different molecular pathways disrupted in 16p duplications and deletions, one needs to also consider that we could be missing some common pathways that are only expressed in the brain. In this regard, it is notable that 7q11.23 cases cluster with the 16p11.2del cases in terms of global gene-expression changes in lymphoblasts. Within the 7q11.23 duplications, we found that STX1A (MIM 186590), CLIP2 (MIM 603432), and LIMK1 (MIM 601329) are upregulated, but we do not see alterations in GTF2I (MIM 601679) and CYLN2 (MIM 603432), which were previously shown to be dysregulated in 7q11.23 duplications by qPCR.79 This might be due to differences in techniques or the phenotypes assayed, and further studies in larger samples will permit more precise expression-phenotype correlations. We hypothesize that the observed expression differences are most likely related to the phenotypic differences observed in reciprocal 7q11.23 events and provide a starting point for connecting specific genes to phenotypes in subjects with 7q11.23 CNVs.
We also provide the molecular correlates of a clinical phenotype, head circumference, in ASDs.48–53 This is especially interesting because 16p11.2 deletions (associated with macrocephaly) are highly penetrant for ASDs, whereas 16p11.2 duplications (associated with microcephaly) are less penetrant for ASDs. Here, in 16p11.2 events, we demonstrate a significant correlation between head circumference and expression of several genes within the CNVs; one such gene is TAOK2, which showed the largest correlation. TAOK2 interacts with the JNK mitogen-activated protein kinase pathway,80 which has been shown to control survival, proliferation, and differentiation of cells composing the central and peripheral nervous systems.81 This provides a biologically plausible link between this gene and a brain-growth phenotype; this link can be tested in neural tissues and model organisms in future studies.
In summary, we present the largest genome-wide expression-profiling study on ASDs and integrate this transcriptional data with genomic data. Each of these datasets, gene expression and CNVs, is complementary, and they are more powerful together than they are alone. These data highlight the utility of this approach for the prioritization of mutations and specific genes for further downstream functional or mutational analysis—an approach that should have widespread utility given the proliferation of genome sequencing and analysis of structural variation. This is especially true for rare, nonrecurrent variants for which standard statistical tests of association are underpowered. We show that the intersection of such events with expression permits a statistical analysis of individual events and facilitates the prioritization of individual rare CNVs. These results elucidate the genome-wide functional impact of CNVs and might help to explain complex phenotypes related to brain growth, such as head circumference, and in doing so, they will help to link genotype to phenotype in complex neuropsychiatric disorders, such as ASDs.
Acknowledgments
We gratefully acknowledge the resources provided by Simons genetic consortium and Simons investigators, and we sincerely thank the families who have participated in the Simons Simplex study. This work is supported by grant R08741 M09R10124 (to D.H.G., M.S., and B.D.) and Pilot Grant 20104829 (to D.H.G.) from the Simons Foundation, the Autism Center of Excellence Network grant 5R01 MH081754-04 (to D.H.G. [principal investigator] and M.S.) from the National Institute of Mental Health, Wellcome Trust grant 077014/Z/05/Z (to M.H.), and the Dennis Weatherstone predoctoral fellowship from Autism Speaks (to R.L). We are also grateful to Neelroop Parikshak, Jamee Bomar, and Jeremy Miller for critical reading of the manuscript and to Lauren Kawaguchi for her help as a laboratory manager.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Allen Brain Atlas, http://human.brain-map.org/
DAVID Bioinformatics Database, http://david.abcc.ncifcrf.gov/
GeneGo, http://www.genego.com/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
SFARI, http://sfari.org/
Accession Numbers
The NCBI Gene Expression Omnibus82 accession number for the microarray data reported in this paper is GSE 37772.
References
- 1.Geschwind D.H. Advances in autism. Annu. Rev. Med. 2009;60:367–380. doi: 10.1146/annurev.med.60.053107.121225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miles J.H. Autism spectrum disorders—a genetics review. Genet. Med. 2011;13:278–294. doi: 10.1097/GIM.0b013e3181ff67ba. [DOI] [PubMed] [Google Scholar]
- 3.Jorde L.B., Hasstedt S.J., Ritvo E.R., Mason-Brothers A., Freeman B.J., Pingree C., McMahon W.M., Petersen B., Jenson W.R., Mo A. Complex segregation analysis of autism. Am. J. Hum. Genet. 1991;49:932–938. [PMC free article] [PubMed] [Google Scholar]
- 4.Bolton P.F., Pickles A., Murphy M., Rutter M. Autism, affective and other psychiatric disorders: Patterns of familial aggregation. Psychol. Med. 1998;28:385–395. doi: 10.1017/s0033291797006004. [DOI] [PubMed] [Google Scholar]
- 5.Ronald A., Hoekstra R.A. Autism spectrum disorders and autistic traits: A decade of new twin studies. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2011;156B:255–274. doi: 10.1002/ajmg.b.31159. [DOI] [PubMed] [Google Scholar]
- 6.Pinto D., Pagnamenta A.T., Klei L., Anney R., Merico D., Regan R., Conroy J., Magalhaes T.R., Correia C., Abrahams B.S. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sebat J., Lakshmi B., Malhotra D., Troge J., Lese-Martin C., Walsh T., Yamrom B., Yoon S., Krasnitz A., Kendall J. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marshall C.R., Noor A., Vincent J.B., Lionel A.C., Feuk L., Skaug J., Shago M., Moessner R., Pinto D., Ren Y. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moreno-De-Luca D., Mulle J.G., Kaminsky E.B., Sanders S.J., Myers S.M., Adam M.P., Pakula A.T., Eisenhauer N.J., Uhas K., Weik L., SGENE Consortium. Simons Simplex Collection Genetics Consortium. GeneSTAR Deletion 17q12 is a recurrent copy number variant that confers high risk of autism and schizophrenia. Am. J. Hum. Genet. 2010;87:618–630. doi: 10.1016/j.ajhg.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bourgeron T., Leboyer M., Delorme R. [Autism: More evidence of a genetic cause] Bull Acad Natl Med. 2009;193:299–304. discussion 304–295. [PubMed] [Google Scholar]
- 11.Jamain S., Quach H., Betancur C., Råstam M., Colineaux C., Gillberg I.C., Soderstrom H., Giros B., Leboyer M., Gillberg C., Bourgeron T., Paris Autism Research International Sibpair Study Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 2003;34:27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.State M.W. The genetics of child psychiatric disorders: Focus on autism and Tourette syndrome. Neuron. 2010;68:254–269. doi: 10.1016/j.neuron.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McClellan J., King M.C. Genomic analysis of mental illness: A changing landscape. JAMA. 2010;303:2523–2524. doi: 10.1001/jama.2010.869. [DOI] [PubMed] [Google Scholar]
- 14.Geschwind D.H. Autism: Many genes, common pathways? Cell. 2008;135:391–395. doi: 10.1016/j.cell.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Helbig I., Mefford H.C., Sharp A.J., Guipponi M., Fichera M., Franke A., Muhle H., de Kovel C., Baker C., von Spiczak S. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat. Genet. 2009;41:160–162. doi: 10.1038/ng.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weiss L.A., Shen Y., Korn J.M., Arking D.E., Miller D.T., Fossdal R., Saemundsen E., Stefansson H., Ferreira M.A., Green T., Autism Consortium Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 17.Glessner J.T., Wang K., Cai G., Korvatska O., Kim C.E., Wood S., Zhang H., Estes A., Brune C.W., Bradfield J.P. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanders S.J., Ercan-Sencicek A.G., Hus V., Luo R., Murtha M.T., Moreno-De-Luca D., Chu S.H., Moreau M.P., Gupta A.R., Thomson S.A. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Treadwell-Deering D.E., Powell M.P., Potocki L. Cognitive and behavioral characterization of the Potocki-Lupski syndrome (duplication 17p11.2) J. Dev. Behav. Pediatr. 2010;31:137–143. doi: 10.1097/DBP.0b013e3181cda67e. [DOI] [PubMed] [Google Scholar]
- 20.Stranger B.E., Forrest M.S., Dunning M., Ingle C.E., Beazley C., Thorne N., Redon R., Bird C.P., de Grassi A., Lee C. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848–853. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abrahams B.S., Geschwind D.H. Advances in autism genetics: On the threshold of a new neurobiology. Nat. Rev. Genet. 2008;9:341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishimura Y., Martin C.L., Vazquez-Lopez A., Spence S.J., Alvarez-Retuerto A.I., Sigman M., Steindler C., Pellegrini S., Schanen N.C., Warren S.T., Geschwind D.H. Genome-wide expression profiling of lymphoblastoid cell lines distinguishes different forms of autism and reveals shared pathways. Hum. Mol. Genet. 2007;16:1682–1698. doi: 10.1093/hmg/ddm116. [DOI] [PubMed] [Google Scholar]
- 23.Coppola G., Karydas A., Rademakers R., Wang Q., Baker M., Hutton M., Miller B.L., Geschwind D.H. Gene expression study on peripheral blood identifies progranulin mutations. Ann. Neurol. 2008;64:92–96. doi: 10.1002/ana.21397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Voineagu I., Huang L., Winden K., Lazaro M., Haan E., Nelson J., McGaughran J., Nguyen L.S., Friend K., Hackett A. CCDC22: A novel candidate gene for syndromic X-linked intellectual disability. Mol. Psychiatry. 2012;17:4–7. doi: 10.1038/mp.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oldham M.C., Konopka G., Iwamoto K., Langfelder P., Kato T., Horvath S., Geschwind D.H. Functional organization of the transcriptome in human brain. Nat. Neurosci. 2008;11:1271–1282. doi: 10.1038/nn.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Voineagu I., Wang X., Johnston P., Lowe J.K., Tian Y., Horvath S., Mill J., Cantor R.M., Blencowe B.J., Geschwind D.H. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller J.A., Horvath S., Geschwind D.H. Divergence of human and mouse brain transcriptome highlights Alzheimer disease pathways. Proc. Natl. Acad. Sci. USA. 2010;107:12698–12703. doi: 10.1073/pnas.0914257107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gold D.L., Wang J., Coombes K.R. Inter-gene correlation on oligonucleotide arrays: How much does normalization matter? Am. J. Pharmacogenomics. 2005;5:271–279. doi: 10.2165/00129785-200505040-00007. [DOI] [PubMed] [Google Scholar]
- 29.Johnson W.E., Li C., Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 30.Bonferroni C.E. Teoria statistica delle classi e calcolo delle probabilità. Pubblicazioni del R Istituto Superiore di Scienze Economiche e Commerciali di Firenze. 1936;8:3–62. [Google Scholar]
- 31.Zhang J., Feuk L., Duggan G.E., Khaja R., Scherer S.W. Development of bioinformatics resources for display and analysis of copy number and other structural variants in the human genome. Cytogenet. Genome Res. 2006;115:205–214. doi: 10.1159/000095916. [DOI] [PubMed] [Google Scholar]
- 32.Liang K.Y., Zeger S.L. Longitudinal Data-Analysis Using Generalized Linear-Models. Biometrika. 1986;73:13–22. [Google Scholar]
- 33.Smyth G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article3. [DOI] [PubMed] [Google Scholar]
- 34.Roche A.F., Mukherjee D., Guo S.M., Moore W.M. Head circumference reference data: Birth to 18 years. Pediatrics. 1987;79:706–712. [PubMed] [Google Scholar]
- 35.Johnson M.B., Kawasawa Y.I., Mason C.E., Krsnik Z., Coppola G., Bogdanović D., Geschwind D.H., Mane S.M., State M.W., Sestan N. Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron. 2009;62:494–509. doi: 10.1016/j.neuron.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodier P.M., Ingram J.L., Tisdale B., Nelson S., Romano J. Embryological origin for autism: Developmental anomalies of the cranial nerve motor nuclei. J. Comp. Neurol. 1996;370:247–261. doi: 10.1002/(SICI)1096-9861(19960624)370:2<247::AID-CNE8>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 37.Ploeger A., Raijmakers M.E., van der Maas H.L., Galis F. The association between autism and errors in early embryogenesis: What is the causal mechanism? Biol. Psychiatry. 2010;67:602–607. doi: 10.1016/j.biopsych.2009.10.010. [DOI] [PubMed] [Google Scholar]
- 38.Walsh C.A., Morrow E.M., Rubenstein J.L. Autism and brain development. Cell. 2008;135:396–400. doi: 10.1016/j.cell.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barnby G., Abbott A., Sykes N., Morris A., Weeks D.E., Mott R., Lamb J., Bailey A.J., Monaco A.P., International Molecular Genetics Study of Autism Consortium Candidate-gene screening and association analysis at the autism-susceptibility locus on chromosome 16p: Evidence of association at GRIN2A and ABAT. Am. J. Hum. Genet. 2005;76:950–966. doi: 10.1086/430454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Caliskan M., Cusanovich D.A., Ober C., Gilad Y. The effects of EBV transformation on gene expression levels and methylation profiles. Hum. Mol. Genet. 2011;20:1643–1652. doi: 10.1093/hmg/ddr041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gilman S.R., Iossifov I., Levy D., Ronemus M., Wigler M., Vitkup D. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron. 2011;70:898–907. doi: 10.1016/j.neuron.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Henrichsen C.N., Vinckenbosch N., Zöllner S., Chaignat E., Pradervand S., Schütz F., Ruedi M., Kaessmann H., Reymond A. Segmental copy number variation shapes tissue transcriptomes. Nat. Genet. 2009;41:424–429. doi: 10.1038/ng.345. [DOI] [PubMed] [Google Scholar]
- 43.Bucan M., Abrahams B.S., Wang K., Glessner J.T., Herman E.I., Sonnenblick L.I., Alvarez Retuerto A.I., Imielinski M., Hadley D., Bradfield J.P. Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet. 2009;5:e1000536. doi: 10.1371/journal.pgen.1000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang N., Lee I., Marcotte E.M., Hurles M.E. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet. 2010;6:e1001154. doi: 10.1371/journal.pgen.1001154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Celestino-Soper P.B., Shaw C.A., Sanders S.J., Li J., Murtha M.T., Ercan-Sencicek A.G., Davis L., Thomson S., Gambin T., Chinault A.C. Use of array CGH to detect exonic copy number variants throughout the genome in autism families detects a novel deletion in TMLHE. Hum. Mol. Genet. 2011;20:4360–4370. doi: 10.1093/hmg/ddr363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Celestino-Soper P.c.B.S., Violante S., Crawford E.L., Luo R., Lionel A.C., Delaby E., Cai G., Sadikovic B., Lee K., Lo C. A common X-linked inborn error of carnitine biosynthesis may be a risk factor for nondysmorphic autism. Proc. Natl. Acad. Sci. USA. 2012;109:7974–7981. doi: 10.1073/pnas.1120210109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Horev G., Ellegood J., Lerch J.P., Son Y.E., Muthuswamy L., Vogel H., Krieger A.M., Buja A., Henkelman R.M., Wigler M., Mills A.A. Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism. Proc. Natl. Acad. Sci. USA. 2011;108:17076–17081. doi: 10.1073/pnas.1114042108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bijlsma E.K., Gijsbers A.C., Schuurs-Hoeijmakers J.H., van Haeringen A., Fransen van de Putte D.E., Anderlid B.M., Lundin J., Lapunzina P., Pérez Jurado L.A., Delle Chiaie B. Extending the phenotype of recurrent rearrangements of 16p11.2: Deletions in mentally retarded patients without autism and in normal individuals. Eur. J. Med. Genet. 2009;52:77–87. doi: 10.1016/j.ejmg.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 49.Fernandez B.A., Roberts W., Chung B., Weksberg R., Meyn S., Szatmari P., Joseph-George A.M., Mackay S., Whitten K., Noble B. Phenotypic spectrum associated with de novo and inherited deletions and duplications at 16p11.2 in individuals ascertained for diagnosis of autism spectrum disorder. J. Med. Genet. 2010;47:195–203. doi: 10.1136/jmg.2009.069369. [DOI] [PubMed] [Google Scholar]
- 50.Hanson E., Nasir R.H., Fong A., Lian A., Hundley R., Shen Y., Wu B.L., Holm I.A., Miller D.T., 16p11.2 Study Group Clinicians Cognitive and behavioral characterization of 16p11.2 deletion syndrome. J. Dev. Behav. Pediatr. 2010;31:649–657. doi: 10.1097/DBP.0b013e3181ea50ed. [DOI] [PubMed] [Google Scholar]
- 51.Kumar R.A., Marshall C.R., Badner J.A., Babatz T.D., Mukamel Z., Aldinger K.A., Sudi J., Brune C.W., Goh G., Karamohamed S. Association and mutation analyses of 16p11.2 autism candidate genes. PLoS ONE. 2009;4:e4582. doi: 10.1371/journal.pone.0004582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shinawi M., Liu P., Kang S.H., Shen J., Belmont J.W., Scott D.A., Probst F.J., Craigen W.J., Graham B.H., Pursley A. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J. Med. Genet. 2010;47:332–341. doi: 10.1136/jmg.2009.073015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McCarthy S.E., Makarov V., Kirov G., Addington A.M., McClellan J., Yoon S., Perkins D.O., Dickel D.E., Kusenda M., Krastoshevsky O., Wellcome Trust Case Control Consortium Microduplications of 16p11.2 are associated with schizophrenia. Nat. Genet. 2009;41:1223–1227. doi: 10.1038/ng.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miller D.T., Nasir R., Sobeih M.M., Shen Y., Wu B.L., Hanson E. 16p11.2 Microdeletion 2009 Sep 22. In: Pagon R.A., Bird T.D., Dolan C.R., Stephens K., Adam M.P., editors. GeneReviews. University of Washington, Seattle; Seattle, WA: 1993. [Google Scholar]
- 55.Kumar R.A., Sudi J., Babatz T.D., Brune C.W., Oswald D., Yen M., Nowak N.J., Cook E.H., Christian S.L., Dobyns W.B. A de novo 1p34.2 microdeletion identifies the synaptic vesicle gene RIMS3 as a novel candidate for autism. J. Med. Genet. 2010;47:81–90. doi: 10.1136/jmg.2008.065821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cai C., Langfelder P., Fuller T.F., Oldham M.C., Luo R., van den Berg L.H., Ophoff R.A., Horvath S. Is human blood a good surrogate for brain tissue in transcriptional studies? BMC Genomics. 2010;11:589. doi: 10.1186/1471-2164-11-589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dolmetsch R., Geschwind D.H. The human brain in a dish: The promise of iPSC-derived neurons. Cell. 2011;145:831–834. doi: 10.1016/j.cell.2011.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iossifov I., Ronemus M., Levy D., Wang Z., Hakker I., Rosenbaum J., Yamrom B., Lee Y.H., Narzisi G., Leotta A. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sanders S.J., Murtha M.T., Gupta A.R., Murdoch J.D., Raubeson M.J., Willsey A.J., Ercan-Sencicek A.G., Dilullo N.M., Parikshak N.N., Stein J.L. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Neale B.M., Kou Y., Liu L., Ma'ayan A., Samocha K.E., Sabo A., Lin C.F., Stevens C., Wang L.S., Makarov V. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.O'Roak B.J., Vives L., Girirajan S., Karakoc E., Krumm N., Coe B.P., Levy R., Ko A., Lee C., Smith J.D. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pobbe R.L., Pearson B.L., Blanchard D.C., Blanchard R.J. Oxytocin receptor and Mecp2(308/Y) knockout mice exhibit altered expression of autism-related social behaviors. Physiol Behav. 2012 doi: 10.1016/j.physbeh.2012.02.024. Published online March 3, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Choy E., Yelensky R., Bonakdar S., Plenge R.M., Saxena R., De Jager P.L., Shaw S.Y., Wolfish C.S., Slavik J.M., Cotsapas C. Genetic analysis of human traits in vitro: Drug response and gene expression in lymphoblastoid cell lines. PLoS Genet. 2008;4:e1000287. doi: 10.1371/journal.pgen.1000287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Itsara A., Wu H., Smith J.D., Nickerson D.A., Romieu I., London S.J., Eichler E.E. De novo rates and selection of large copy number variation. Genome Res. 2010;20:1469–1481. doi: 10.1101/gr.107680.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Girirajan S., Rosenfeld J.A., Cooper G.M., Antonacci F., Siswara P., Itsara A., Vives L., Walsh T., McCarthy S.E., Baker C. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat. Genet. 2010;42:203–209. doi: 10.1038/ng.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Conrad D.F., Pinto D., Redon R., Feuk L., Gokcumen O., Zhang Y., Aerts J., Andrews T.D., Barnes C., Campbell P., Wellcome Trust Case Control Consortium Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hehir-Kwa J.Y., Wieskamp N., Webber C., Pfundt R., Brunner H.G., Gilissen C., de Vries B.B., Ponting C.P., Veltman J.A. Accurate distinction of pathogenic from benign CNVs in mental retardation. PLoS Comput. Biol. 2010;6:e1000752. doi: 10.1371/journal.pcbi.1000752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Novak M.J., Sweeney M.G., Li A., Treacy C., Chandrashekar H.S., Giunti P., Goold R.G., Davis M.B., Houlden H., Tabrizi S.J. An ITPR1 gene deletion causes spinocerebellar ataxia 15/16: A genetic, clinical and radiological description. Mov. Disord. 2010;25:2176–2182. doi: 10.1002/mds.23223. [DOI] [PubMed] [Google Scholar]
- 69.Schorge S., van de Leemput J., Singleton A., Houlden H., Hardy J. Human ataxias: A genetic dissection of inositol triphosphate receptor (ITPR1)-dependent signaling. Trends Neurosci. 2010;33:211–219. doi: 10.1016/j.tins.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Settembre C., Annunziata I., Spampanato C., Zarcone D., Cobellis G., Nusco E., Zito E., Tacchetti C., Cosma M.P., Ballabio A. Systemic inflammation and neurodegeneration in a mouse model of multiple sulfatase deficiency. Proc. Natl. Acad. Sci. USA. 2007;104:4506–4511. doi: 10.1073/pnas.0700382104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Benderska N., Becker K., Girault J.A., Becker C.M., Andreadis A., Stamm S. DARPP-32 binds to tra2-beta1 and influences alternative splicing. Biochim. Biophys. Acta. 2010;1799:448–453. doi: 10.1016/j.bbagrm.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee J.A., Tang Z.Z., Black D.L. An inducible change in Fox-1/A2BP1 splicing modulates the alternative splicing of downstream neuronal target exons. Genes Dev. 2009;23:2284–2293. doi: 10.1101/gad.1837009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Martin C.L., Duvall J.A., Ilkin Y., Simon J.S., Arreaza M.G., Wilkes K., Alvarez-Retuerto A., Whichello A., Powell C.M., Rao K. Cytogenetic and molecular characterization of A2BP1/FOX1 as a candidate gene for autism. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2007;144B:869–876. doi: 10.1002/ajmg.b.30530. [DOI] [PubMed] [Google Scholar]
- 74.Morrow E.M., Yoo S.Y., Flavell S.W., Kim T.K., Lin Y., Hill R.S., Mukaddes N.M., Balkhy S., Gascon G., Hashmi A. Identifying autism loci and genes by tracing recent shared ancestry. Science. 2008;321:218–223. doi: 10.1126/science.1157657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Newbury D.F., Warburton P.C., Wilson N., Bacchelli E., Carone S., Lamb J.A., Maestrini E., Volpi E.V., Mohammed S., Baird G., Monaco A.P., International Molecular Genetic Study of Autism Consortium Mapping of partially overlapping de novo deletions across an autism susceptibility region (AUTS5) in two unrelated individuals affected by developmental delays with communication impairment. Am. J. Med. Genet. A. 2009;149A:588–597. doi: 10.1002/ajmg.a.32704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Laumonnier F., Roger S., Guérin P., Molinari F., M'rad R., Cahard D., Belhadj A., Halayem M., Persico A.M., Elia M. Association of a functional deficit of the BKCa channel, a synaptic regulator of neuronal excitability, with autism and mental retardation. Am. J. Psychiatry. 2006;163:1622–1629. doi: 10.1176/ajp.2006.163.9.1622. [DOI] [PubMed] [Google Scholar]
- 77.Pellerin L. Food for thought: The importance of glucose and other energy substrates for sustaining brain function under varying levels of activity. Diabetes Metab. 2010;36(Suppl 3):S59–S63. doi: 10.1016/S1262-3636(10)70469-9. [DOI] [PubMed] [Google Scholar]
- 78.Okamoto S., Sherman K., Bai G., Lipton S.A. Effect of the ubiquitous transcription factors, SP1 and MAZ, on NMDA receptor subunit type 1 (NR1) expression during neuronal differentiation. Brain Res. Mol. Brain Res. 2002;107:89–96. doi: 10.1016/s0169-328x(02)00440-0. [DOI] [PubMed] [Google Scholar]
- 79.Van der Aa N., Rooms L., Vandeweyer G., van den Ende J., Reyniers E., Fichera M., Romano C., Delle Chiaie B., Mortier G., Menten B. Fourteen new cases contribute to the characterization of the 7q11.23 microduplication syndrome. Eur. J. Med. Genet. 2009;52:94–100. doi: 10.1016/j.ejmg.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 80.Moore T.M., Garg R., Johnson C., Coptcoat M.J., Ridley A.J., Morris J.D. PSK, a novel STE20-like kinase derived from prostatic carcinoma that activates the c-Jun N-terminal kinase mitogen-activated protein kinase pathway and regulates actin cytoskeletal organization. J. Biol. Chem. 2000;275:4311–4322. doi: 10.1074/jbc.275.6.4311. [DOI] [PubMed] [Google Scholar]
- 81.Wagner B., Sibilia M. Methods to study MAP kinase signalling in the central nervous system. Methods Mol. Biol. 2010;661:481–495. doi: 10.1007/978-1-60761-795-2_30. [DOI] [PubMed] [Google Scholar]
- 82.Barrett T., Edgar R. Gene expression omnibus: microarray data storage, submission, retrieval, and analysis. Methods Enzymol. 2006;411:352–369. doi: 10.1016/S0076-6879(06)11019-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.