

Abstract
Potocki-Shaffer syndrome (PSS) is a contiguous gene disorder due to the interstitial deletion of band p11.2 of chromosome 11 and is characterized by multiple exostoses, parietal foramina, intellectual disability (ID), and craniofacial anomalies (CFAs). Despite the identification of individual genes responsible for multiple exostoses and parietal foramina in PSS, the identity of the gene(s) associated with the ID and CFA phenotypes has remained elusive. Through characterization of independent subjects with balanced translocations and supportive comparative deletion mapping of PSS subjects, we have uncovered evidence that the ID and CFA phenotypes are both caused by haploinsufficiency of a single gene, PHF21A, at 11p11.2. PHF21A encodes a plant homeodomain finger protein whose murine and zebrafish orthologs are both expressed in a manner consistent with a function in neurofacial and craniofacial development, and suppression of the latter led to both craniofacial abnormalities and neuronal apoptosis. Along with lysine-specific demethylase 1 (LSD1), PHF21A, also known as BHC80, is a component of the BRAF-histone deacetylase complex that represses target-gene transcription. In lymphoblastoid cell lines from two translocation subjects in whom PHF21A was directly disrupted by the respective breakpoints, we observed derepression of the neuronal gene SCN3A and reduced LSD1 occupancy at the SCN3A promoter, supporting a direct functional consequence of PHF21A haploinsufficiency on transcriptional regulation. Our finding that disruption of PHF21A by translocations in the PSS region is associated with ID adds to the growing list of ID-associated genes that emphasize the critical role of transcriptional regulation and chromatin remodeling in normal brain development and cognitive function.
Introduction
In many regions of the genome, microdeletions1,2 or balanced translocations3–6 are associated with phenotypic abnormalities and are presumably caused by haploinsufficiency of the various genes involved. Potocki-Shaffer syndrome (PSS [MIM 601224]) is a rare contiguous gene-deletion syndrome caused by heterozygous interstitial microdeletions of chromosomal region 11p11-p12 and is characterized by developmental defects that include intellectual disability (ID), craniofacial anomalies (CFAs), multiple exostoses (MIM 133701), and parietal foramina (MIM 609597).7,8 Genes responsible for the latter two phenotypes in this chromosomal region have been identified: Deletion of EXT2 (MIM 608210) causes multiple exostoses,9 and deletion of ALX4 (MIM 605420) causes parietal foramina.10,11 However, the cause of the ID and abnormal craniofacial development has remained uncertain. Through identification of two independent subjects with balanced translocations and support from a third published translocation subject,12,13 we have uncovered evidence that haploinsufficiency of a single gene, PHF21A (also known as BHC80 [MIM 608325]), at 11p11.2 is associated with ID and CFA phenotypes. This evidence was complemented by comparative deletion mapping of PSS subjects with diverse phenotypes that positioned PHF21A within the critical region associated with ID and CFAs, functional in vitro analysis of PHF21A in cells from translocation subjects, and generation and rescue of zebrafish phenotypes through the suppression of phf21a with morpholino oligonucleotides (MOs). PHF21A specifically binds unmethylated histone H3 lysine 4 (H3K4me0) and participates in the lysine-specific demethylase 1 (LSD1 [MIM 609132]) demethylase complex, implicating it as a regulatory protein in histone-methylation dynamics14 and suggesting that disruption of this process might underlie the ID and CFA phenotypes in these translocation individuals.
Subjects and Methods
Human Subjects
DGAP012 and MCN1762 (Table 1) were ascertained through the Developmental Genome Anatomy Project (DGAP) and Mendelian Cytogenetic Network, respectively, and blood samples were obtained for initiation of a lymphoblastoid cell line.17 PSS subjects PSS02, PSS08, PSS10, and PSS-Romeike were previously described.7,15,18 Where possible, all subjects were tested for copy-number variants (CNVs) by array comparative genomic hybridization (CGH) with the use of Agilent (Santa Clara, CA) 244K arrays as described (Table 2).19 DGAP012 displayed one 156 kb deletion CNV (chr13: 48,430,969–48,587,500; hg18) that is located at 13q14.2 and has not been reported previously and that involves FNDC3A, which is expressed in spermatids and Leydig cells and produces male sterility when homozygously inactivated.20 All human studies were performed under informed-consent protocols approved by the Partners HealthCare System Human Research Committee.
Table 1.
Phenotypic Comparison of Individuals with Balanced Translocations and Their Breakpoints
|
Individual |
Typical PSS phenotyped | |||
|---|---|---|---|---|
| DGAP012a | MCN1762b | GILLEc | ||
| Intellectual disability | + | + | + | + |
| Facial dysmorphism | + | + | + | + |
| Narrow nose | + | + | + | + |
| Downturned mouth | + | +e | ND | + |
| Large ears | + | − | − | + |
| Brachycephaly | + | + | mild craniofacial asymmetry and thin corpus callosum | + |
| Microcephaly | + | + | hypoplasia of inferior cerebellar vermis | + |
| Myopia | + | + | ND | + |
| Strabismus | + | − | nystagmus | + |
| Heart defect | + | − | ND | + |
| Hypotonia | + | + | + | + |
| Small hands and feet | + | − | ND | + |
| Multiple exostoses | − | − | − | + |
| Parietal foramina | − | − | − | + |
| Tapering fingers | − | − | ND | + |
| Digitalized thumbs | + | − | ND | − |
| Retinal dystrophy | + | − | iris hypoplasia, superior atypical coloboma, and foveal hypoplasia | − |
| Revised karyotype | t(11;19)(p11.2;p13.2)dn | t(1;11)(p21.1;p11.2)dn | t(X;11)(p22.2;p11.2)dnf | |
| Location of breakpoint in PHF21A | intron 14 | intron 5 | breakpoint mapped within PHF21A | |
| Disruption of PHF21A (confirmation method) | truncated (FISH and breakpoint cloning) | truncated (FISH and breakpoint cloning) | disrupted (FISH); breakpoint-spanning BAC clone at 11p11.2 is RP11-618K13; fine mapped with 10 kb PCR product | |
| Breakpoint at 11p11.2 (Human Genome Browser hg18) | chr11: 45,921,645–45,921,646 | chr11: 46,020,273–46,020,274 | chr11: 45,907,446–46,099,561 | |
The following abbreviations are used: PSS, Potocki-Shaffer syndrome; ND, not determined; and FISH, fluorescence in situ hybridization.
DGAP012 was assessed at age 15 months.
MCN1762 was assessed at ages 3.5 years and 42 years.
GILLE was assessed only at ages 4 and 8 months when her phenotype was thought to suggest Gillespie syndrome.12,13
Typical PSS phenotypes from reported PSS subjects,15 including patients 1, 2, and 3 in Wuyts et al.16
Not visible in Figure 4 as a result of laughing expression.
Table 2.
PSS Subjects with Deletions Tested for Inclusion of PHF21A
| Individual ID | Phenotype | Karyotype | Deletion Size | Deletion of PHF21A (Confirmation Method) | Deletion by Array CGH (Human Genome Browser hg18) |
|---|---|---|---|---|---|
| PSS02 | Potocki-Shaffer syndrome with ID and CFAs | del(11)(p11.2p12) | 5.6 Mb | deleted (FISH and CGH) | chr11: 40,434,231–46,041,804 |
| PSS08 | Potocki-Shaffer syndrome with ID and CFAs | del(11)(p11.2p13) | 15.2 Mb | deleted (FISH and CGH) | chr11: 31,009,420–46,204,605 |
| PSS10 | Potocki-Shaffer syndrome with ID and CFAs | del(11)(p11.2p12) | N/A | deleted (FISH) | DNA unavailable because primary cells did not survive |
| PSS Romeike18 | Potocki-Shaffer syndrome with ID and CFAs | del(11)(p11.12p12) | 11.9 Mb | deleted (CGH) | chr11: 38,824,655–50,638,770 |
| GM03316 | ID and CFAs | t(X;11)(q11.1;p11.2)dn del(11)(p11.2p12) |
3.6 Mb | deleted (FISH and CGH) | chr11: 43,125,403–46,706,549 |
| GC14361 | Potocki-Shaffer syndrome with ID and CFAs | del(11)(p11.12p12) | 13.8 Mb | deleted (FISH and CGH) | chr11: 33,430,130–47,276,580 |
Deletions of all subjects except PSS10 (for whom a microarray could not be performed) are also depicted as blue bars in Figure 3, and PHF21A FISH results are shown in the Figure 3 inset for all subjects except PSS Romeike. The following abbreviations are used: ID, intellectual disability, CFAs, craniofacial anomalies; FISH, fluorescence in situ hybridization; and CGH, comparative genomic hybridization.
DGAP012
At birth, this white male was small but normally proportioned and suffered from supraventricular tachycardia, hyperbilirubinemia, and hypoglycemia, all of which resolved. Chromosome analysis revealed an apparently balanced chromosomal translocation with the karyotype 46,XY,t(11;19)(p11.2;p13.3)dn, which was revised by molecular analysis here to 46,XY,t(11;19)(p11.2;p13.2)dn. The family history was unremarkable, and parental chromosomes were normal. At 1 year of age, the subject displayed bifrontal biparietal atrophy on a computed tomography (CT) scan, and at 15 months of age, he showed significant global developmental delay, digitalized thumbs, brachycephaly, microcephaly (a head circumference of 46 cm; tenth percentile), a small downturned mouth, mild midfacial hypoplasia, a flat midface, a narrow nasal bridge, a very small nose, large ears, bilateral epiblepharon (an extra skin fold was medially under each lower lid) without trichiasis, small hands and feet, and an absence of emotional expression. He also displayed hand flapping and had feeding problems prior to the age of 3 years.
A magnetic resonance image (MRI) at the age of 2 years revealed prominent cerebrospinal fluid, which might represent volume loss. At the age of 5 years, DGAP012 displayed hypotonia and dystonic movement and could not walk and showed significant nearsightedness and astigmatism (−3.00 +3.00 × 90 in both eyes), as well as some diffuse pigmentary mottling suggesting a possible retinal dystrophy.
MCN1762
This white female, aged 42 years, was delivered at term after an uneventful pregnancy but was lethargic and had feeding difficulties. At age 3.5 years, her gross motor development was normal, but her speech development was delayed, and she displayed hyperactivity, poor concentration, and mild myopia (−2.00 [right eye], −2.50 [left eye]). A verbal intelligence quotient (IQ) test (Terman-Merril) indicated a delay of 1 year, and a nonverbal IQ test (Leiter) was normal. The following dysmorphic features were noted: brachycephaly, microcephaly, a long narrow nose, mild midfacial hypoplasia, a downturned mouth, thin lips, and prominent ear lobes and, in childhood only, downslanting palpebral fissures and epicanthal folds. GTG-banded karyotyping revealed an apparently balanced reciprocal translocation, 46,XX,t(1;11)(p13;p11)dn, which was revised by molecular analysis to 46,XX,t(1;11)(p21.1p11.2)dn. Her parents and two siblings are healthy, and parental karyotypes are normal. MCN1762 attended a school for special needs and is able to read. Her linear growth (to 173 cm) and pubertal development were normal, and she has developed truncal obesity (see Figure 4C). She now has mild ID but manages to live independently and have a sheltered part-time job.
Figure 4.
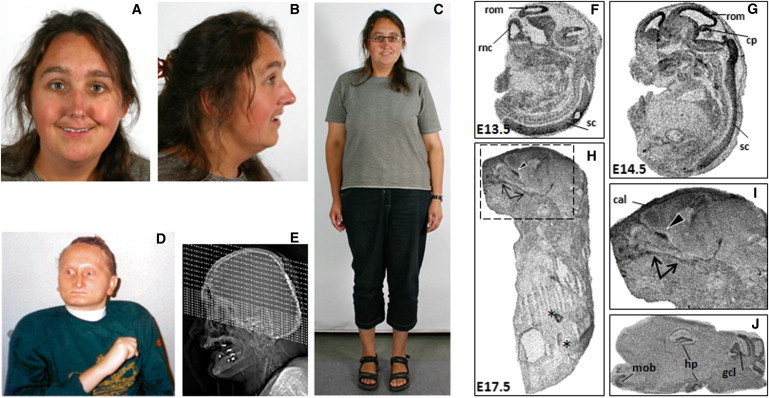
Human Craniofacial Anomalies and Murine Phf21a Expression in Craniofacial and Brain Regions
(A–E) Human craniofacial anomalies with PHF21A truncation and PHF21A deletion.
(A–C) Female subject MCN1762 with balanced translocation t(1;11) exhibits ID and CFAs that include brachycephaly, microcephaly, a long narrow nose, mild midfacial hypoplasia, thin lips, and prominent ear lobes.
(D) The PSS male proband (PSS-Romeike) with a deletion including PHF21A is shown at the age of 31 years. He shows microcephaly, brachycephaly, a broad forehead, a long narrow nose, a hypoplastic mandible, very thin lips, hypotelorism, and dysplastic low set ears, in addition to ID and no speech, among other phenotypes.
(E) A topogram of a cerebral CT of individual PSS-Romeike at the age of 33 years, shortly before he died, shows a hypoplastic mandible and the parietal foramen at the back of the head.
(F–J) Phf21a expression during murine craniofacial development.
(F and G) Expression of Phf21a at embryonic days (E) 13.5 and 14.5. Highest transcript abundance is seen in the developing CNS with [35S]-UTP labeled in situ probes. The following abbreviations are used: rom, roof of midbrain; rnc, roof of neopallial cortex; sc, spinal cord; and cp, intraventricular portion of cerebellar primordiu.
(H and I) At E17.5, prominent signals were detected with Phf21a antisense probes in bones of the facial skeleton. The palatine bone is marked by arrows, and the orbitosphenoidal bone is marked by a black arrowhead. Signals in the intestine were also detected with the sense probes and are most likely not specific (these are marked by asterisks). The following abbreviation is used: cal, calvaria.
(J) Sagittal sections of the adult mouse brain showed expression of Phf21a within the hippocampal formation, the cerebellum, and the olfactory bulb. Three different Phf21a antisense probes gave consistent results. The following abbreviations are used: mob, main olfactory bulb; hp, hippocampus; and gcl, granule cell layer of the cerebellum.
GM03316
This female Venezuelan subject was 3 years old when her blood was submitted to the National Institute of General Medical Sciences Human Genetic Cell Repository at the Coriell Institute in January of 1978 (prior to identification of PSS) and showed an apparently balanced translocation: t(X;11)(q11.1;p11.2)dn. The clinical symptoms were listed as ID (quantitative intelligence [Gq] = 60) with a strikingly unusual dysmorphology syndrome including epicanthus, hypertelorism, oblique palpebral fissures, trigonocephaly, and micrognathia. At 5 years of age, her vocabulary was progressing well, and she had a good memory, but her Gq corresponded to that of a 3-year-old girl. Her principal problem was an inability to concentrate. She could feed herself when she wished and had gained control of her sphincters both day and night by age 3.5 years. She was shy and easily frightened and was clumsy with both hands and legs.
GC14361
This 2.25-year-old male from Bangladesh had a history of static encephalopathy and developmental delay, which were first noted when he was 6 months old. He displayed microcephaly, short stature, a small phallus, a unilateral absent testis, and dysmorphic features, including a short forehead, prominent biparietal foramina, a midline parietal cortical defect, a flat midface, a flat occiput, sensorineural hearing loss, epicanthal folds, protuberant ears, a bulbous nasal tip that continued below the columella, a depressed nasal root, a small mouth and small chin (micrognathia), hypotonia, a slight pectus excavatum, recurrent otitis media, and slender fingers.
Breakpoint Mapping, Cloning, and Characterization
In brief, bacterial artificial chromosomes (BACs), followed by fosmid or cosmid probes chosen on the basis of the human genome map, were used for fluorescence in situ hybridization (FISH) of metaphase chromosomes as previously described.6 For breakpoint fine mapping by array painting, metaphase chromosomes were flow sorted,21,22 and DNA was extracted, amplified, labeled, and hybridized to a custom array (Agilent, Santa Clara, CA) with an average spacing of three unique probes per 1 kb as described previously.4 The array design has been made public at the eArray website (name: Translok4; design number: 022990). Standard DNA blotting with PCR-generated probes was used for delineating the breakpoints, which were then isolated with suppression PCR and sequenced with an ABI 3730XL DNA Analyzer (Applied Biosystems, Foster City, CA). This suppression-PCR method exploits a specially designed adaptor to walk in unknown genomic DNA regions from known adjacent sequences.23 Specific steps for breakpoint mapping and cloning in each case are the following.
DGAP012 with t(11;19)(p11.2;p13.2)
On chromosome 11, BAC clones RP11-618K13, CTD-2254P23, and RP11-142O14 all span the breakpoint (Figure S1A and Table S1, available online) that lies in the 14 kb between BAC clones CTD-2580L7 and CTD-2353H9. Four PCR probes (D012-A, D012-B, D012-C, and D012-D; see Table S3 for primers) detected junction fragments with various enzymes (e.g., Figure S1C). This Southern analysis narrowed down the breakpoint to 729 bp (Figure S1A).
We reported previously that FISH with BAC CTD-3193O13 spanned the chromosome 19 breakpoint24 (Figure S1B and Table S1). Additional FISH with five cosmids and three restriction fragments narrowed down the breakpoint region to 11 kb between the telomeric end of a 30 kb restriction fragment and the telomeric end of RP11-585H16 (Figure S1B). Three PCR probes (D012-E, D012-F, and D012-G; primers in Table S3) detected junction fragments (e.g., Figure S1D), narrowing the breakpoint to 3,441 bp at 19p13.2 (Figure S1B).
For suppression PCR, adaptors 1 and 2 were ligated to ScaI-digested DGAP012 genomic DNA (Table S3).23 The ligated DNA was diluted 1:10 with pure water and used as a template for suppression nested PCR (two-step PCR in which first boost PCR is followed by nested PCR) with the chromosome 19 primers (Table S3) designed from the 3,441 bp deduced breakpoint region (Figure S1B) and adaptor primers (Table S3). This suppression PCR generated products composed of sequences from both chromosomes 11 and 19 and thereby identified an unknown chromosome 11 sequence that confirmed the 729 bp breakpoint region at 11p11.2 by Southern analysis (Figure S1A).
The der(19) junction fragment was amplified by nested PCR with the following primer pairs: chr11 forward 1 + chr19 reverse 1 for boost PCR and chr11 forward 2 + chr19 reverse 2 for nested PCR. The der(11) junction fragment was amplified by nested PCR with the following primer pairs: chr19 forward 1 + chr11 reverse 1 for boost PCR and chr19 forward 2 + chr11 reverse 2 for nested PCR (Table S3).
MCN1762 with t(1;11)(p21.1;p11.2)
We first performed FISH with BAC clones RP11-992G23 and RP11-425L10 (which hybridize distally and proximally to PHF21A, respectively) in 11p11.2 to confirm localization of the translocation to the PHF21A region. Then, guided by array painting, we performed FISH with additional BACs and subsequently with fosmids and identified RP11-177H1 as a BAC spanning the breakpoint lying within the 37 kb segment covered by fosmid G248P88722D5 (Figure 1D and Table S2). From the array-painting results, breakpoint regions were refined to 11.4 kb in 1p21.1 and 93 bp in 11p11.2, resulting in the revision of the previously designated karyotype to 46,XY,t(1;11)(p21.1;p11.2)dn. We then used PCR to amplify junction fragments by using primers in the refined regions of chromosomes 1 and 11 by array painting.
Figure 1.
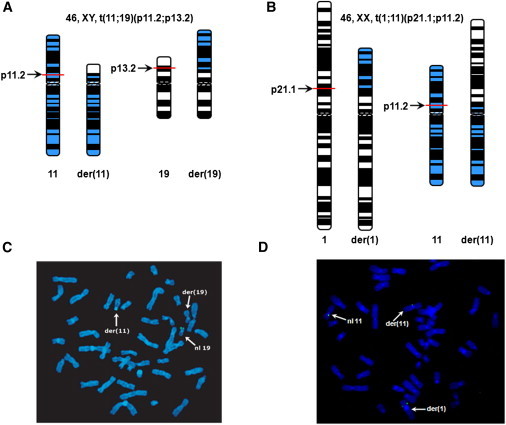
Ideograms of Balanced Chromosome Translocations and FISH Mapping of the Chromosomal Breakpoints
(A and B) Ideograms illustrating the t(11;19)(p11.2;p13.2)dn karyotype in DGAP012 and the t(1;11)(p21.1;p11.2)dn karyotype in MCN1762; this latter karyotype was revised from the original karyotypic assessment, t(1;11)(p13;p11)dn.
(C) FISH analysis with a 30 kb SnaBI restriction fragment (signal shown in green) from BAC CTD-3193O13, which has previously been reported as a breakpoint-crossing clone.24 Probe hybridization is seen on the normal 19, der(19), and der(11) chromosomes, indicating that this 30 kb restriction fragment spans the chromosome 19 breakpoint region (Figure S1B).
(D) FISH analysis with 37 kb fosmid G248P88722D5 spanning the breakpoints on the normal 11, der(1), and der(11) chromosomes, indicating that the translocation breakpoint of the chromosome 11 is located within the sequence of this genomic clone.
The junction fragment of ∼0.8 kb from der(1) was amplified by normal PCR from one primer pair of chr11 forward and chr1 reverse and was confirmed by sequencing and subsequent BLAST searching. The ∼650 bp junction fragment of der(11) was amplified by primer set chr1 forward and chr11 reverse (Table S3). Partial sequences from chromosomes 1 and 11 of this junction fragment were confirmed by sequencing and BLAST searching.
Mutation Screening of PHF21A in Various Subjects with Partially Overlapping Phenotypes
We performed mutation analysis by direct sequencing of exonic PCR products to exclude an alteration in the nontranslocated allele of PHF21A in DGAP012 and MCN1762, as well as in 200 additional subjects with various phenotypes: 25 with features of Saethre-Chotzen syndrome (MIM 101400) and without mutations in TWIST (MIM 601622) or hotspot regions of FGFR1 (MIM 136350), FGFR2 (MIM 176943), and FGFR3 (MIM 134934); 14 with undiagnosed syndromes and isolated features of Rubinstein-Taybi syndrome (MIM 180849) and without CREBBP (MIM 600140) mutations;25 16 with micropenis or hypogonadotropic hypogonadism and ID and no mutations in known genes involved in Kallmann syndrome or hypogonadotropic hypogonadism; five with metopic craniosynostosis of unknown etiology; 19 with typical or atypical Smith-Magenis syndrome (MIM 182290) and no mutations in RAI1 (MIM 607642); 25 with Kleefstra syndrome (MIM 610203) and no mutations in EHMT1 (MIM 607001);2 and 96 with Cornelia de Lange syndrome (MIM 122470, 610759, and 300590) and no mutations in NIPBL (MIM 608667), SMC3 (MIM 606062), or SMC1 (MIM 300040). We screened the entire PHF21A coding region and intron-exon boundaries comprising 16 exons defined from RefSeq accession numbers NM_001101802.1 and NM_016621.3. We found no potentially pathogenic variants. We also screened exons 2–6 of ELAVL1 (RefSeq NM_001419.2) in 18 subjects with features of Saethre-Chotzen syndrome, 12 subjects with isolated features of Rubinstein-Taybi syndrome, 16 subjects with micropenis or hypogonadotropic hypogonadism and ID, and five subjects with metopic craniosynostosis; again, we detected no potentially pathogenic variants.
RNA and ChIP Analysis in Lymphoblasts
For analysis of expression of neuronal genes in lymphoblastoid cell RNA, cells were harvested and RNAs were prepared with Trizol reagent (Sigma, St. Louis, MO). Lymphoblastoid cell cDNA samples were obtained by oligo dT and Superscript III (Invitrogen, San Diego, CA) with primers designed to cross multiple introns for the exclusion of genomic amplification. RT-PCR was carried out for 34 cycles by annealing at 60°C for 30 s and elongation at 72°C for 30 s with Platinum PCR SuperMix (Invitrogen).
LSD1 chromatin immunoprecipitation (ChIP) experiments were done as previously described.26,27 Primer pairs for the SCN3A (MIM 182391) promoter target and control oligos designed in an intergenic region between ACTG1 (MIM 102560) and FSCN2 (MIM 607643) are listed in Table S5.
Immunoblot Analysis
Whole cell protein extracts were prepared from harvested lymphoblastoid cells and were lysed on ice for 30 min in RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, and 0.1% SDS) containing a protease inhibitor mixture (Roche) and 1 mM phenylmethylsulfonyl fluoride. Total lysates were then cleared by centrifugation at 14,000 × g for 30 min, and supernatants were collected. Protein concentration was determined by a Bio-Rad (detergent compatible) protein assay. Fifty micrograms of protein extract was mixed with 4 × SDS sample buffer, boiled for 2 min, and subjected to 10% SDS-PAGE. After electrophoresis, proteins were transferred to Schleicher & Schuell nitrocellulose membranes (Whatman, UK) and incubated for 30 min in blocking solution containing 5% nonfat powdered milk in TBS-T (50 mM Tris-HCl, 150 mM NaCl, pH 7.4, and 0.1% Tween 20). Blots were probed overnight at 4°C with antibodies against an N-terminal 93 residue polypeptide of mouse Phf21a28 or α-tubulin (Sigma), rinsed four times for 10 min in TBS-T, and incubated for 1 hr at room temperature with a horseradish peroxidase-conjugated anti-rabbit antibody. After extensive washing for 30 min, membranes were processed with an enhanced chemiluminescence (ECL) substrate kit (New England Biolabs, Beverly, MA) and exposed to autoradiographic film (Hyperfilm ECL, Amersham Bioscience, Piscataway, NJ).
Immunostaining
Cells were grown on glass coverslips overnight. After being washed with PBS, cells were fixed in 4% paraformaldehyde and 0.5% Triton X-100 for 10 min. Coverslips were washed three times with PBS and incubated in block-permeabilization solution (15% goat serum [Sigma], 0.2% fish skin gelatin [Sigma], 0.03% NaN3, and 0.5% Triton X-100 in PBS) for 10 min. Cells were rinsed and incubated with blocking solution (15% goat serum, 0.2% fish skin gelatin, and 0.03% NaN3 in PBS) overnight. After this, cells were incubated with a PHF21A antibody28 in blocking solution for 2 hr at 37°C. Coverslips were washed three times with PBS and incubated for 1.5 hr with an Alexa Fluor 488-conjugated secondary antibody in blocking solution. Washing three times with PBS removed the excess antibody. DNA was stained with DAPI (Invitrogen). Images of fixed cells were collected with an LSM 510 META confocal microscope (Zeiss).
Mouse In Situ Hybridization Analysis
The primers used for amplification of three independent probes of the murine Phf21a transcript (RefSeq NM_138755.2) are listed in Table S6. Because multiple splice variants for Phf21a are listed in the Ensembl Browser (Ensembl Mouse, based on the National Center for Biotechnology Information [NCBI] m37 mouse assembly), probes were designed to detect the majority of the known isoforms. Probes were labeled with [α-35S]-UTP for hybridization on 15 μm cryosections. The day of plug was not counted for the specification of embryonic stages. No specific signals were detected with the respective sense probes except as indicated in the results section.
Fish Stocks and Maintenance
Zebrafish were maintained at 28.5°C under a 14 hr light/10 hr dark cycle in 1/3 Ringer's solution. Transgenic fish Tg[flk1:GFP] (kindly provided by Dr. Tao P. Zhong) and Tg[huC:EGFP]29 were used in overexpression or knockdown experiments. Embryos older than 24 hr postfertilization (hpf) were usually incubated in 0.003% 1-phenyl-2-thiourea (PTU, Sigma) for the inhibition of pigmentation. Embryos at appropriate stages were fixed with 4% paraformaldehyde in PBS.
Zebrafish phf21a Constructs
Zebrafish phf21a was isolated from the 24 hpf zebrafish cDNA library by RT-PCR and was first cloned in a pGEM-T easy vector (Promega, Madison, USA) and then subcloned into the EcoRI site in the pCS2+ multipurpose expression vector. For the construction of the phf21a-RFP fusion reporter, specific enzyme-linked primers were designed for PCR amplification. PCR primers are listed in Table S7. PCR products were subcloned into the ClaI site in a pCS2+ RFP vector.
Whole-Mount In Situ Hybridization and Alcian-Blue and Acridine-Orange Staining
Antisense digoxigenin-labeled RNA probes for dlx2a, ngn1, huC, and phf21a were produced with a DIG-RNA labeling kit (Roche, Germany) according to the manufacturer's instructions. Whole-mount in situ hybridization was performed with digoxigenin-labeled probes as previously described.30 Cartilage staining was carried out with Alcian blue.31 For the detection of apoptotic cells, embryos were placed in 10 μg/ml acridine orange (Sigma) for 30 min and were washed in egg water.
Microinjection of mRNA and Antisense MOs
Synthetic capped mRNAs for PHF21A and phf21a were transcribed in vitro with the linearized plasmid DNA as a template. mRNA was dissolved in 0.2% phenol red (as a tracking dye) and then microinjected into 1- to 2-cell-stage embryos. Antisense MOs for phf21a MO 5′-GCGTCATAAATGATATTTACCTGTG-3′ and standard control MO 5′-CCTCTTACCTCAGTTACAATTTATA-3′ were synthesized by Gene Tools (Corvallis, OR, USA). Each morpholino was resuspended in 1 × Danieau buffer (58 mM NaCl, 0.7 mM KCl, 0.4 mM MgSO4, 0.6 mM Ca(NO3)2, 5.0 mM HEPES, and pH 7.6) and injected into 1- to 2-cell-stage embryos at the concentration of 5 ng/embryo.
Results
PHF21A Is Disrupted in Unrelated Subjects with Chromosomal Translocations, ID, and CFAs
DGAP is a collaborative effort to identify genes of developmental importance through the study of individuals with apparently balanced chromosomal abnormalities and developmental defects.32 Identification of multiple cases in whom the same gene is disrupted in independent subjects with de novo translocations and similar phenotypes provides particularly strong evidence of the causative nature of the lesion. Through DGAP, we identified a subject (DGAP012) with an apparently balanced de novo translocation between chromosomes 11 and 19; this translocation resulted in a 46,XY,t(11;19)(p11.2;p13.2)dn karyotype (Figure 1A). A second subject, MCN1762 (MCN19730002-227), identified through the Mendelian Cytogenetic Network database, had an apparently balanced de novo translocation between chromosomes 1 and 11; this was initially reported as 46,XX,t(1;11)(p13;p11)dn (Figure 1B). They both display evidence of ID with CFAs, as well as other typical PSS features, except for multiple exostoses and parietal foramina, as summarized in Table 1, suggesting that the disruption in each case might affect the same gene in or near the PSS region in 11p11.2.
To map precisely the translocation in DGAP012, we first used FISH to bracket a candidate region and then to define a breakpoint-crossing BAC (Figure 1C and Figures S1A and S1B).24 After DNA blotting (Figures S1C and S1D) to refine the breakpoints, we used suppression cloning23 and targeted PCR for subsequent isolation and sequencing of junction fragments. Full details of the breakpoint-cloning steps are given in the Subjects and Methods section. The chromosome 19 breakpoint lies within a SINE/Alu sequence in intron 5 of ELAVL1, whereas the chromosome 11 breakpoint interrupts a LINE/L2 repetitive element in intron 14 of PHF21A (Figure 2A). At the der(19) breakpoint, there was a 5 nt CTCCT deletion of chromosome 11 sequence and a 5 nt TTCAG deletion of chromosome 19 sequence, whereas at the der(11) breakpoint, there was no loss or gain of nucleotides as a result of the translocation (Figure 2C). The breakpoints were also confirmed independently with a multiplexed targeted-capture and sequencing approach as previously described.33
Figure 2.
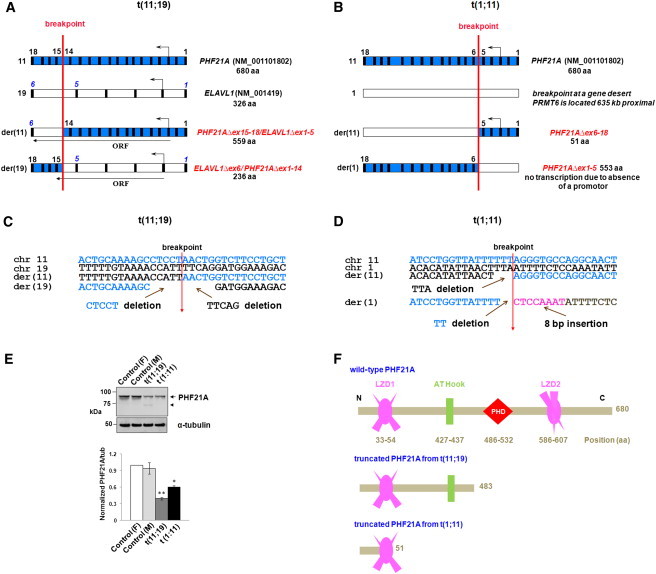
Mapping of the Breakpoints in Two Balanced Translocations
(A and B) Disruption of PHF21A by two translocations. A schematic diagram of the DGAP012 breakpoints (not to scale) shows, in blue, PHF21A on chromosome 11 (exons 1–18) and, in white, either (A) ELAVL1 on chromosome 19 (exons 1–6) or (B) chromosome 1. All exons are depicted as vertical black boxes, and the start and direction of transcription are indicated by an arrow above the corresponding exons. The translocation occurred at the site of the red vertical line. In DGAP012, the translocation produced der(11) and der(19) chromosomes that encode potential fusion transcripts (A). Note that the stop codon of the potential PHF21AΔex15-18/ELAVL1Δex1-5 fusion gene is at the same location as wild-type ELAVL1 because there is no frameshift, whereas ELAVL1Δex6/ PHF21AΔex1-14 has a frameshift with a premature stop codon in the new exon 6 (equivalent to exon 15 of PHF21A). The translocation in MCN1762 does not predict any potential fusion product because the breakpoint on chromosome 1 is located in a gene desert.
(C and D) Genomic DNA sequence from the normal chromosomes and at the breakpoints on derivative chromosomes. In DGAP012, at the der(19) breakpoint, there was a 5 nt CTCCT deletion of chromosome 11 sequence and a 5 nt TTCAG deletion of chromosome 19 sequence, whereas at the der(11) breakpoint, there was no loss or gain of sequence (C). In MCN1762, the junction sequences revealed a 3 bp TTA deletion of chromosome 1 sequence on der(11) and a 2 bp TT deletion of chromosome 11 sequence and an 8 bp CTCCAAAT insertion on der(1). Details of the mapping of both breakpoints in DGAP012 and MCN1762 are described in Figure S1 as well as in the Subjects and Methods section.
(E) Immunoblot analysis of PHF21A levels in DGAP012 and MCN1762 and in controls. The PHF21A antibody against an N-terminal 93 residue polypeptide28 recognizes the ∼92 kDa PHF21A in both female (“F”) and male (“M”) controls, as well as in DGAP012 and MCN1762 lymphoblastoid cell line extracts. It shows notably reduced protein levels due to disruption of PHF21A (arrow) in both DGAP012 and MCN1762. A 73 kDa protein (arrowhead) was noted in DGAP012 and is likely to be a product of the PHF21AΔex15-18/ELAVL1Δex1-5 fusion gene, which deletes the critical plant homeodomain (PHD) finger domain. α-tubulin was used as an internal loading control. The bar graph shows the mean and ± standard deviations from three independent experiments (∗p < 0.001, ∗∗p < 0.0001). The subcellular localization of PHF21A with the same antibody is described in Figure S3.
(F) PHF21A functional domains in wild-type and theoretical truncated proteins in two balanced translocation subjects. PHF21A contains two leucine zipper domains (LZD1 and LZD2), one AT-hook domain, and one PHD zinc finger domain. The amino acid positions of all domains are indicated as numbers below the domain structures. Note that if any protein were produced from the truncated PHF21A of DGAP012 or MCN1762, it would lack the PHD finger domain essential for binding H3K4me0.14
For the second subject, MCN1762, we used FISH (Figure 1D and Table S2) and array painting followed by PCR amplification to isolate the breakpoints, and this resulted in the revision of the previously designated karyotype to 46,XY,t(1;11)(p21.1;p11.2)dn (Figure 1B). The chromosome 1 breakpoint lies within a small 48 bp nonrepetitive sequence surrounded by LINE/L1 sequences in a region devoid of annotated genes; this region is 635 kb distal from the 5′ end of PRMT6 (MIM 608274). The chromosome 11 breakpoint is in intron 5 of PHF21A (Figure 2B). At the der(1) breakpoint, there was a 2 bp TT deletion of chromosome 1 sequence and an 8 bp CTCCAAAT insertion, whereas at the der(11) breakpoint, there was a 3 bp TTA deletion of chromosome 1 sequence (Figure 2D).
Immunofluorescence in cultured cells with an antibody specific to the amino-terminal segment of PHF21A revealed that the majority of the protein resides in the nuclei (Figure S3), consistent with a role for PHF21A in nuclear processes, such as chromatin association and transcriptional regulation. As expected from the sites of the translocations in both DGAP012 and MCN1762, disruption of PHF21A resulted in reduced protein levels of full-length PHF21A (as detected by immunoblot analysis) (Figure 2E). In both subjects, the PHF21A promoter could theoretically drive expression of a truncated PHF21A either alone or, in the case of DGAP012, as part of a fusion protein (Figure S2). However, in neither case would such a protein product contain the plant homeodomain (PHD) required for binding H3K4me014 (Figure 2F).
Localization of the chromosome 11 breakpoint from each subject within the same gene with consequent reduction in the protein product strongly supports a role for disruption of PHF21A in generating the ID and CFA phenotypes. This possibility was further supported by a third translocation subject, a young girl who displayed both ID and CFAs in conjunction with phenotypes reminiscent of Gillespie syndrome (MIM 206700). In this female subject, the translocation directly disrupted both PHF21A and the X chromosome gene ARHGAP6 (MIM 300118), encoding Rho-GTPase-activating protein 6.12,13 The precise breakpoints in this case were not determined (Table 1). The phenotypes of all three translocation subjects are compared in Table 1.
PHF21A Maps to the Critical PSS Interval Associated with ID and CFAs
Mapping of the translocation sites in PHF21A places them in proximity to the 11p11.2 genomic deletion region associated with PSS, in which phenotypic manifestations depend upon the precise location and extent of the deletion.15 The full phenotypic spectrum of PSS is manifested when deletions are at least 2.1 Mb in size,15 in which case they span the segment from D11S1393 to D11S1385/D11S1319 and contain 16 annotated genes and (Figure 3). The three translocation subjects share phenotypes with PSS subjects, but the notable exceptions are multiple exostoses and parietal foramina (Table 1), which are known to be caused by two genes (EXT2 and ALX4, respectively) distal to PHF21A. As an example, Figure 4 displays one of our translocation subjects and a PSS subject who both have CFAs that include microcephaly (Figures 4A, 4B, and 4D), brachycephaly (Figures 4A, 4B, and 4D), midfacial hypoplasia (Figure 4B), and a hypoplastic mandible (Figures 4D and 4E). Notably, neither of the two translocation subjects investigated here shows a second alteration on the nontranslocated chromosome by exon sequencing of PHF21A.
Figure 3.
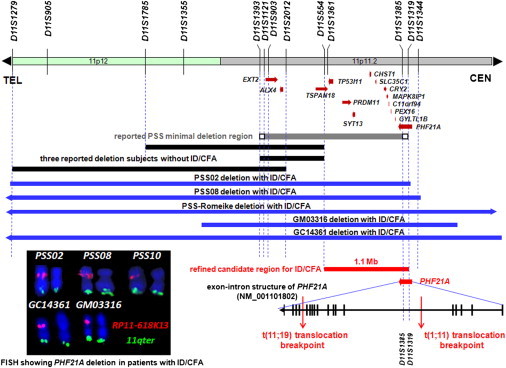
PHF21A Is Disrupted in DGAP012 and MCN1762 and Maps within the Refined Interval Associated with ID and CFAs
The minimal deletion region associated with the full spectrum of PSS phenotypes, compiled from a consensus of ten PSS subjects, is shown as a gray line encompassing deleted markers and flanked by small white boxes leading to markers D11S1393 and D11S1319, which are at the end of each box and are definitely not deleted.15 The PSS-associated genes EXT2 and ALX4, together with PHF21A, all map to this deletion region. Black lines show regions of deletion in three subjects showing neither ID nor CFAs.34–36 These deletion regions, subtracted from the above gray line, predict the ID and CFA candidate region (shown as a red line), a segment encompassing ∼1.1 Mb and containing 12 annotated genes between D11S554 and D11S1319. The proximal end of the ID and CFA candidate region overlaps with the terminal 98 kb of PHF21A. The blue lines of PSS subjects with ID and CFAs show unanimous deletion of PHF21A. At the bottom right, the breakpoints of DGAP012 and MCN1762 are shown. PHF21A is disrupted in both of these balanced translocation subjects with ID and CFAs but without multiple exostoses and parietal foramina. All together, the above genotype-phenotype analyses, combined with these two translocation cases, support a causative role for PHF21A in ID and CFAs. In the lower left corner is FISH mapping showing deletion of PHF21A in the five 11p11.2-deletion ID and CFA subjects shown above. FISH was carried out with BAC RP11-618K13 (red), which encompasses the 3′ portion of PHF21A, for three PSS subjects (PSS02, PSS08, and PSS10) whose centromeric PSS deletion boundaries had not been fully delineated15 and for two additional 11p11.2-deletion subjects (GC14361 and GM03316). Both chromosomes 11 from the same metaphase spread are shown, indicating the absence of PHF21A from one chromosome in each subject. Clone GS-770G7 from 11q25 (green) was used as a positive chromosome 11q control.
A critical reassessment of the in silico comparative deletion mapping of published deletion subjects, including those who display multiple exostoses and parietal foramina without ID or CFAs,34–36 excludes the distal portion of the PSS interval. This leaves a ∼1.1 Mb ID and CFA candidate interval that is between D11S554 and D11S1319 and encompasses 12 annotated genes (TSPAN18, TP53I11, PRDM11, SYT13, CHST1, SLC35C1, CRY2, MAPK8IP1, C11orf94, PEX16, GYLTL1B, and PHF21A) (Figure 3). PHF21A maps at the centromeric end of this candidate interval, and markers D11S1385 and D11S1319 are located within intron 5 (Figure 3).
To test whether PHF21A disruption occurs in PSS with ID and CFAs, we investigated five subjects clinically diagnosed with PSS and one subject with a PSS-like phenotype in association with a 11p11.2 deletion whose extent had not been previously resolved (Table 2). For five subjects (PSS02, PSS08, PSS10, GC14361, and GM03316), we were able to perform FISH with BAC clone RP11-618K13, which contains the telomeric 65 kb of PHF21A and spans the breakpoint in DGAP012. This BAC revealed heterozygous deletions in all five subjects (Figure 3, inset). In addition, we carried out array-CGH analysis on PSS02, PSS08, GC14361, GM03316, and one additional PSS subject (PSS-Romeike; Figure 4) reported recently,18 and this confirmed the loss of PFH21A in the first four subjects and also established it for the final subject (Figure 3 and Table 2). The coordinates of the deletions found by array CGH are listed in Table 2. Thus, PHF21A is hemizygous in all six subjects, five of whom were clinically diagnosed with PSS and all of whom display both ID and CFA phenotypes comparable to those of the translocation subjects.
To test the frequency of PHF21A hemizygosity in apparently normal individuals, we obtained a CNV dataset for 13,991 independent control individuals (collected across multiple studies37–42) from the International Schizophrenia Consortium and from Cooper et al.43 In this collection, the only structural variations (SVs) that crossed an exon of PHF21A were two identical 590 bp duplications that overlapped portions of exon 1 and intron 1 (the coding sequence of PHF21A begins in exon 3). We also examined the Database of Genomic Variants (DGV), which shows six SVs reported in this region; these include four small deletions (444–5,230 bp) within PHF21A introns (one in intron 1, two in intron 5, and one in intron 6) and one example of the aforementioned 590 bp 5′ UTR duplication. Data from the 1,000 Genomes Project also shows multiple CNVs at each of the exon 1 and intron 5 locations, as well as a single 545 bp CNV in the 3′ UTR.44 In addition to these presumably benign SVs, there is a single report in the DGV of a large 75 kb deletion encompassing six genes, including a portion of PHF21A. We are not in a position to validate that deletion or to confirm the absence of phenotype; however, there is precedent for even well-established disease-associated CNVs being nonpenetrant in some individuals.
Murine Phf21a Is Expressed in the CNS and Cranial Bones
To determine whether the pattern of expression of PHF21A supports a role in craniofacial and neuronal development, we performed in situ hybridization experiments for the orthologous mouse gene. Predominant expression of Phf21a is detected in the developing CNS at early stages. At mouse embryonic days (E) 13.5 and 14.5, the roof of the neopallidal cortex and thus the developing cerebral cortex, as well as the roof of the midbrain and the spinal cord, showed the highest expression levels of Phf21a. The intraventricular portion of the cerebellar primordium also expressed Phf21a at E14.5 (Figure 4G). At early embryonic stages, facial bone and viscerocranial ossification initiates and, with ongoing ossification, high levels of Phf21a transcripts were found at E17.5 in the palatine bone (Figure 4H and magnification in Figure 4I, marked by arrows) and the orbitosphenoidal bone (Figure 4H, I black arrowhead), as well as in the calvaria. Signals observed in bone with Phf21a antisense probes were essentially restricted to cranial bones, suggesting a particular function for Phf21a in craniofacial development. In the adult mouse brain, the most abundant expression of Phf21a was observed in the neuronal layers of the hippocampus, the granule cell layer of the cerebellum, and the main olfactory bulb (Figure 4J). All together, these findings indicate that expression of Phf21a is consistent with an important role in the CNS and craniofacial skeletal development and in adult neuronal function. Interestingly, a single report of a mouse knockout for Phf21a described no gross morphological abnormality, although the potential for CFAs was not specifically evaluated. Neonatal mice died as a result of an inability to suckle properly; this inability was interpreted as a likely defect in neuronal control of milk-sucking behavior.45
Suppression of Zebrafish phf21a Expression Causes CFAs and Neuronal Apoptosis
To directly test the developmental importance of PHF21A, we isolated the zebrafish phf21a ortholog, examined its expression pattern, and performed gain- and loss-of-function experiments in this model organism. The zebrafish phf21a is highly related to human and mouse PHF21A proteins: It exhibits an AT-hook domain, a PHD, and two coiled-coil domains (data not shown). Using RT-PCR, we confirmed that zebrafish phf21a showed maternal and zygotic transcripts during embryonic development (Figure S4A). Whole-mount in situ hybridization analyses revealed phf21a transcripts ubiquitously distributed throughout the embryo during the stages of cleavage, blastula, gastrula, and early segmentation. Expression in the head region was increased from later somitogenesis and continued to 24 and 48 hpf (Figures S4B–S4J).
To investigate the function of phf21a in zebrafish development, we tested the effect of phf21a knockdown by antisense MO. Injection of the phf21a MO, but not of a standard control MO or no MO, caused a small-head phenotype and facial dysmorphism with a pronounced defect in growth of the lower jaw at 3 days postfertilization (dpf); these features are reminiscent of the microcephaly and dysmorphism seen in the translocation subjects (Figures 5A–5C and 5Q).
Figure 5.
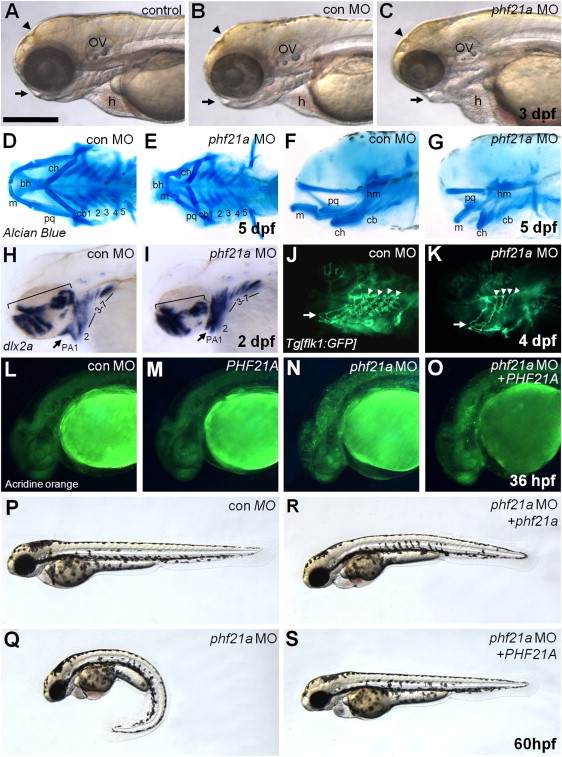
PHF21A Regulates Zebrafish Neuronal Cell Survival and Craniofacial Development
(A–C) A noninjected control embryo (A), an embryo injected with a control MO (B), and an embryo injected with a phf21a MO (C). Knockdown of phf21a causes a reduction in head size, a change of head and face shape (arrowhead), and a major reduction of the jaw (arrow) at 3 dpf; these features are reminiscent of the microcephaly and craniofacial dysmorphism seen in the translocation subjects. The scale bar in (A) represents 300 μm. The following abbreviations are used: ov, otic vesicle; and h, heart.
(D–G) Cartilage staining of embryos injected with the control MO (D and F) or phf21a MO (E and G). Compared with that of the control embryo, Meckel's and palatoquadrate cartilage in the phf21a-MO-injected embryos are severely distorted in their size and shape. Five-day-old embryos are shown in ventral (D and E) or lateral (F and G) views. The following abbreviations are used: ch, ceratohyal; bh, basihyal cartilage; m, Meckel's cartilage; pq, palatoquadrate cartilage; and cb 1–5, ceratobranchial cartilage 1–5.
(H and I) dlx2a expression in the control (H) and the phf21a morphant (I) at 2 dpf. phf21a morphants fail to expand dlx2a-expressing pharyngeal-arch progenitor cells (arrow). The scale bar in (H) represents 300 μm, and the scale bar in (I) represents 230 μm. The following abbreviation is used: PA 1–7, pharyngeal arches 1–7.
(J and K) Formation and patterning of arch-associated blood vessels in the control (J) and the phf21a morphant (K). A lateral view of the flk1:GFP transgenic line at 4 dpf is shown. In phf21a-MO-injected embryos, aortic arches (arrowheads) form normally but fail to swing to a more anterior position (compare the position of arrows in J and K). Also, the vessels associated with gill filaments develop poorly in morphants.
(L–O) Effects on neuronal cell survival. Compared with the control MO (L) and PHF21A-mRNA-injected embryos (M), phf21a-MO-injected embryos (N) show a dramatically increased number of acridine-orange-positive apoptotic cells. The apoptotic phenotype of the phf21a morphant is rescued by coinjection of PHF21A mRNA (O).
(P–S) The phf21a-MO causes craniofacial, morphological, and growth defects in the developing zebrafish embryo. Reintroduction of wild-type phf21a/PHF21A rescues the zebrafish phenotype. A control-MO-injected embryo is shown in (P). Knockdown of phf21a causes ventral curvature of the body and a small-head phenotype (Q). These phenotypes of the phf21a MO are rescued by coinjection of either zebrafish phf21a mRNA (R) or human PHF21A mRNA (S). Expression of phf21a during zebrafish early development is described in Figure S4.
We examined the head structure of phf21a morphants in more detail by using Alcian blue to visualize the extent of cartilage development in larval fishes. At 5 dpf, Meckel's and palatoquadrate cartilages were severely distorted in their size and shape in phf21a morphants (Figures 5D–5G). Such defects were already manifest during early stages given that we also observed defects in dlx2a-positive pharyngeal-arch-cartilage progenitor cells in phf21a morphants at 2 dpf (Figures 5H and 5I). Defects in cranial-cartilage formation were also observed for the zebrafish headless mutation, which is known to be involved in the signaling pathway of vertebrate head formation and patterning.31 To investigate further whether these defects also involve other arch-associated structures, we injected the phf21a MO into flk1:GFP transgenic zebrafish, in which the vascular endothelial cells were visualized by green fluorescent protein (GFP) fluorescence.46 At 4 dpf, the aortic arches of the phf21a-MO-injected flk1:GFP transgenic embryos were found to be hypoplastic: They showed poor development of capillary networks associated with pharyngeal arches (Figures 5J and 5K). In vertebrates, Meckel's and palatoquadrate cartilages form the embryonic jaw apparatus.47,48 Thus, it would be interesting to examine whether the Phf21a/Bhc80-deficient mice, which display a failure to suckle,45 might also have a defect in jaw structure.
We also examined the effects of gain or loss of phf21a function on neuronal development but did not see any prominent change (Figure S5). However, injection of the phf21a MO, but not a standard control MO or PHF21a mRNA, caused apoptosis in the developing brain region at 36 hpf (Figures 5L–5N). Importantly, this apoptosis and the small-head phenotype can be rescued by introduction of wild-type human PHF21A mRNA (Figure 5O), suggesting that the ID phenotype in humans might be due to a requirement for PHF21A in the function of neuronal cell survival in the developing brain.
Overall, the phf21a MO caused craniofacial, morphological, and growth defects in the developing zebrafish embryo, as depicted by the notable ventral curvature of the body and small-head phenotype (Figure 5Q) relative to that of the control (Figure 5P). The body axis of the zebrafish normally straightens from its early curvature in the pharyngula developmental stage (24–48 hr), during which phf21a is strongly expressed in the spinal cord (Figures S4G and S4I). Therefore, phf21a depletion in the spinal cord might impair the straightening process, resulting in persistent spinal and tail curvature. Two processes that have previously been implicated in such ventral curvature are dorsal midline development and cilia development, but the relationship of this phenotype to human PSS is unclear. Notably, injection of either wild-type phf21A or PHF21A mRNA rescued both the spinal curvature and small-head phenotypes (Figures 5R and 5S), demonstrating that they result from a lack of a conserved function of the protein.
Disruption of PHF21A in the Translocation Subjects Derepresses SCN3A
Along with LSD1, PHF21A is known to be a component of the CoREST-related protein complex, BRAF-HDAC complex (BHC), which participates in neuron-specific gene repression presumably by regulating histone-demethylation activity.26,27 PHF21A specifically recognizes unmethylated histone H3 lysine 4 residues and is required for LSD1-mediated transcriptional repression and LSD1 occupancy at target promoters.14 To investigate whether LSD1-mediated transcriptional repression is functionally altered by the PHF21A-disrupting translocations, we first examined transcription levels of several reported LSD1 targets, including SCN1A, SCN3A, and SYN1, in lymphoblastoid cell lines derived from normal males, normal females, and translocation subjects DGAP012 and MCN1762. We found that SYN1 and SCN1A have high and moderate levels, respectively, of expression, even in normal lymphoblastoid cells, indicating that they are not epigenetically repressed like they are in HeLa cells.26,27 However, SCN3A mRNA was expressed at a lower level as measured by RT-PCR in lymphoblastoid cells from normal controls, suggesting that this LSD1 target gene is transcriptionally repressed. An increase of SCN3A transcript was readily detected in DGAP012 and MCN1762 cells (Figures 6A and 6B), indicating that a single functional allele of PHF21A might not be sufficient for effective repression of SCN3A transcription. This is consistent with the fact that correct dosage of PHF21A is important for its function. In support of this hypothesis, ChIP results showed a significant reduction of LSD1 binding to the SCN3A promoter in DGAP012 and MCN1762 cells compared to normal control cells (Figure 6C). These findings indicate that SCN3A might be repressed by the LSD1 corepressor complex through a mechanism similar to that reported in HeLa cells14,26 and that PHF21A might be similarly required for LSD1 promoter occupancy in lymphoblasts. This functional disruption of PHF21A in DGAP012 and MCN1762 lymphoblasts is probably mirrored by alterations in gene regulation in many tissues, including the CNS.
Figure 6.
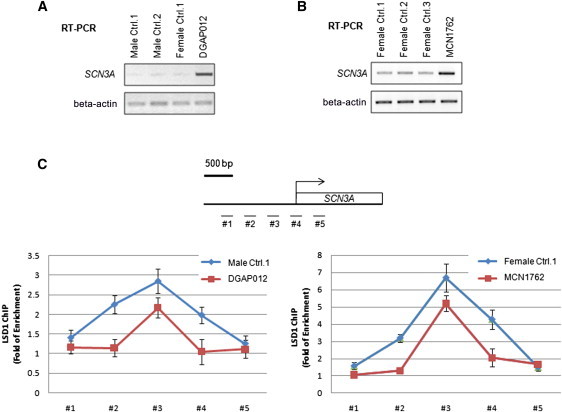
SCN3A Transcriptional Derepression and Reduced LSD1 Promoter Occupancy in DGAP012 and MCN1762 Lymphoblastoid Cells
SCN3A transcription is derepressed in affected individuals DGAP012 and MCN1762. Semiquantitative RT-PCR reactions were carried out in separate experiments for comparing the levels of SCN3A transcript (top box) in normal male and female controls with those of DGAP012 (A) and MCN1762 (B). Beta-actin (lower box) was used as a control in both experiments.
(C) LSD1 occupancy at the SCN3A promoter was compared in separate experiments between a normal male (#1 in left panel) and DGAP012 and between a normal female (#1 in right panel) and MCN1762 with the use of a ChIP assay. Five sets of PCR primers spanning the SCN3A promoter were used. These assays revealed reduced LSD1 occupancy at the SCN3A promoter in DGAP012 and MCN1762. The two panels cannot be directly compared because the two sets of assays were carried out at different times with different reagents (Abcam ab-17721 in left panel and Cell Signaling 2184 in right panel) and detection methods (radioactive PCR in left panel and SYBR Green real-time PCR in right panel). Error bars represent the standard error of the mean calculated on the basis of three independent experiments. An intragenic region upstream of the ACTG promoter not bound by LSD1 was used as a reference region in the ChIP assay for normalization.
Discussion
PSS is a contiguous gene syndrome involving ID and CFAs, along with other distinctive features, including eye abnormalities (severe myopia, nystagmus, and strabismus), skeletal anomalies (small hands and feet and tapering fingers), multiple exostoses, and enlarged parietal foramina.7,15,16,36,49,50 The challenging aspect of positional cloning in such contiguous gene syndromes is assigning individual phenotypes to individual genes in the deleted region. Because the size and location of contiguous-gene-syndrome deletions can vary from individual to individual, comparison of overlapping regions for defining a minimal candidate region associated with a particular phenotype has often preceded candidate-gene analysis for identifying the associated gene.51,52 These strategy and linkage analyses, respectively, led to two genes implicated in the pathogenesis of PSS: ALX4, associated with enlarged parietal foramina,53 and EXT2, responsible for multiple exostoses.9 However, these approaches have not identified the gene(s) responsible for the ID and CFA phenotypes. Attempts to identify the gene(s) underlying the latter PSS phenotypes have been hampered by the relatively large size of the minimal candidate interval (∼2.1 Mb).15 Both categories of clinical features are individually relatively common: ID affects ∼2%–3% of humans,54 and CFAs are present in ∼1/3 of human congenital defects.55 In many cases, these phenotypes manifest together, suggesting a common underlying etiology. Our reinterpretation of the PSS candidate region for ID and CFAs to ∼1.1 Mb on the basis of published subjects with neither ID nor CFAs34–36 and the identification of two independent translocation subjects with breakpoints at the proximal end of this region suggest that disruption of a single gene, PHF21A, is responsible for both ID and CFAs. This finding is supported by a third translocation case from the literature.12,13
Haploinsufficiency of PHF21A is the probable cause of ID and CFAs in all cases that we studied given that we detected no additional PHF21A mutation on the nontranslocated alleles in DGAP012 and MCN1762 and no clear differences between the critical ID and CFA features of these subjects and those of the PSS subjects with interstitial deletions (Tables 1 and 2). In MCN1762, PHF21A was the only gene disrupted, whereas in DGAP012, ELAVL1 was also truncated. ELAVL1 encodes a protein that contains three RNA-binding domains and binds cis-acting AU-rich elements. It destabilizes mRNAs and thereby regulates gene expression.56,57 Because both subjects show comparable phenotypes, it is unlikely that ELAV1 haploinsufficiency or fusion proteins contribute strongly to the critical PSS-like phenotypes (Table 1), although they might be responsible for unusual features, such as possible retinal dystrophy and a digitized thumb, in DGAP012. The third translocation subject was only examined as a very young girl and displayed some phenotypes (bilateral superior coloboma, foveal hypoplasia, inferior cerebellar hypoplasia, and ID) thought to be suggestive of Gillespie syndrome (partial aniridia, cerebellar ataxia, and ID). Gillespie syndrome is known to be genetically heterogeneous and is caused in some cases by lesions in PAX6 (MIM 607108). In this subject, it is likely that the disruption of PHF21A contributed to the ID and craniofacial dysmorphism in common with the other two translocation subjects (Table 1). The disparate phenotypes might then have been contributed by disruption of ARHGAP6 or by an independent lesion elsewhere. Notably, mice homozygous for a targeted null mutation of ARHGAP6 do not exhibit any detected phenotypic or behavioral abnormalities.58 A strict PHF21A dosage requirement for normal craniofacial and neurodevelopment is also supported by our findings in the zebrafish system where phf21a suppression produced abnormalities in the development of the head, face, and jaw, as well as increased neuronal apoptosis. Importantly, these deficiencies were rescued by the human PHF21A, indicating a conserved developmental function.
The discovery that translocations disrupting PHF21A are associated with abnormal craniofacial and intellectual development adds to the evidence that regulation of gene expression through chromatin modifications is crucial to both processes. To date, three genes, NSD1 (MIM 606681), KDM5C (MIM 314690), and PHF8 (MIM 300560), encoding proteins that have PHD domains and bind methylated histone tails have been found in syndromes with ID and distinctive craniofacial features. NSD1, which is deficient in Sotos syndrome, specifically binds methylated H3K4 and H3K9 via domains PHD1, PHD4, PHD5, and PHD6;59 SMCX/JARID1C (encoded by KDM5C) of nonsyndromic XLID binds histone H3K9me3 via its PHD1 domain;60 and PHF8 of Siderius-Hamel syndrome binds histone H3K4me3 via its PHD.61 At least the latter two act as demethylases by targeting H3K4me2 and H3K4me3 in the case of SMCX/JARID1C60 and H3K9me1/2 and H4K20me1 in the case of PHF8.61,62
Unlike the above proteins, PHF21A is neither a methyltransferase nor a demethylase but instead specifically binds histone H3K4 when it is not methylated.14 This suggests that both recognition of the unmodified state of histone tails and binding of proteins to methylated histone tails are critical for maintaining the appropriate balance and control of particular chromatin modifications for the support of normal intellectual and craniofacial development. Although the PSS-associated ID and CFAs appear to be due to haploinsufficiency of PHF21A, it has not been possible to ascertain and screen a large series of nontranslocation subjects with comparable phenotypes for PHF21A mutations. Therefore, we cannot state with certainty that missense, nonsense, splicing, or other mutations in PHF21A would lead to the same ID and CFA phenotypes. Indeed, it is conceivable that other types of genetic lesions in PHF21A could actually be associated with other developmental phenotypes; we were able to identify 200 individuals with ID and/or CFAs but without the full constellation of phenotypes exhibited by our translocation subjects, and we performed a mutation screen of PHF21A. We did not detect any truncating or missense mutations that have implicated particular PHD domains in NSD1 in binding to their methylated targets and that could thus aid in structure-function experiments. This is not surprising given the frequency of these two major phenotypes and the extent of genetic heterogeneity underlying each of them, but more extensive mutation analysis of subjects with various manifestations of ID, CFAs, and additional phenotypes seen in our translocation subjects might prove valuable to understanding the functional domains of PHF21A.
PHF21A (BHC80) is known to participate in the six-subunit BHC, which also comprises BRAF35 (MIM 605535), HDAC1 (MIM 601241), HDAC2 (MIM 605164), CoREST (MIM 607675), and LSD1 (BHC110); the latter is a histone demethylase that targets H3K4me2.26 This complex interacts with the promoters of genes, such as synapsin and sodium-channel genes, to mediate repression of these neuron-specific genes through the cis-regulatory element known as repressor element 1 or neural restrictive silencer (RE1/NRS).63 Specific binding of PHF21A to H3K4me0 is required for optimal LSD1 promoter occupancy in vivo and for LSD1-mediated gene repression.14 Our data showing derepression of the neural gene SCN3A in lymphoblasts from the translocation subjects as a consequence of reduced levels of PHF21A are consistent with this role for the protein. Repression of neuronal-specific genes is of fundamental importance in the development of both neuronal and nonneuronal tissues,63 so the failure of this particular function in the translocation subjects might have contributed to their ID and CFAs.
Another interesting XLID candidate, ZMYM3 (MIM 300061), encodes a zinc finger protein that is predominantly expressed in the brain and that is a component of transcriptional corepressor complexes that also contain LSD1 (BHC110) and HDAC2.64,65 The 5′ UTR of ZMYM3 is disrupted by a presumably balanced t(X;13) karyotype in a female with ID and preferential inactivation of the normal X chromosome.66 In addition, Kleefstra syndrome, characterized by ID and CFAs comparable to PSS, has been associated with disruption of EHMT1, encoding euchromatin histone methyltransferase 1, which acts as a methyl transferase to modify H3K9 and has been reported as a component of the E2F6 transcription repressor complex and of a CtBP repressor complex that also contains LSD1.2,67–69 The parallels between PHF21A, ZMYM3, and EHMT1 suggest that other X-linked and autosomal loci underlying ID and/or CFAs might encode proteins that participate in complexes involving LSD1 or potentially other demethylases or methyltransferases. Our finding that decreased dosage of PHF21A, a histone-binding protein that interacts with and is required for the histone-demethylase activity of LSD1, leads to both ID and CFAs provides the proof of principle for investigation of other regulators of histone modification as genetic factors in ID and/or CFAs. Indeed, the recent finding of haploinsufficiency of ARID1B (MIM 614556), encoding an E3-ubiquitin-ligase component that functions with the chromatin-remodeling switch/sucrose nonfermentable complex,70–74 suggests that genes involved with other aspects of chromatin modification might also contribute to ID and CFAs and that ultimately, human mutations affecting both regulatory and enzymatic components of histone-modification complexes might represent important tools for delineating the chromatin-regulation features that are critical for normal craniofacial and neurological development and cognitive function.
Acknowledgments
We are grateful to DGAP012 and MCN1762, as well as their family members, for their cooperation and participation in this study. We are also indebted to Amy Bosco, Heather L. Ferguson, and Chantal Kelly for obtaining informed consent and clinical information; to Joanne Sutherland, genetic counselor at the Hospital for Sick Children, Toronto, for her assistance in obtaining samples; to Shigeki Iwase and Tadashi Baba for the PHF21A antibody; to Mary Anne Anderson and Tammy Gillis in the Center for Human Genetic Research Tissue Culture Facility and Genomics Resource for technical assistance; to Ian Krantz, Stephanie Seminara, and Simeon A. Boyadjiev for providing samples of affected individuals; and to Ines Müller and Corinna Menzel for technical assistance. This work was supported by a grant from the Next-Generation BioGreen 21 Program (PJ00812701 to C.H.K.), Republic of Korea, a grant from the Deutsche Forschungsgemeinschaft (KU 1240/5-1 to K.K.), the Danish National Research Foundation (N.T.), the Lundbeck Foundation (N.T. and A.S.), National Institutes of Health grants NCI118487 (to Y.S.) and RO1 GM071004 (to Y.S.), and United States Public Health Service grants GM061354 (Developmental Genome Anatomy Project to C.C.M. and J.F.G.) and HD065286 (to J.F.G.). Part of this work was financed by the European Union's Seventh Framework Program under grant agreement number 241995, project GENCODYS, and the German Federal Ministry of Education and Research through the German Mental Retardation Network (grant 01GS08161 to H.H.R.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Agilent Technologies, https://earray.chem.agilent.com
Developmental Genome Anatomy Project, http://dgap.harvard.edu
Mendelian Cytogenetics Network Online Database, http://www.mcndb.org/index.jsp
NCBI and GenBank, http://www.ncbi.nlm.nih.gov
NIGMS Human Genetic Cell Repository, http://ccr.coriell.org/nigms/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Koolen D.A., Vissers L.E., Pfundt R., de Leeuw N., Knight S.J., Regan R., Kooy R.F., Reyniers E., Romano C., Fichera M. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat. Genet. 2006;38:999–1001. doi: 10.1038/ng1853. [DOI] [PubMed] [Google Scholar]
- 2.Kleefstra T., Brunner H.G., Amiel J., Oudakker A.R., Nillesen W.M., Magee A., Geneviève D., Cormier-Daire V., van Esch H., Fryns J.P. Loss-of-function mutations in euchromatin histone methyl transferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome. Am. J. Hum. Genet. 2006;79:370–377. doi: 10.1086/505693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalscheuer V.M., Tao J., Donnelly A., Hollway G., Schwinger E., Kübart S., Menzel C., Hoeltzenbein M., Tommerup N., Eyre H. Disruption of the serine/threonine kinase 9 gene causes severe X-linked infantile spasms and mental retardation. Am. J. Hum. Genet. 2003;72:1401–1411. doi: 10.1086/375538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kalscheuer V.M., FitzPatrick D., Tommerup N., Bugge M., Niebuhr E., Neumann L.M., Tzschach A., Shoichet S.A., Menzel C., Erdogan F. Mutations in autism susceptibility candidate 2 (AUTS2) in patients with mental retardation. Hum. Genet. 2007;121:501–509. doi: 10.1007/s00439-006-0284-0. [DOI] [PubMed] [Google Scholar]
- 5.Kim H.G., Ahn J.W., Kurth I., Ullmann R., Kim H.T., Kulharya A., Ha K.S., Itokawa Y., Meliciani I., Wenzel W. WDR11, a WD protein that interacts with transcription factor EMX1, is mutated in idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am. J. Hum. Genet. 2010;87:465–479. doi: 10.1016/j.ajhg.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim H.G., Kishikawa S., Higgins A.W., Seong I.S., Donovan D.J., Shen Y., Lally E., Weiss L.A., Najm J., Kutsche K. Disruption of neurexin 1 associated with autism spectrum disorder. Am. J. Hum. Genet. 2008;82:199–207. doi: 10.1016/j.ajhg.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Potocki L., Shaffer L.G. Interstitial deletion of 11(p11.2p12): A newly described contiguous gene deletion syndrome involving the gene for hereditary multiple exostoses (EXT2) Am. J. Med. Genet. 1996;62:319–325. doi: 10.1002/(SICI)1096-8628(19960329)62:3<319::AID-AJMG22>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 8.Shaffer L.G., Hecht J.T., Ledbetter D.H., Greenberg F. Familial interstitial deletion 11(p11.12p12) associated with parietal foramina, brachymicrocephaly, and mental retardation. Am. J. Med. Genet. 1993;45:581–583. doi: 10.1002/ajmg.1320450512. [DOI] [PubMed] [Google Scholar]
- 9.Stickens D., Clines G., Burbee D., Ramos P., Thomas S., Hogue D., Hecht J.T., Lovett M., Evans G.A. The EXT2 multiple exostoses gene defines a family of putative tumour suppressor genes. Nat. Genet. 1996;14:25–32. doi: 10.1038/ng0996-25. [DOI] [PubMed] [Google Scholar]
- 10.Mavrogiannis L.A., Antonopoulou I., Baxová A., Kutílek S., Kim C.A., Sugayama S.M., Salamanca A., Wall S.A., Morriss-Kay G.M., Wilkie A.O. Haploinsufficiency of the human homeobox gene ALX4 causes skull ossification defects. Nat. Genet. 2001;27:17–18. doi: 10.1038/83703. [DOI] [PubMed] [Google Scholar]
- 11.Wu Y.Q., Badano J.L., McCaskill C., Vogel H., Potocki L., Shaffer L.G. Haploinsufficiency of ALX4 as a potential cause of parietal foramina in the 11p11.2 contiguous gene-deletion syndrome. Am. J. Hum. Genet. 2000;67:1327–1332. doi: 10.1016/s0002-9297(07)62963-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dollfus H., Joanny-Flinois O., Doco-Fenzy M., Veyre L., Joanny-Flinois L., Khoury M., Jonveaux P., Abitbol M., Dufier J.L. Gillespie syndrome phenotype with a t(X;11)(p22.32;p12) de novo translocation. Am. J. Ophthalmol. 1998;125:397–399. doi: 10.1016/s0002-9394(99)80157-3. [DOI] [PubMed] [Google Scholar]
- 13.Fantes J.A., Boland E., Ramsay J., Donnai D., Splitt M., Goodship J.A., Stewart H., Whiteford M., Gautier P., Harewood L. FISH mapping of de novo apparently balanced chromosome rearrangements identifies characteristics associated with phenotypic abnormality. Am. J. Hum. Genet. 2008;82:916–926. doi: 10.1016/j.ajhg.2008.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lan F., Collins R.E., De Cegli R., Alpatov R., Horton J.R., Shi X., Gozani O., Cheng X., Shi Y. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature. 2007;448:718–722. doi: 10.1038/nature06034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wakui K., Gregato G., Ballif B.C., Glotzbach C.D., Bailey K.A., Kuo P.L., Sue W.C., Sheffield L.J., Irons M., Gomez E.G. Construction of a natural panel of 11p11.2 deletions and further delineation of the critical region involved in Potocki-Shaffer syndrome. Eur. J. Hum. Genet. 2005;13:528–540. doi: 10.1038/sj.ejhg.5201366. [DOI] [PubMed] [Google Scholar]
- 16.Wuyts W., Waeber G., Meinecke P., Schüler H., Goecke T.O., Van Hul W., Bartsch O. Proximal 11p deletion syndrome (P11pDS): Additional evaluation of the clinical and molecular aspects. Eur. J. Hum. Genet. 2004;12:400–406. doi: 10.1038/sj.ejhg.5201163. [DOI] [PubMed] [Google Scholar]
- 17.Anderson M.A., Gusella J.F. Use of cyclosporin A in establishing Epstein-Barr virus-transformed human lymphoblastoid cell lines. In Vitro. 1984;20:856–858. doi: 10.1007/BF02619631. [DOI] [PubMed] [Google Scholar]
- 18.Romeike B.F., Wuyts W. Proximal chromosome 11p contiguous gene deletion syndrome phenotype: Case report and review of the literature. Clin. Neuropathol. 2007;26:1–11. doi: 10.5414/npp26001. [DOI] [PubMed] [Google Scholar]
- 19.Miller D.T., Shen Y., Wu B.L. Oligonucleotide microarrays for clinical diagnosis of copy number variation. Curr. Protoc. Hum. Genet. 2008;58:8.12.1–8.12.17. doi: 10.1002/0471142905.hg0812s58. [DOI] [PubMed] [Google Scholar]
- 20.Obholz K.L., Akopyan A., Waymire K.G., MacGregor G.R. FNDC3A is required for adhesion between spermatids and Sertoli cells. Dev. Biol. 2006;298:498–513. doi: 10.1016/j.ydbio.2006.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arkesteijn G., Jumelet E., Hagenbeek A., Smit E., Slater R., Martens A. Reverse chromosome painting for the identification of marker chromosomes and complex translocations in leukemia. Cytometry. 1999;35:117–124. doi: 10.1002/(sici)1097-0320(19990201)35:2<117::aid-cyto2>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 22.Chen W., Kalscheuer V., Tzschach A., Menzel C., Ullmann R., Schulz M.H., Erdogan F., Li N., Kijas Z., Arkesteijn G. Mapping translocation breakpoints by next-generation sequencing. Genome Res. 2008;18:1143–1149. doi: 10.1101/gr.076166.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siebert P.D., Chenchik A., Kellogg D.E., Lukyanov K.A., Lukyanov S.A. An improved PCR method for walking in uncloned genomic DNA. Nucleic Acids Res. 1995;23:1087–1088. doi: 10.1093/nar/23.6.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheung V.G., Nowak N., Jang W., Kirsch I.R., Zhao S., Chen X.N., Furey T.S., Kim U.J., Kuo W.L., Olivier M., BAC Resource Consortium Integration of cytogenetic landmarks into the draft sequence of the human genome. Nature. 2001;409:953–958. doi: 10.1038/35057192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bartsch O., Schmidt S., Richter M., Morlot S., Seemanová E., Wiebe G., Rasi S. DNA sequencing of CREBBP demonstrates mutations in 56% of patients with Rubinstein-Taybi syndrome (RSTS) and in another patient with incomplete RSTS. Hum. Genet. 2005;117:485–493. doi: 10.1007/s00439-005-1331-y. [DOI] [PubMed] [Google Scholar]
- 26.Shi Y., Lan F., Matson C., Mulligan P., Whetstine J.R., Cole P.A., Casero R.A., Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 27.Shi Y.J., Matson C., Lan F., Iwase S., Baba T., Shi Y. Regulation of LSD1 histone demethylase activity by its associated factors. Mol. Cell. 2005;19:857–864. doi: 10.1016/j.molcel.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 28.Iwase S., Januma A., Miyamoto K., Shono N., Honda A., Yanagisawa J., Baba T. Characterization of BHC80 in BRAF-HDAC complex, involved in neuron-specific gene repression. Biochem. Biophys. Res. Commun. 2004;322:601–608. doi: 10.1016/j.bbrc.2004.07.163. [DOI] [PubMed] [Google Scholar]
- 29.Park H.C., Kim C.H., Bae Y.K., Yeo S.Y., Kim S.H., Hong S.K., Shin J., Yoo K.W., Hibi M., Hirano T. Analysis of upstream elements in the HuC promoter leads to the establishment of transgenic zebrafish with fluorescent neurons. Dev. Biol. 2000;227:279–293. doi: 10.1006/dbio.2000.9898. [DOI] [PubMed] [Google Scholar]
- 30.Kim C.H., Ueshima E., Muraoka O., Tanaka H., Yeo S.Y., Huh T.L., Miki N. Zebrafish elav/HuC homologue as a very early neuronal marker. Neurosci. Lett. 1996;216:109–112. doi: 10.1016/0304-3940(96)13021-4. [DOI] [PubMed] [Google Scholar]
- 31.Kim C.H., Oda T., Itoh M., Jiang D., Artinger K.B., Chandrasekharappa S.C., Driever W., Chitnis A.B. Repressor activity of Headless/Tcf3 is essential for vertebrate head formation. Nature. 2000;407:913–916. doi: 10.1038/35038097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Higgins A.W., Alkuraya F.S., Bosco A.F., Brown K.K., Bruns G.A., Donovan D.J., Eisenman R., Fan Y., Farra C.G., Ferguson H.L. Characterization of apparently balanced chromosomal rearrangements from the developmental genome anatomy project. Am. J. Hum. Genet. 2008;82:712–722. doi: 10.1016/j.ajhg.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Talkowski M.E., Ernst C., Heilbut A., Chiang C., Hanscom C., Lindgren A., Kirby A., Liu S., Muddukrishna B., Ohsumi T.K. Next-generation sequencing strategies enable routine detection of balanced chromosome rearrangements for clinical diagnostics and genetic research. Am. J. Hum. Genet. 2011;88:469–481. doi: 10.1016/j.ajhg.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hall C.R., Wu Y., Shaffer L.G., Hecht J.T. Familial case of Potocki-Shaffer syndrome associated with microdeletion of EXT2 and ALX4. Clin. Genet. 2001;60:356–359. doi: 10.1034/j.1399-0004.2001.600506.x. [DOI] [PubMed] [Google Scholar]
- 35.Mavrogiannis L.A., Taylor I.B., Davies S.J., Ramos F.J., Olivares J.L., Wilkie A.O. Enlarged parietal foramina caused by mutations in the homeobox genes ALX4 and MSX2: From genotype to phenotype. Eur. J. Hum. Genet. 2006;14:151–158. doi: 10.1038/sj.ejhg.5201526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wuyts W., Di Gennaro G., Bianco F., Wauters J., Morocutti C., Pierelli F., Bossuyt P., Van Hul W., Casali C. Molecular and clinical examination of an Italian DEFECT11 family. Eur. J. Hum. Genet. 1999;7:579–584. doi: 10.1038/sj.ejhg.5200339. [DOI] [PubMed] [Google Scholar]
- 37.Ferreira M.A., O'Donovan M.C., Meng Y.A., Jones I.R., Ruderfer D.M., Jones L., Fan J., Kirov G., Perlis R.H., Green E.K., Wellcome Trust Case Control Consortium Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat. Genet. 2008;40:1056–1058. doi: 10.1038/ng.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O'Donovan M.C., Craddock N., Norton N., Williams H., Peirce T., Moskvina V., Nikolov I., Hamshere M., Carroll L., Georgieva L., Molecular Genetics of Schizophrenia Collaboration Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat. Genet. 2008;40:1053–1055. doi: 10.1038/ng.201. [DOI] [PubMed] [Google Scholar]
- 39.Purcell S.M., Wray N.R., Stone J.L., Visscher P.M., O'Donovan M.C., Sullivan P.F., Sklar P., International Schizophrenia Consortium Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith E.N., Bloss C.S., Badner J.A., Barrett T., Belmonte P.L., Berrettini W., Byerley W., Coryell W., Craig D., Edenberg H.J. Genome-wide association study of bipolar disorder in European American and African American individuals. Mol. Psychiatry. 2009;14:755–763. doi: 10.1038/mp.2009.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sullivan P.F., Lin D., Tzeng J.Y., van den Oord E., Perkins D., Stroup T.S., Wagner M., Lee S., Wright F.A., Zou F. Genomewide association for schizophrenia in the CATIE study: Results of stage 1. Mol. Psychiatry. 2008;13:570–584. doi: 10.1038/mp.2008.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.International Schizophrenia Consortium Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cooper G.M., Coe B.P., Girirajan S., Rosenfeld J.A., Vu T.H., Baker C., Williams C., Stalker H., Hamid R., Hannig V. A copy number variation morbidity map of developmental delay. Nat. Genet. 2011;43:838–846. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iwase S., Shono N., Honda A., Nakanishi T., Kashiwabara S., Takahashi S., Baba T. A component of BRAF-HDAC complex, BHC80, is required for neonatal survival in mice. FEBS Lett. 2006;580:3129–3135. doi: 10.1016/j.febslet.2006.04.065. [DOI] [PubMed] [Google Scholar]
- 46.Choi J., Dong L., Ahn J., Dao D., Hammerschmidt M., Chen J.N. FoxH1 negatively modulates flk1 gene expression and vascular formation in zebrafish. Dev. Biol. 2007;304:735–744. doi: 10.1016/j.ydbio.2007.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schilling T.F., Piotrowski T., Grandel H., Brand M., Heisenberg C.P., Jiang Y.J., Beuchle D., Hammerschmidt M., Kane D.A., Mullins M.C. Jaw and branchial arch mutants in zebrafish I: Branchial arches. Development. 1996;123:329–344. doi: 10.1242/dev.123.1.329. [DOI] [PubMed] [Google Scholar]
- 48.Wilkie A.O., Morriss-Kay G.M. Genetics of craniofacial development and malformation. Nat. Rev. Genet. 2001;2:458–468. doi: 10.1038/35076601. [DOI] [PubMed] [Google Scholar]
- 49.Bartsch O., Wuyts W., Van Hul W., Hecht J.T., Meinecke P., Hogue D., Werner W., Zabel B., Hinkel G.K., Powell C.M. Delineation of a contiguous gene syndrome with multiple exostoses, enlarged parietal foramina, craniofacial dysostosis, and mental retardation, caused by deletions in the short arm of chromosome 11. Am. J. Hum. Genet. 1996;58:734–742. [PMC free article] [PubMed] [Google Scholar]
- 50.Yamamoto T., Akaboshi S., Ninomiya H., Nanba E. DEFECT 11 syndrome associated with agenesis of the corpus callosum. J. Med. Genet. 2001;38:E5. doi: 10.1136/jmg.38.2.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dodé C., Levilliers J., Dupont J.M., De Paepe A., Le Dû N., Soussi-Yanicostas N., Coimbra R.S., Delmaghani S., Compain-Nouaille S., Baverel F. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat. Genet. 2003;33:463–465. doi: 10.1038/ng1122. [DOI] [PubMed] [Google Scholar]
- 52.Vissers L.E., van Ravenswaaij C.M., Admiraal R., Hurst J.A., de Vries B.B., Janssen I.M., van der Vliet W.A., Huys E.H., de Jong P.J., Hamel B.C. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet. 2004;36:955–957. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- 53.Wuyts W., Cleiren E., Homfray T., Rasore-Quartino A., Vanhoenacker F., Van Hul W. The ALX4 homeobox gene is mutated in patients with ossification defects of the skull (foramina parietalia permagna, OMIM 168500) J. Med. Genet. 2000;37:916–920. doi: 10.1136/jmg.37.12.916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gécz J. The molecular basis of intellectual disability: Novel genes with naturally occurring mutations causing altered gene expression in the brain. Front. Biosci. 2004;9:1–7. doi: 10.2741/1199. [DOI] [PubMed] [Google Scholar]
- 55.Tassabehji M., Hammond P., Karmiloff-Smith A., Thompson P., Thorgeirsson S.S., Durkin M.E., Popescu N.C., Hutton T., Metcalfe K., Rucka A. GTF2IRD1 in craniofacial development of humans and mice. Science. 2005;310:1184–1187. doi: 10.1126/science.1116142. [DOI] [PubMed] [Google Scholar]
- 56.Ma W.J., Furneaux H. Localization of the human HuR gene to chromosome 19p13.2. Hum. Genet. 1997;99:32–33. doi: 10.1007/s004390050305. [DOI] [PubMed] [Google Scholar]
- 57.Ma W.J., Cheng S., Campbell C., Wright A., Furneaux H. Cloning and characterization of HuR, a ubiquitously expressed Elav-like protein. J. Biol. Chem. 1996;271:8144–8151. doi: 10.1074/jbc.271.14.8144. [DOI] [PubMed] [Google Scholar]
- 58.Prakash S.K., Paylor R., Jenna S., Lamarche-Vane N., Armstrong D.L., Xu B., Mancini M.A., Zoghbi H.Y. Functional analysis of ARHGAP6, a novel GTPase-activating protein for RhoA. Hum. Mol. Genet. 2000;9:477–488. doi: 10.1093/hmg/9.4.477. [DOI] [PubMed] [Google Scholar]
- 59.Pasillas M.P., Shah M., Kamps M.P. NSD1 PHD domains bind methylated H3K4 and H3K9 using interactions disrupted by point mutations in human sotos syndrome. Hum. Mutat. 2011;32:292–298. doi: 10.1002/humu.21424. [DOI] [PubMed] [Google Scholar]
- 60.Iwase S., Lan F., Bayliss P., de la Torre-Ubieta L., Huarte M., Qi H.H., Whetstine J.R., Bonni A., Roberts T.M., Shi Y. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell. 2007;128:1077–1088. doi: 10.1016/j.cell.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 61.Feng W., Yonezawa M., Ye J., Jenuwein T., Grummt I. PHF8 activates transcription of rRNA genes through H3K4me3 binding and H3K9me1/2 demethylation. Nat. Struct. Mol. Biol. 2010;17:445–450. doi: 10.1038/nsmb.1778. [DOI] [PubMed] [Google Scholar]
- 62.Qi H.H., Sarkissian M., Hu G.Q., Wang Z., Bhattacharjee A., Gordon D.B., Gonzales M., Lan F., Ongusaha P.P., Huarte M. Histone H4K20/H3K9 demethylase PHF8 regulates zebrafish brain and craniofacial development. Nature. 2010;466:503–507. doi: 10.1038/nature09261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hakimi M.A., Bochar D.A., Chenoweth J., Lane W.S., Mandel G., Shiekhattar R. A core-BRAF35 complex containing histone deacetylase mediates repression of neuronal-specific genes. Proc. Natl. Acad. Sci. USA. 2002;99:7420–7425. doi: 10.1073/pnas.112008599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hakimi M.A., Dong Y., Lane W.S., Speicher D.W., Shiekhattar R. A candidate X-linked mental retardation gene is a component of a new family of histone deacetylase-containing complexes. J. Biol. Chem. 2003;278:7234–7239. doi: 10.1074/jbc.M208992200. [DOI] [PubMed] [Google Scholar]
- 65.Scheer M.P., van der Maarel S., Kübart S., Schulz A., Wirth J., Schweiger S., Ropers H., Nothwang H.G. DXS6673E encodes a predominantly nuclear protein, and its mouse ortholog DXHXS6673E is alternatively spliced in a developmental- and tissue-specific manner. Genomics. 2000;63:123–132. doi: 10.1006/geno.1999.6027. [DOI] [PubMed] [Google Scholar]
- 66.van der Maarel S.M., Scholten I.H., Huber I., Philippe C., Suijkerbuijk R.F., Gilgenkrantz S., Kere J., Cremers F.P., Ropers H.H. Cloning and characterization of DXS6673E, a candidate gene for X-linked mental retardation in Xq13.1. Hum. Mol. Genet. 1996;5:887–897. doi: 10.1093/hmg/5.7.887. [DOI] [PubMed] [Google Scholar]
- 67.Kleefstra T., Smidt M., Banning M.J., Oudakker A.R., Van Esch H., de Brouwer A.P., Nillesen W., Sistermans E.A., Hamel B.C., de Bruijn D. Disruption of the gene Euchromatin Histone Methyl Transferase1 (Eu-HMTase1) is associated with the 9q34 subtelomeric deletion syndrome. J. Med. Genet. 2005;42:299–306. doi: 10.1136/jmg.2004.028464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ogawa H., Ishiguro K., Gaubatz S., Livingston D.M., Nakatani Y. A complex with chromatin modifiers that occupies E2F- and Myc-responsive genes in G0 cells. Science. 2002;296:1132–1136. doi: 10.1126/science.1069861. [DOI] [PubMed] [Google Scholar]
- 69.Shi Y., Sawada J., Sui G., Affar B., Whetstine J.R., Lan F., Ogawa H., Luke M.P., Nakatani Y., Shi Y. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature. 2003;422:735–738. doi: 10.1038/nature01550. [DOI] [PubMed] [Google Scholar]
- 70.Halgren C., Kjaergaard S., Bak M., Hansen C., El-Schich Z., Anderson C., Henriksen K., Hjalgrim H., Kirchhoff M., Bijlsma E. Corpus callosum abnormalities, intellectual disability, speech impairment, and autism in patients with haploinsufficiency of ARID1B. Clin. Genet. 2011 doi: 10.1111/j.1399-0004.2011.01755.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li X.S., Trojer P., Matsumura T., Treisman J.E., Tanese N. Mammalian SWI/SNF—a subunit BAF250/ARID1 is an E3 ubiquitin ligase that targets histone H2B. Mol. Cell. Biol. 2010;30:1673–1688. doi: 10.1128/MCB.00540-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hoyer J., Ekici A.B., Endele S., Popp B., Zweier C., Wiesener A., Wohlleber E., Dufke A., Rossier E., Petsch C. Haploinsufficiency of ARID1B, a member of the SWI/SNF-a chromatin-remodeling complex, is a frequent cause of intellectual disability. Am. J. Hum. Genet. 2012;90:565–572. doi: 10.1016/j.ajhg.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Santen G.W., Aten E., Sun Y., Almomani R., Gilissen C., Nielsen M., Kant S.G., Snoeck I.N., Peeters E.A., Hilhorst-Hofstee Y. Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat. Genet. 2012;44:379–380. doi: 10.1038/ng.2217. [DOI] [PubMed] [Google Scholar]
- 74.Tsurusaki Y., Okamoto N., Ohashi H., Kosho T., Imai Y., Hibi-Ko Y., Kaname T., Naritomi K., Kawame H., Wakui K. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat. Genet. 2012;44:376–378. doi: 10.1038/ng.2219. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.