

Abstract
INTRODUCTION
Gorlin syndrome (GS), also known as nevoid basal cell carcinoma syndrome (NBCCS), is a rare hereditary, autosomal dominant disease that affects various systems. Its prevalence is estimated at 1/57,000 to 1/256,000 of the population. It is characterized by basal cell carcinomas, multiple odontogenic keratocysts, skeletal abnormalities and ovarian fibroma, among other disorders.
PRESENTATION OF CASE
To report the case of a young patient with Gorlin syndrome and bilateral ovarian fibroma.
DISCUSSION
A 20-year old patient with Gorlin syndrome presented with facial asymmetry, broad nasal root, dental abnormalities, micrognathism, convergent strabismus, multiple pigmented lesions on the trunk and face, pectus excavatum, kyphoscoliosis and a palpable mass in the abdomen occupying the entire pelvic region.
CONCLUSION
Gorlin–Goltz syndrome is a hereditary pathology that includes numerous clinical manifestations. Diagnosis is clinical and genetic confirmation is unnecessary.
Keywords: Gorlin–Goltz syndrome, Nevoid basal cell carcinoma syndrome, Keratocysts, Ovarian fibroma
1. Introduction
Gorlin syndrome (GS) or nevoid basal cell carcinoma syndrome (NBCCS) was first reported in 1894 by Jarish, who applied a vast nomenclature to report the case of a patient with multiple basal cell carcinomas, scoliosis and learning difficulties.1 Binkley and Johnson in 1951 and Howell and Caro in 1959 associated the presence of basal cell nevi and other developmental abnormalities and disorders with this syndrome.1,2 In 1960, Gorlin and Goltz defined the triad composed of multiple basal cell carcinomas, odontogenic cysts and the presence of skeletal abnormalities as constituting part of this syndrome.1–6 In 1997, Kimonis proposed certain criteria on the basis of which a diagnosis of Gorlin syndrome could be made (Table 1).2,16,22
Table 1.
Diagnostic criteria for Gorlin–Goltz syndrome.
| Major criteria |
| • Multiple basal cell carcinomas (more than two) prior to 30 years of age or more than 10 basal cell nevi. |
| • Odontogenic keratocysts (confirmed histologically) or polyostotic bone cyst. |
| • Plantar or palmar pits (three or more). |
| • Ectopic, lamellar or early (prior to 20 years of age) calcification of the falx cerebri and/or cerebelli. |
| • Family history of nevoid basal cell carcinoma syndrome. |
| Minor criteria |
| • Congenital skeletal malformations: Sprengel deformity, pectus excavatum or carinatum, kyphoscoliosis, frontal and temporal protuberances. |
| • Macrocephalia. |
| • Cardio or ovarian fibroma. |
| • Medulloblastoma. |
| • Lymphomesenteric cysts. |
| • Other congenital abnormalities: cleft lip, polydactilia, microophthalmia, cataract, coloboma. |
Source: Lo Muzio L. Nevoid basal cell carcinoma syndrome (Gorlin syndrome). Orphanet Journal of Rare Diseases 2008 [16].
Gorlin syndrome is a rare hereditary, autosomal-dominant disease7,9,23 with high penetrance and varying phenotypic expression, affecting various systems.2,3,10,11 Recently, some studies have associated this syndrome with a mutation in the patched (PTCH) gene, a tumor suppressor gene situated on the long arm of chromosome 9 (9q22.3-q31).3,5,7,10,23 Prevalence is estimated at 1/57,000 to 1/256,000 of the population and there is no difference between genders. It may affect any ethnic group; however, the majority of reports refer to Caucasians.1,8,9
Diagnosis is based on clinical findings, the most striking being basal cell carcinomas (BCC), odontogenic keratocysts, palmoplantar pits,2,5,10,12 ectopic calcifications including calcification of the falx cerebri, and certain types of tumor such as ovarian fibroids, which may be unilateral or bilateral. Other neurological, ophthalmological, endocrine and genital manifestations may also be present.4,8,11,13
The objective of the present study was to report the case of a young patient with Gorlin syndrome and bilateral ovarian fibroma.
2. Case report
MFA, a 20-year old, dark-skinned female patient consulted at the general gynecology outpatient clinic of the Hospital Santa Casa de Misericórdia in Vitória, Espírito Santo, Brazil. She had undergone menarche at 14 years of age, had her last menstrual period on April 12, 2010, had never been pregnant and was complaining of a swelling in the abdomen.14
2.1. Previous medical history
She had been born by uncomplicated Cesarean section. She spoke and walked at 18 months. Her upper teeth were not substituted until 8 years of age, and at 10 years of age she was submitted to surgery for the removal of her incisors and dental cysts. Anatomopathology detected the presence of odontogenic keratocysts in the mandibular and maxillary regions. In August 2009, she complained of pain in her lower abdomen and was submitted to computed tomography on November 23, 2009, which revealed a pelvic mass measuring 18 × 20 cm in a posterior position in relation to the uterus and directed anterolaterally to the left. Two exophytic nodules were also found measuring 4 × 4 cm in the right iliac fossa with absence of a clear cleavage plane between these tumors and the small bowel loops. The nodules had a hyperdense appearance. The patient was submitted to right oophorectomy. Histology revealed ovarian tissue with stromal proliferation and no atypia, as well as areas of necrosis, corresponding to an ovarian fibroma.
2.2. Physical examination
Physical examination revealed: facial asymmetry, a broad nose root, dental abnormalities (Fig. 1A), micrognathism, convergent strabismus, multiple pigmented lesions on her trunk and face, pectus excavatum (Fig. 1B), kyphoscoliosis, a palpable, firm, mobile mass in the abdomen occupying the entire pelvis and extending as far as the umbilicus, provoking only mild pain at palpation.
Fig. 1.
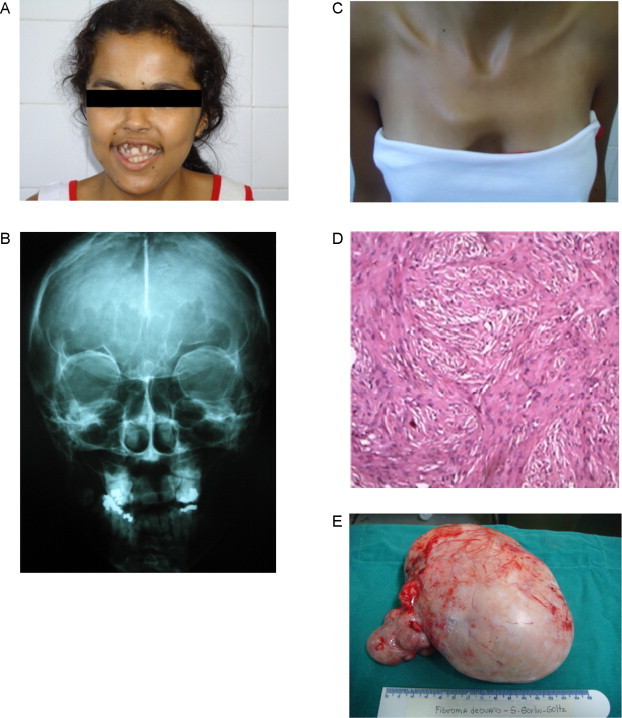
(A) Gorlin syndrome. Facial asymmetry, dental abnormalities. (B) Gorlin syndrome. Pectus excavatum. (C) Gorlin syndrome. Calcification of the falx cerebri. (D) Gorlin syndrome. Histology of the calcified ovarian fibroma. (E) Gorlin syndrome. Left ovarian fibroma.
2.3. Supplementary tests
Chest X-ray: Pectus excavatum, congenital elevation of the scapula (with the presence of a winged scapula) (Sprengel deformity), incomplete fusion of the upper vertebrae from C7 to D1, morphostructural abnormality in the posterior region of the third costal arch of the left hemithorax, with thinning of an area of bone and bilateral widening of the anterior portions of the first costal arches.
Spine X-ray: Incomplete fusion of the posterior vertebrae at C7 and T1.
Brain X-ray: Signs of bilamellar calcification of the falx cerebri (Fig. 1C).
Lumbosacral spine X-ray: Incomplete fusion of the posterior arch at S1.
Computed tomography of the abdomen: Performed on March 5, 2010. Eutopic hydronephrotic kidneys retaining normal shape, contour and dimensions. Heterogenous, voluminous mass measuring approximately 20 × 15 cm, with interspersed cystic and solid areas, uneven borders, occupying a large portion of the pelvic cavity, with its epicenter in the uteroovarian region, creating a mass effect characterized by compression of the left posterolateral wall of the bladder. The mass compressed and encompassed the ureters bilaterally, with consequent hydronephrosis, a distended bladder with non-opacification and compression of the left posterolateral wall.
Normal tumor markers: Alpha-fetoprotein 1.3 mg/ml; beta-hCG negative and CA 125 12 U/ml.14
On April 12, 2010, a double J catheter was inserted due to the hydronephrosis and on April 13, 2010 the patient was submitted to exploratory laparotomy, which revealed a voluminous tumor of approximately 20 cm in diameter on her left ovary, with a solid cystic consistency and a smooth, whitish surface. The tumor was adhered to a small bowel loop and occupied the entire pelvic cavity as far as the umbilicus. The left ovary was removed and the right ovary was not visualized. Histopathology revealed a stromal proliferation without atypia, with areas of calcification corresponding to a calcified ovarian fibroma (Fig. 1D and E).
3. Discussion
Gorlin syndrome is a rare autosomal-dominant disease,13,15,23 with high penetrance and varying phenotypic expression.2,10 Its prevalence is estimated at 1/57,000 to 1/256,000 of the population. It generally appears in adolescence,3,16 and there is no difference in prevalence between males and females; however, those affected are predominantly Caucasians.2,11,16,18
The etiopathogenesis of this disease remains unclear; however, mutation of the PTCH tumor-suppressing gene located on the long arm of chromosome 9 is believed to be responsible for the development of multiple malformations and for the variety of manifestations.1,5,6,10,11,15,18,23
The major issue in Gorlin syndrome is the difficulty in recognizing the condition at an early stage, since many of the clinical signs are absent in childhood and due to the variability in its phenotypic expression, there is no single component that is present in all patients.1 The principal clinical characteristics consist of basal cell carcinomas (BCC), odontogenic keratocysts and palmoplantar pits.1,2,16 According to Hilgert et al., the basal cell carcinomas appear between puberty and 35 years of age, ranging in number from one to thousands and principally affecting the epithelium of the chest and cervicofacial regions.1 According to Medeiros and Ferreira, the carcinomas in this syndrome may occur at any moment between childhood and old age, unlike the usual development of these tumors, which do not generally occur in children.11
In 1997, Kimonis et al. documented 105 individuals with nevoid basal cell carcinoma syndrome: 48 males and 57 females of 4 months to 87 years of age. Eighty percent of the Caucasians and 38% of the African-Americans had at least one basal cell carcinoma (BCC), with the first tumor occurring at a mean age of 23 years and 21 years, respectively.22
Since this syndrome involves various organs and systems and since these patients are at a greater risk of developing malignancies, the presence of basal cell carcinomas was investigated in the patient in the present report. None were found up to the present moment; however, the patient will be followed up by a multidisciplinary team and will be submitted to periodic tests.
Odontogenic keratocysts are present in up to 75% of patients and relapses may occur in around 60%.4,9,16 The keratocysts are generally asymptomatic and are usually detected at radiology.3 Most are situated in the canine and premolar regions. They may result in abnormalities in the dental arch and may replace the teeth permanently. Kimonis et al. found cysts of the jaw in 74% of 105 individuals in their sample, with the first tumor appearing prior to 20 years of age in 80% of cases.22 In the patient in question, Gorlin syndrome was diagnosed at 10 years of age when she underwent surgery to remove multiple odontogenic keratocysts, confirmed by histopathology.
Palmoplantar pits appear in 80% of patients. Kimonis et al. reported a finding of palmoplantar pits in 87% of patients with NBCCS.22
These three principal characteristics are associated with skeletal abnormalities such as pectus excavatum, kyphoscoliosis and a variety of neurological, ophthalmological, endocrine and genital manifestations.1,11,13,16
The principal clinical characteristics and diagnostic criteria are described in Table 1, with two major criteria or one major criterion and two minor criteria being required for confirmation of diagnosis.11,12,16,18
In the present case, diagnosis presented an abundance of clinical characteristics: major criteria such as odontogenic keratocysts and bilamellar calcification of the falx cerebri, in addition to various other minor criteria such as skeletal abnormalities (Sprengel deformity, pectus excavatum and kyphoscoliosis), radiological abnormalities (fused and elongated vertebral bodies) and bilateral ovarian fibroma, which was the factor that motivated the patient to visit the clinic.
Ovarian fibroma constitutes 4% of all ovarian tumors.8,19,20 Its symptomatology varies, with the majority of patients being asymptomatic20; however, it may lead to adnexal torsion, causing intense pain.21 This ovarian tumor may be very large and may become calcified; however, it rarely becomes malignant. According to Seracchioli et al., around 90% of ovarian fibromas are unilateral; however, when they present bilaterally, when they are calcified and have a nodular shape, Gorlin syndrome should be investigated.8
In their review, Kimonis et al. identified the presence of 9 ovarian fibromas using ultrasonography, i.e. in 17% of patients in that sample, with a mean age of 30 years.22 What is noteworthy in the present case is the early and bilateral presentation of the ovarian fibroma, which was calcified, a condition rarely reported according to Seracchioli et al.8
Treatment of this syndrome must be multidisciplinary and will depend on the systems affected.10,11,16,18 Prognosis depends on the malignant progression of the lesions; however, life expectancy is no different from that of the general population.6,16,17
4. Conclusion
The clinical case described above represents an example of Gorlin–Goltz syndrome in which many of the manifestations of this disease were present: odontogenic keratocysts, kyphoscoliosis, fused vertebral bodies, pectus excavatum and bilateral ovarian fibroma. Diagnosis is clinical and there is no need for genetic confirmation.
Conflict of interest statement
All the authors do not have any conflicts of interest.
Funding
This study was sponsored by the authors.
Ethical approval
Written informed consent was obtained from the patient for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.
Author contributions
Fernanda Pirschner – data analysis, study design; Pollyana Marçal Bastos – data collections; George Luiz Contarato – data analysis, writing; Anna Carolina Bon Lima Bimbato – writing, study design; Antônio Chambô Filho – data analysis.
References
- 1.Hilgert R., Fonseca L.A., Felipe F.F. Gorlin's Syndrome: report of family involvement. Revista de Cirurgia e Traumatologia Buco-maxilo-facial Camaragibe. 2010;10:39–44. [Google Scholar]
- 2.Dávalos C.P., Vélez J.U., Mori Estévez A.D. Gorlin's Syndrome. A case. Revista Habanera de Ciencias Médicas de la Habana. 2008;7(1) [Google Scholar]
- 3.Carbia S.G., Marrero M., Glorio R., Etchart C., La Forgia M. Nevoid basal-cell carcinoma syndrome (Gorlin syndrome) Archivos Argentinos de Dermatología. 2009;15:209–212. [Google Scholar]
- 4.Dowling P.A., Fleming P., Saunders I.D., Gorlin R.J., Napier S.S. Odontogenic keratocysts in a 5-year-old: initial manifestations of nevoid basal cell carcinoma syndrome. Pediatric Dentistry. 2000;22:53–55. [PubMed] [Google Scholar]
- 5.Leonardi R., Caltabiano M., Lo Muzio L., Gorlin R.J., Bucci P., Pannone G. Bilateral hyperplasia of the mandibular coronoid processes in patients with nevoid basal cell carcinoma syndrome: an undescribed sign. American Journal of Medical Genetics. 2002;110:400–403. doi: 10.1002/ajmg.10432. [DOI] [PubMed] [Google Scholar]
- 6.Moret Y., González J.M. Basal cell nevus syndrome. Acta Odontologica Venezolana. 2004;42:53–56. [Google Scholar]
- 7.Ragge N.K., Salt A., Collin J.R., Michalski A., Farndon P.A. Gorlin syndrome: the PTCH gene links ocular developmental defects and tumour formation. British Journal of Ophthalmology. 2005;89:988–991. doi: 10.1136/bjo.2004.061390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seracchioli R., Bagnoli A., Colombo F.M., Missiroli S., Venturoli S. Conservative treatment of recurrent ovarian fibromas in a young patient affected by Gorlin syndrome. Human Reproduction. 2001;16:1261–1263. doi: 10.1093/humrep/16.6.1261. [DOI] [PubMed] [Google Scholar]
- 9.Stolz A.S., Oliveira M.O., Ferreira F.V., Uggeri C.M. Preservação pós-operatória de paciente com Síndrome de Gorlin–Goltz (Síndrome do carcinoma basocelular nevoide) Revista Sul-Brasileira de Odontologia. 2010;7:226–230. [Google Scholar]
- 10.De-Domingo B., González F., Lorenzo P. Gorlin syndrome (nevoid basal cell carcinoma syndrome) Archivos de la Sociedad Espanola de Oftalmologia. 2008;83:321–324. doi: 10.4321/s0365-66912008000500008. [DOI] [PubMed] [Google Scholar]
- 11.Medeiros L., Ferreira J.C. Síndrome de Gorlin–Goltz: revisão bibliográfica a propósito de um caso clínico. Revista Portuguesa de Estomatologia e Cirurgia Maxilofacial. 2006;47:25–32. [Google Scholar]
- 12.Hokama M., Guimarães E.L. The Goltz–Gorlin syndrome: a case report. Pediatria (São Paulo) 2005;27:61–64. [Google Scholar]
- 13.Leiva A.L., Terry O.S., Manso B.L. Maxillar neurofibroma. A case presentation. Revista Electrónica de las Ciencias Médicas en Cienfuegos. 2007;5(3) [Google Scholar]
- 14.Almeida J.R., Pedrosa N.L., Leite J.B., Fleming T.R., Carvalho V.H., Cardoso A.A. Tumor markers: a literature review. Revista Brasileira de Cancerologia. 2007;53:305–316. [Google Scholar]
- 15.Amlashi S.F., Riffaud L., Brassier G., Morandi X. Nevoid basal cell carcinoma syndrome: relation with desmoplastic medulloblastoma in infancy. A population-based study and review of the literature. Cancer. 2003;98:618–624. doi: 10.1002/cncr.11537. [DOI] [PubMed] [Google Scholar]
- 16.Lo Muzio L. Nevoid basal cell carcinoma syndrome (Gorlin syndrome) Orphanet Journal of Rare Diseases. 2008;3:32. doi: 10.1186/1750-1172-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Torriani M.A., Moojen Z.E., Zambrano C.B., Hosni E.S., Pires M.S. Síndrome de Gorlin – relato de caso. Revista Internacional de Estomatologia. 2004;1:51–56. [Google Scholar]
- 18.Valiati A.A., Pavelecini M., Netto R., Pereira Filho G., Fauri M.A., Lima L.P. Basal cell nevus syndrome: case report. Arquivos Catarinenses de Medicina. 2009;38(Suppl 1) [Google Scholar]
- 19.Monge A.H., Sánchez L.R., Hernández M.R., Pineda R.P., Muñoz L.A. Fibroma edematoso de ovario con extensa degeneración quística. Reporte de un caso y revisión de la bibliografía. Ginecologia y Obstetricia de Mexico. 2009;77:244–249. [PubMed] [Google Scholar]
- 20.De Gracia R., González E. Fibroma ovárico en mujer joven: reporte de un caso. Revista Centroamericana de Obstetricia y Ginecología. 2009;14(2) [Google Scholar]
- 21.Mateo Corbalán C., González Sanz N., Cusiné López L., Pons Ferré L.E., Rubio del Caño M., Pla Hierro J. Fibroma ovárico torsionado. Progresos de Obstetricia y Ginecología. 2006;49:401–405. [Google Scholar]
- 22.Kimonis V., Goldstein A., Pastakia B. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. American Journal of Medical Genetics. 1997;69:299. [PubMed] [Google Scholar]
- 23.Boutet N., Bignon Y.J., Drouin-Garraud V., Sarda P., Longy M., Lacombe D. Spectrum of PTCH1 mutations in French patients with Gorlin syndrome. Journal of Investigative Dermatology. 2003;121:478–481. doi: 10.1046/j.1523-1747.2003.12423.x. [DOI] [PubMed] [Google Scholar]