

Abstract
Global activation of MAP kinases has been reported in both human and experimental heart failure. Chronic remodeling of the surviving ventricular wall after myocardial infarction (MI) involves both myocyte loss and fibrosis; we hypothesized that this cardiomyopathy involves differential shifts in pro- and anti-apoptotic MAP kinase signaling in cardiac myocyte (CM) and non-myocyte. Cardiomyopathy after coronary artery ligation in mice was characterized by echocardiography, ex vivo Langendorff preparation, histologic analysis and measurements of apoptosis. Phosphorylation (activation) of signaling molecules was analyzed by Western blot, ELISA and immunohistochemistry. Post-MI remodeling involved dramatic changes in the phosphorylation of both stress-activated MAP (SAP) kinase p38 as well as ERK, a known mediator of cell survival, but not of SAP kinase JNK or the anti-apoptotic mediator of PI3K, Akt. Phosphorylation of p38 rose early after MI in the infarct, whereas a more gradual rise in the remote myocardium accompanied a rise in apoptosis in that region. In both areas, ERK phosphorylation was lowest early after MI and rose steadily thereafter, though infarct phosphorylation was consistently higher. Immunostaining of p-ERK localized to fibrotic areas populated primarily by non-myocytes, whereas staining of p38 phosphorylation was stronger in areas of progressive CM apoptosis. Relative segregation of CMs and non-myocytes in different regions of the post-MI myocardium revealed signaling patterns that imply cell type-specific changes in pro- and anti-apoptotic MAP kinase signaling. Prevention of myocyte loss and of LV remodeling after MI may therefore require cell type-specific manipulation of p38 and ERK activation.
Keywords: MYOCARDIAL INFARCTION, MAP KINASE, REMODELING, APOPTOSIS
The molecular regulation of cardiac myocyte (CM) fate is increasingly recognized as an important target for novel therapeutic interventions in heart failure. Phenomena such as ischemic preconditioning underscore the impact that alterations of intra- and extra-cellular biochemistry can have on the ability of CMs to withstand otherwise lethal insults. In recent years, genetic models involving changes in myocardial expression and/or activation of just a single enzyme–or a single class of enzyme isoforms–have also demonstrated that the activity of these molecules alone can alter both CM fate as well as the course of left ventricular (LV) remodeling [Mehrhof et al., 2001; Braz et al., 2002; Bueno et al., 2007].
Two signaling cascades have been particularly well-studied both in cultured myocardial cells and in experimental models of LV remodeling: the phosphatidylinositol-3 kinase (PI3K) and mitogen activated protein (MAP) kinase pathways. These pathways, in fact, share an interesting quality: different elements of each pathway have been associated either with (1) promotion of CM apoptosis and progression of pathologic hypertrophy to dilated cardiomyopathy or (2) protection of CMs from apoptosis and establishment of non-pathologic, or “physiologic,” hypertrophy. In the case of the PI3K cascade, this dichotomy has been at times observed with regard to the gamma and alpha isoforms of PI3K itself [Mehrhof et al., 2001; Doukas et al., 2006; Bueno et al., 2007]; in MAP kinase signaling, the so-called stress activated kinases p38 and JNK have been more commonly associated with pathologic dilatation and CM apoptosis whereas ERK has been found to play an anti-apoptotic role in promoting physiologic hypertrophy [Jones et al., 2000; Braz et al., 2002; Ferrandi et al., 2004; Ren et al., 2005].
Most of these myocardial signaling data, however, have been collected in the context of global cardiomyopathy, often induced either by genetic models or by experimental pressure overload. In order to extend these data further toward the design of translational strategies for human intervention, we studied signaling patterns associated with chronic ventricular remodeling after myocardial infarction (MI), in which cardiac stresses and patterns of remodeling are quite distinct from these other experimental settings. Our model of post-MI remodeling not only mimics changes that occur in the most common form of human heart failure, but also allows a unique opportunity to study relatively segregated populations of CMs and cardiac non-myocytes in the in vivo contexts of the remote myocardium and the infarct, respectively. Based on our preliminary results, we hypothesized that different patterns of MAP kinase signaling are induced in CMs and non-myocytes during the evolution of post-MI cardiomyopathy. Our findings suggest that heterogeneity of cellular responses should be considered both in the analysis of data from human and animal tissues and in the design of future molecular interventions.
METHODS
MOUSE CORONARY LIGATION
Male C57Bl/6 mice (25 g) were anesthetized with isoflurane prior to intubation with a 24-gauge catheter. Inhalation anesthesia was then instituted with 1.5% isoflurane using a rodent ventilator (Harvard) at 115 breaths/min. A left lateral thoracotomy incision was placed at the level of the fourth interspace and a 7.0 polypropylene suture was used to ligate the left anterior descending artery (LAD) at approximately 1/3 the distance from the base to the apex of the heart. The left chest was then closed and the animal was recovered after extubation in a light-warmed incubator. Animals were subsequently sacrificed at time points ranging from 1 to 12 weeks after infarction as indicated below; at sacrifice, some hearts were fixed and sectioned for histologic analysis, while others were snap frozen for protein extraction. All procedures conformed with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996), and were approved by the Institutional Animal Care and Use Committee of the San Francisco Veterans Affairs Medical Center.
WESTERN BLOTTING AND ELISA
At the time of harvest of specimens for protein extraction, the left ventricle was dissected from the remainder of the heart. The infarct area was identified visually and dissected from the remainder of the myocardium. In addition, myocardial tissue from the free wall and septum clearly distant from the area of infarction was collected as remote myocardium. Any attempt to accurately isolate the irregular borderzone from these small mouse hearts was felt to introduce a prohibitive sampling error. Specimens were snap frozen in liquid nitrogen. Myocardial tissues were homogenized in a lysis buffer containing 0.13 M KCL, 1 mM EDTA, 1 mM EGTA, 1 mM Na3(VO4), 5 mM NaF, 20 mM HEPES, and Protease inhibitor cocktail tablet (Roche Diagnostics, Indianapolis, IN) and centrifuged at 35,000 rpm for 30 min. Supernatants were separated by gel electrophoresis, blotted onto a PVDF membrane (Invitrogen), and detected by phospho-ERK1/2 and phospho-p38 Abs (Cell Signaling, Danvers, MA) and chemiluminescence (Pierce) after incubation with horse-radish peroxidase-labeled secondary antibody according to the manufacturer’s instructions. For ELISA, activities of phospho-ERK1/2, phospho-Akt, phospho-JNK, phospho-p38α and phospho-p38γ were assayed using commercially available ELISA kits (R&D Systems) according to the manufacture’s instructions.
ECHOCARDIOGRAPHY
Transthoracic echocardiography was performed in conscious mice using an Acuson Sequoia 512 machine and a 13-MHz probe. A two-dimensional short-axis view of the left ventricle was obtained at the level of the papillary muscles, that is, in myocardium remote to the infarction, and two-dimensional M-mode tracings were also recorded. LV fractional shortening was calculated as (LVDd –LVDs)/LVDdX100, where LVDd = LV diastolic dimension and LVDs = LV systolic dimension [Sahn et al., 1978; Kanno et al., 2002].
LANGENDORFF PREPARATION
Mice were heparinized (500 U/kg i.p.) and anesthetized with pentobarbital sodium (60 mg/kg i.p.). Hearts were rapidly excised, washed in ice-cold arresting solution (120 mmol/L NaCl, 30 mmol/L KCl), and cannulated via the aorta on a 20-gauge stainless steel blunt needle. Hearts were perfused at 70 mmHg on a modified Langendorff apparatus using Krebs–Henseleit solution containing 1 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 1.2 mM KH2PO4, 24 mM NaHCO3, 5.5 mM glucose, 5.0 mM Na pyruvate, 0.5 mM EDTA, and bubbled with 95% O2–5% CO2 at 37°C. Platinum electrodes were connected to a Grass Instrument stimulus generator and were used to pace hearts at 360 beats/min. LV pressure was measured by placing into the ventricle a fluid-filled balloon that was coupled to a pressure transducer. The balloon volume was gradually increased at 5 ml intervals and pressure curves were recorded until a plateau of developed pressure (systolic–diastolic) was achieved. Pressure measurements were then repeated in the presence of norepinephrine (10 μM). Developed pressures and dP/dt were extrapolated from the pressure tracings [Turnbull et al., 2003].
MEASUREMENT OF APOPTOSIS
Terminal deoxynucleotidyl Transferase Biotin-dUTP Nick End Labeling (TUNEL) for detection of apoptotic nuclei was performed according to the manufacturer’s protocol (Roche). Apoptotic indices were calculated by dividing the number of apoptotic nuclei in three thin sections by the total number of nuclei as counterstained with hematoxylin. In situ ligase assay (ApopTag® Peroxidase In Situ Oligo Ligation (ISOL) Kit, Chemicon) was used to verify the apoptotic index measured by TUNEL assay. Negative controls were performed by omitting the enzyme TdT (for TUNEL) or the T4 DNA ligase (for the ligase assay); positive controls were performed by pretreating the sections with 10 U/ml DNase I for 20 min at 37°C.
HISTOLOGY AND HISTOCHEMISTRY
Mouse hearts were arrested in diastole, and embedded in paraffin or frozen medium. For Sirius red staining, paraffin sections were stained for 1 h in saturated picric acid with 0.1% Sirius Red F3BA (Sigma). After staining, slides were washed twice in acidified water (0.01 N HCl), and dehydrated. Photomicrographic images were then acquired and collagen content (fibrosis) was analyzed using Image J software (NIH); fibrosis is expressed as the percent of total tissue surface area per field stained with Sirius red. To measure cell proliferation, paraffin sections were stained with primary antibody against PCNA using the ABC kit (Santa Cruz Biotechnology), and counterstained with hematoxylin; proliferative index is reported as PCNA-positive nuclei as a percent of total nuclei. Omission of primary antibodies and staining with non-immune IgG served as negative controls. For fluorescence staining, cryo-sections was pre-incubated in phosphate-buffered saline containing 0.3% Triton X-100 for 30 min and then in phosphate-buffered saline containing 2% bovine serum albumin, 0.1% Triton X-100, and 2% serum were treated with primary antibodies at 4°C overnight. Primary antibodies were against phospho-ERK1/2, phospho-p38, and myosin heavy chain (MF20). After washing, secondary antibodies were incubated at room temperature for 1 h. Fluorescent-tagged secondary antibodies were applied at 1:200 dilutions (Molecular Probes). Nuclei were counterstained with diamidino-2-phenylindole (DAPI). After additional washes, the sections were mounted in Vectashield (Vector Laboratory). Images were captured using the SPOT imaging system (Diagnostic Instruments, Inc.) and Nikon E400 microscopy (Nikon, Inc.).
CARDIAC MYOCYTE AND NON-MYOCYTE ISOLATION AND CULTURE
Adult myocytes and non-myocytes were isolated from C57Bl/6 male mice as described [Zhou et al., 2000]. Hearts were digested with 4 ml/min Langendorff perfusion of collagenase. Myocytes and non-myocytes were separated from the digested pellet by differential centrifugation, and were plated in 35 mm dishes in MEM plus 10% FCS with 2% CO2 at 37°C. Myocytes were cultured in MEM containing 1 mM BDM overnight, and were then stimulated for 5 min with norepinephrine (2 μM) prior to harvest and preparation of cell lysate. In a separate experiment, myocytes were stimulated for 10 min with 1 μM epinephrine (EPI) with or without the pretreatment of beta-blocker (1 μM propranolol (PROP)) for 10 min. Non-myocytes were serum starved overnight prior to norepinephrine stimulation.
STATISTICS
Values are reported as mean ± SEM. Comparisons among groups were made using ANOVA, followed by Neuman–Keuls post-hoc testing. P-values < 0.05 were considered statistically significant, with Bonferroni correction where appropriate.
RESULTS
CHRONIC POST-MI CARDIOMYOPATHY
Mid-LAD ligation in C57Bl/6 mice resulted in infarction of approximately one-third of the left ventricle. Ex vivo Langendorff preparations at the time of harvest documented a persistent decline in LV function over 8 weeks post-MI, as manifest both in a decline in maximum developed pressure and a large increase in the volume required to achieve half-maximum pressure (Table I). Post-MI LV remodeling was also characterized by an increase in LVEDV and LVESV, and by a persistent decline in fractional shortening in the remote myocardium as assessed by serial echocardiography in conscious mice (Table I). Histologic analysis revealed increased fibrosis in the remote myocardium late after MI, and PCNA staining, observed among non-myocytes, was more prominent in areas of fibrosis at weeks 8–12 (Table I).
TABLE I.
Functional and Histologic Parameters of Post-MI Remodeling in the Remote (Uninfarcted) Myocardium After LAD Ligation in Mice
| Sham | Earlya | Latea | |
|---|---|---|---|
| LVMDP (mmHg) | 111.7±1.7 | 94.3±3.4§ | 85.2±22.1 |
| LV volume at 1/2 LVMDP (μl) | 9.2±0.8 | 13.0±1.7 | 29.6±6.9† |
| LVESV (μl) | 5.9±0.6 | 14.1±1.6 | 18.2±3.5† |
| LVEDV (μl) | 35.2±2.2 | 45.1±1.7 | 53.3±2.1† |
| Fractional shortening (%) | 55±2.0 | 50±1§ | 44±1§ |
| Fibrosis (% Sirius red stain) | 4.3±0.5 | 6.1±0.8 | 7.3±0.7† |
| Proliferation index (% total cells) | 1.5±0.1 | 1.9±0.1† | 2.9±0.2§ |
| Apoptosis rate (% total cells) | 0.08±0.05 | 1.02±0.39§ | 2.27±0.40§ |
LVMDP, maximum developed pressure in the left ventricle; LVESV, left ventricular end systolic volume; LVEDV, left ventricular end diastolic volume.
P<0.05,
P<0.01.
Early, 2–4 weeks post-MI; Late≥8 weeks post-MI.
A persistent increase in myocardial apoptotic index was also observed during the progression of late post-MI cardiomyopathic remodeling of the remote myocardium. TUNEL staining detected rates of myocardial apoptosis that continued to increase throughout the duration of follow up in post-MI hearts (Table I). In situ ligase assay was utilized in select specimens to confirm the TUNEL data, since the latter can detect label nicks, gaps, single-stranded DNA, 3′-recessed ends or 3′-overhanging ends longer than one base from necrotic as opposed to apoptotic nuclei. In all instances ligase assay data were consistent with TUNEL results.
In summary, this mouse model of chronic infarction recapitulated progressive, late-stage LV remodeling marked by a steady increase in myocardial apoptosis and a decline in global LV function.
SIGNALING PATTERNS DURING POST-MI REMODELING
Substantial changes were observed in MAP kinase phosphorylation (i.e., activation) during chronic post-MI remodeling, particularly with regard to p38 and ERK. Progressive increase in p38 phosphorylation was observed via Western blot of lysates from the remote myocardium of chronic MI hearts (Fig. 1A). ELISA for both alpha and gamma isoforms, two major isoforms whose expression has been well documented in the myocardium, suggested a steady increase in phosphorylation in the remote myocardium over time. In contrast, p38 phosphorylation rose quickly in specimens from the infarcted regions, and either remained constant or declined slightly at later time points (Fig. 1B).
Fig. 1.

A: Representative Western blots indicating phosphorylation (i.e., activation) of p38 MAPK in the remote myocardium during post-MI remodeling. Changes in p38α (white bars) and p38γ (gray bars) were measured via ELISA of tissue lysates from remote (B) or infarct/borderzone myocardium (C). n = 4–9, *P < 0.05, week 8 and 12 versus week 1.
Elevations in ERK phosphorylation were also detected both by Western blot and by ELISA in post-MI myocardium (Fig. 2). Although this increase was again observed in the remote myocardium as well as in the infarct, by week 2 ERK phosphorylation remained consistently higher in the infarcted region that wasdominated by cardiac non-myocytes. The rise of ERK phosphorylation in the remote myocardium occurred at later time points, when more extensive fibrosis and non-myocyte proliferation was seen in this region.
Fig. 2.

Representative Western blots (A) and ELISA (B,C) indicated increases in ERK phosphorylation (i.e., activation) during chronic post-MI remodeling that were consistently greater in the infarcted/borderzone region than in the remote myocardium. n = 4–9, *P < 0.05 versus normal control.
In contrast to progressive, substantial increase in p38 phosphorylation during chronic remodeling in the remote myocardium, a much smaller percent drop was observed in JNK phosphorylation (i.e., activation) in the remote myocardium at week 1 after MI (Fig. 3A). JNK phosphorylation then remained relatively stable during the progression of post-MI remodeling in the remote myocardium. JNK phosphorylation increased in the infarct region during the first 2 weeks after MI (Fig. 3B), then remained fairly constant during the period of observation.
Fig. 3.
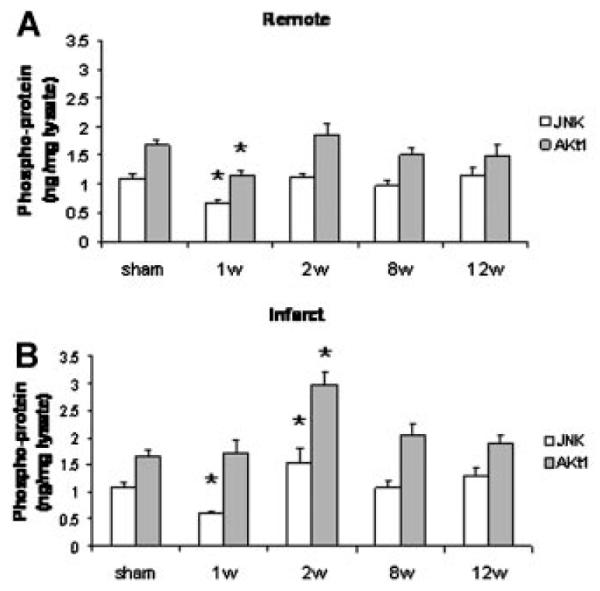
Myocardial JNK (white bars) and Akt (gray bars) activation in the remote (A) and in the infarct/borderzone (B) myocardium after myocardial infarction. n = 4–9, *P < 0.05 vs. normal control.
Phosphorylation (i.e., activation) of key PI3K intermediate Akt, has been identified as an important event in CM survival signaling [Palojoki et al., 2001; McMullen et al., 2003, 2007]. A 30% drop in Akt activation was observed only early after MI in the remote myocardium, and did not change during the later stages of post-MI remodeling, despite an increase in apoptosis and progressive fibrosis in the remote myocardium (Fig. 3). Like JNK, a bump in Akt phosphorylation was detected in the infarct region at 2 weeks, but returned to baseline levels during the later stages of post-MI remodeling.
We hypothesized that the distinct patterns of MAP kinase activation observed in different regions of the post-MI heart reflected different levels of phosphorylation in the different predominant cell types in each region. Immunofluorescent co-staining of sections for either phospho-ERK or phospho-p38 plus myosin heavy chain (a CM marker) revealed an increase in p38 phosphorylation in both myocytes and non-myocytes of the infarct/borderzone (Fig. 4A,D). Interestingly, p-38 phosphorylation was also detected in CMs in the non-ischemic remote myocardium, presumably related to the stress associated with post-MI remodeling in this region (Fig. 4B,E). In contrast, phospho-ERK immunostaining tended to localize in cardiac non-myocytes in areas of fibrosis observed in both the infracted and remote regions of the myocardium (Fig. 4C,F).
Fig. 4.

Immunofluorescent co-staining of phospho-p38 (red, A,B,D,E) and phospho-ERK (red, C,F) together with myosin heavy chain staining using MF-20 (green). Nuclear staining for phospho-p38 was observed in both myocytes (arrows) and non-myocytes (arrowheads), whereas phospho-ERK staining, even in fibrotic areas of the remote myocardium, predominated in non-myocytes (arrowheads). Nuclei are counterstained with DAPI; cardiac non-myocytes appear black. A–C: 400 ×; D–F: 1,600 ×.
To further confirm this distinction in cell signaling patterns and to better understand its pathophysiologic significance, CMs and non-myocytes were isolated from the same mouse hearts and grown in cell culture. We then examined both the patterns of baseline phosphorylation of ERK and p38 in these different cells as well as the response of these different patterns to adrenergic stimulation, known to play a role in the progression of dilated cardiomyopathy [Ungerer et al., 1993; Kaumann et al., 1999; Freeman et al., 2001]. Phospho-ERK was not detectable in lysates of adult mouse myocytes, even after the stress of cell isolation and even after in vitro stimulation with norepinephrine (Fig. 5A). In a set of preliminary experiments, ERK responsiveness in CMs isolated from sham-operated hearts was demonstrated via the addition of the beta-blocker PROP to EPI stimulation (Fig. 5B), suggesting beta adrenergic inhibition of alpha-induced ERK phopsphorylation. Interestingly, p38 activation was detected in these stressed cells, with or without norepinephrine treatment. In contrast, low levels of both ERK and p38 phosphorylation were detected in cultured non-myocytes, and this activation did increase with adrenergic stimulation with norepinephrine.
Fig. 5.
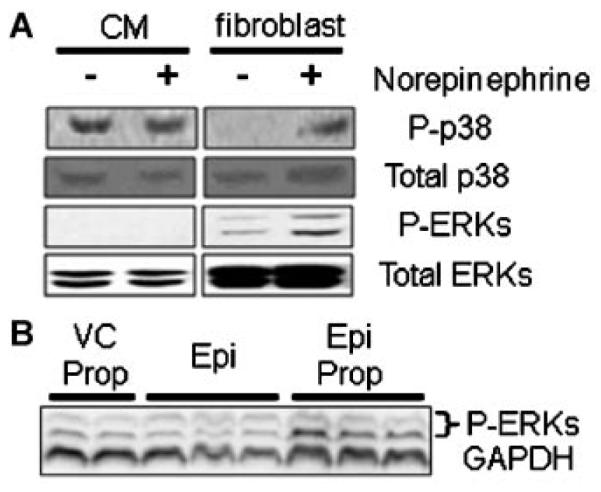
A: Representative Western blot of lystaes from cardiac myocytes and fibroblasts isolated from the same mouse hearts and cultured for 48 h in vitro. Note the identification of p38 activation but not ERK activation in isolated myocytes, both with and without adrenergic stimulation with norepinepherine. In contrast, both ERK and p38 MAPK were activated in cardiac fibroblasts, particularly with adrenergic stimulation. B: Simultaneous adrenergic stimulation with epinephrine (EPI) and blockade of beta receptors with propranolol (PROP) unmasked an apparent alpha adrenergic-induced phosphorylation of ERK in isolated myocytes compared to myocytes treated with vehicle and propranolol alone (VC PROP).
DISCUSSION
In this study, we observed substantial changes in patterns of myocardial MAP kinase activation during the increase in myocardial apoptosis that has been associated in human and animal studies with the progression of post-MI LV remodeling [Palojoki et al., 2001; Abbate et al., 2002; Baldi et al., 2002]. Changes in LV function at the earliest time points after MI may primarily reflect the loss of function in the infarcted region, since fibrosis and other remodeling of the remote myocardium has not yet progressed at those times. It is interesting that changes in p38 and ERK signaling in the remote myocardium were not very pronounced at these early time points despite an early decline in global function, but were more prominent in this region only when more substantial remodeling and a more significant drop in local fractional shortening could be observed.
Different patterns of p38 phosphorylation were observed in the histologically distinct regions of the remote myocardium and the infarct. Phosphorylation of p38 increased steadily in the remote myocardium during the progression of post-MI remodeling, while remaining constant or declining in the infarct/borderzone. Phospho-ERK levels, in contrast, remained higher in the infarct than in the remote myocardium during this process. The relative segregation of CMs and non-myocytes in the remote myocardium and infarcted regions, respectively, supports our hypothesis that progressive remodeling and apoptotic myocyte loss in the remote myocardium may be associated with a cell-type specific increase in the ratio between p38 and ERK signaling in CMs.
In vitro experiments confirmed a pattern of p38 phosphorylation in CMs subjected to the stress of cell isolation, while pro-survival ERK activation in these stressed cells was low or undetectable. Interestingly, whereas p38 phosphorylation in CMs did not increase upon further stimulation with norepinephrine, adrenergic stimulation of cardiac non-myocytes did increase both p38 and ERK activation, suggesting that similar stimulation in vivo might contribute to progressive cardiac fibrosis. Preliminary experiments in our group have suggested that addition of beta blockade to adrenergic stimulation of CMs does result in increased ERK phosphorylation in CMs, but not non-myocytes, suggesting a cell-type specific protective role of alpha adrenergic stimulation that may be masked by more pathologic beta stimulation [Karoor et al., 2004; Tutor et al., 2007].
A report by Qin et al. [2005] also described a steady though much smaller increase in p38 phosphorylation in the remote myocardium after MI in rabbits. In contrast to our findings, however, a decrease in post-MI myocardial ERK phosphorylation was observed in that study. Qin et al., however, did not study MAP kinase phosphorylation in the infarct; our observation that increasing ERK activation was consistently greatest in that region of the myocardium suggested that the increased MEK-1/ERK signaling may be localized to non-myocytes that predominate in areas of scarring and fibrosis. Immuno co-staining of thin sections supported this hypothesized dichotomy in MAP kinase activation between CMs and non-myocytes.
Previously, elegant transgenic models developed by McMullen et al. [2003, 2007] and Nicol et al. [2001] have demonstrated that changes in expression and/or activation of only a single kinase in CMs can either induce stable physiologic ventricular hypertrophy, or lead to pathologic LV remodeling in otherwise healthy mice. The results of this study suggest that strategies for therapeutic manipulation of signaling patterns, particularly in the complex setting of post-MI remodeling and cardiomyopathy, may require cell type-specific intervention. For example, ERK activation in CMs might inhibit apoptotic loss of functional myocardium, but unregulated activation of ERK in cardiac fibroblasts might result in increased fibrosis. Although still controversial, p38 activation has been implicated in the progression of dilated cardiomyopathy, and in the apoptotic loss and dysfunction of CMs [Wang et al., 1998; Liao et al., 2002; Zhang et al., 2003; Nishida et al., 2004; Liu et al., 2006]; recent data have suggested a benefit from non-specific, pharmacologic inhibition of p38 after MI [Engel et al., 2006; Li et al., 2006]. Ongoing studies in our laboratory are exploring CM-specific upregulation of ERK signaling both in hearts at risk for post-MI cardiomyopathy and in already-remodeled hearts that undergo therapeutic surgical reconstruction.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health, Grant numbers:1R01 HL083118; 1K08HL079239 to M.J.M. and HL31113 to P.C.S., and by American Heart Associstion, Grant number: 0465090Y to M.J.M.
Grant sponsor: National Institutes of Health; Grant numbers: 1R01 HL083118, 1K08HL079239; Grant sponsor: American Heart Associstion; Grant number: 0465090Y.
REFERENCES
- Abbate A, Bussani R, Biondi-Zoccai GG, Rossiello R, Silvestri F, Baldi F, Biasucci LM, Baldi A. Persistent infarct-related artery occlusion is associated with an increased myocardial apoptosis at postmortem examination in humans late after an acute myocardial infarction. Circulation. 2002;106:1051–1054. doi: 10.1161/01.cir.0000030936.97158.c4. [DOI] [PubMed] [Google Scholar]
- Baldi A, Abbate A, Bussani R, Patti G, Melfi R, Angelini A, Dobrina A, Rossiello R, Silvestri F, Baldi F, Di Sciascio G. Apoptosis and post-infarction left ventricular remodeling. J Mol Cell Cardiol. 2002;34:165–174. doi: 10.1006/jmcc.2001.1498. [DOI] [PubMed] [Google Scholar]
- Braz JC, Bueno OF, De Windt LJ, Molkentin JD. PKC alpha regulates the hypertrophic growth of cardiomyocytes through extracellular signal-regulated kinase1/2 (ERK1/2) J Cell Biol. 2002;156:905–919. doi: 10.1083/jcb.200108062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueno OF, De Windt LJ, Tymitz KM, Witt SA, Kimball TR, Klevitsky R, Hewett TE, Ceci M, Gallo P, Santonastasi M, Grimaldi S, Latronico MV, Pitisci A, Missol-Kolka E, Scimia MC, Catalucci D, Hilfiker-Kleiner D, Condorelli G. Cardiac-specific overexpression of E40K active Akt prevents pressure overload-induced heart failure in mice by increasing angiogenesis and reducing apoptosis. Cell Death Differ. 2007;14:1060–1062. doi: 10.1038/sj.cdd.4402095. [DOI] [PubMed] [Google Scholar]
- Doukas J, Wrasidlo W, Noronha G, Dneprovskaia E, Fine R, Weis S, Hood J, Demaria A, Soll R, Cheresh D. Phosphoinositide 3-kinase gamma/delta inhibition limits infarct size after myocardial ischemia/reperfusion injury. Proc Natl Acad Sci USA. 2006;103:19866–19871. doi: 10.1073/pnas.0606956103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel FB, Hsieh PC, Lee RT, Keating MT. FGF1/p38 MAP kinase inhibitor therapy induces cardiomyocyte mitosis, reduces scarring, andrescues function after myocardial infarction. Proc Natl Acad Sci USA. 2006;103:15546–15551. doi: 10.1073/pnas.0607382103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrandi C, Ballerio R, Gaillard P, Giachetti C, Carboni S, Vitte PA, Gotteland JP, Cirillo R. Inhibition of c-Jun N-terminal kinase decreases cardiomyocyte apoptosis and infarct size after myocardial ischemia and reperfusion in anaesthetized rats. Br J Pharmacol. 2004;142:953–960. doi: 10.1038/sj.bjp.0705873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman K, Lerman I, Kranias EG, Bohlmeyer T, Bristow MR, Lefkowitz RJ, Iaccarino G, Koch WJ, Leinwand LA. Alterations in cardiac adrenergic signaling and calcium cycling differentially affect the progression of cardiomyopathy. J Clin Invest. 2001;107:967–974. doi: 10.1172/JCI12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SP, Lefer DJ, Peng CF, Kitsis RN, Molkentin JD. The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J. 2000;19:6341–6350. doi: 10.1093/emboj/19.23.6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno S, Lerner DL, Schuessler RB, Betsuyaku T, Yamada KA, Saffitz JE, Kovacs A. Echocardiographic evaluation of ventricular remodeling in a mouse model of myocardial infarction. J Am Soc Echocardiogr. 2002;15:601–609. doi: 10.1067/mje.2002.117560. [DOI] [PubMed] [Google Scholar]
- Karoor V, Vatner SF, Takagi G, Yang G, Thaisz J, Sadoshima J, Vatner DE. Propranolol prevents enhanced stress signaling in Gs alpha cardiomyopathy: Potential mechanism for beta-blockade in heart failure. J Mol Cell Cardiol. 2004;36:305–312. doi: 10.1016/j.yjmcc.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Kaumann A, Bartel S, Molenaar P, Sanders L, Burrell K, Vetter D, Hempel P, Karczewski P, Krause EG. Activation of beta2-adrenergic receptors hastens relaxation and mediates phosphorylation of phospholamban, troponin I, and C-protein in ventricular myocardium from patients with terminal heart failure. Circulation. 1999;99:65–72. doi: 10.1161/01.cir.99.1.65. [DOI] [PubMed] [Google Scholar]
- Li Z, Ma JY, Kerr I, Chakravarty S, Dugar S, Schreiner G, Protter AA. Selective inhibition of p38alpha MAPK improves cardiac function and reduces myocardial apoptosis in rat model of myocardial injury. Am J Physiol Heart Circ Physiol. 2006;291:H1972–H1977. doi: 10.1152/ajpheart.00043.2006. [DOI] [PubMed] [Google Scholar]
- Liao P, Wang SQ, Wang S, Zheng M, Zheng M, Zhang SJ, Cheng H, Wang Y, Xiao RP. p38 mitogen-activated protein kinase mediates a negative inotropic effect in cardiac myocytes. Circ Res. 2002;90:190–196. doi: 10.1161/hh0202.104220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Sadoshima J, Zhai P, Hong C, Yang G, Chen W, Yan L, Wang Y, Vatner SF, Vatner DE. Pressure overload induces greater hypertrophy and mortality in female mice with p38alpha MAPK inhibition. J Mol Cell Cardiol. 2006;41:680–688. doi: 10.1016/j.yjmcc.2006.07.007. [DOI] [PubMed] [Google Scholar]
- McMullen JR, Amirahmadi F, Woodcock EA, Schinke-Braun M, Bouwman RD, Hewitt KA, Mollica JP, Zhang L, Zhang Y, Shioi T, Buerger A, Izumo S, Jay PY, Jennings GL. Protective effects of exercise and phosphoinositide 3-kinase(p110alpha) signaling in dilated and hypertrophic cardiomyopathy. Proc Natl Acad Sci USA. 2007;104:612–617. doi: 10.1073/pnas.0606663104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullen JR, Shioi T, Zhang L, Tarnavski O, Sherwood MC, Kang PM, Izumo S. Phosphoinositide 3-kinase(p110alpha) plays a critical role for the induction of physiological, but not pathological, cardiac hypertrophy. Proc Natl Acad Sci USA. 2003;100:12355–12360. doi: 10.1073/pnas.1934654100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrhof FB, Muller FU, Bergmann MW, Li P, Wang Y, Schmitz W, Dietz R, von Harsdorf R. In cardiomyocyte hypoxia, insulin-like growth factor-I-induced antiapoptotic signaling requires phosphatidylinositol-3-OH-kinase-dependent and mitogen-activated protein kinase-dependent activation of the transcription factor cAMP response element-binding protein. Circulation. 2001;104:2088–2094. doi: 10.1161/hc4201.097133. [DOI] [PubMed] [Google Scholar]
- Nicol RL, Frey N, Pearson G, Cobb M, Richardson J, Olson EN. Activated MEK5 induces serial assembly of sarcomeres and eccentric cardiac hypertrophy. EMBO J. 2001;20:2757–2767. doi: 10.1093/emboj/20.11.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida K, Yamaguchi O, Hirotani S, Hikoso S, Higuchi Y, Watanabe T, Takeda T, Osuka S, Morita T, Kondoh G, Uno Y, Kashiwase K, Taniike M, Nakai A, Matsumura Y, Miyazaki J, Sudo T, Hongo K, Kusakari Y, Kurihara S, Chien KR, Takeda J, Hori M, Otsu K. p38alpha mitogen-activated protein kinase plays a critical role in cardiomyocyte survival but not in cardiac hypertrophic growth in response to pressure overload. Mol Cell Biol. 2004;24:10611–10620. doi: 10.1128/MCB.24.24.10611-10620.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palojoki E, Saraste A, Eriksson A, Pulkki K, Kallajoki M, Voipio-Pulkki LM, Tikkanen I. Cardiomyocyte apoptosis and ventricular remodeling after myocardial infarction in rats. Am J Physiol Heart Circ Physiol. 2001;280:H2726–H2731. doi: 10.1152/ajpheart.2001.280.6.H2726. [DOI] [PubMed] [Google Scholar]
- Qin F, Liang MC, Liang CS. Progressive left ventricular remodeling, myocyte apoptosis, and protein signaling cascades after myocardial infarction in rabbits. Biochim Biophys Acta. 2005;1740:499–513. doi: 10.1016/j.bbadis.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Ren J, Zhang S, Kovacs A, Wang Y, Muslin AJ. Role of p38alpha MAPK in cardiac apoptosis and remodeling after myocardial infarction. J Mol Cell Cardiol. 2005;38:617–623. doi: 10.1016/j.yjmcc.2005.01.012. [DOI] [PubMed] [Google Scholar]
- Sahn DJ, DeMaria A, Kisslo J, Weyman A. Recommendations regarding quantitation in M-mode echocardiography: Results of a survey of echocardiographic measurements. Circulation. 1978;58:1072–1083. doi: 10.1161/01.cir.58.6.1072. [DOI] [PubMed] [Google Scholar]
- Turnbull L, McCloskey DT, O’Connell TD, Simpson PC, Baker AJ. Alpha 1-adrenergic receptor responses in alpha 1AB-AR knockout mouse hearts suggest the presence of alpha 1D-AR. Am J Physiol Heart Circ Physiol. 2003;284:H1104–H1109. doi: 10.1152/ajpheart.00441.2002. [DOI] [PubMed] [Google Scholar]
- Tutor AS, Penela P, Mayor F., Jr. Anti-beta1-adrenergic receptor autoantibodies are potent stimulators of the ERK1/2 pathway in cardiac cells. Cardiovasc Res. 2007;76:51–60. doi: 10.1016/j.cardiores.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Ungerer M, Böhm M, Elce JS, et al. Altered expression of beta-adrenergic receptor kinase and beta 1-adrenergic receptors in the failing human heart. Circulation. 1993;87:454–463. doi: 10.1161/01.cir.87.2.454. [DOI] [PubMed] [Google Scholar]
- Wang Y, Huang S, Sah VP, Ross J, Jr, Brown JH, Han J, Chien KR. Cardiac muscle cell hypertrophy and apoptosis induced by distinct members of the p38 mitogen-activated protein kinase family. J Biol Chem. 1998;273:2161–2168. doi: 10.1074/jbc.273.4.2161. [DOI] [PubMed] [Google Scholar]
- Zhang S, Weinheimer C, Courtois M, Kovacs A, Zhang CE, Cheng AM, Wang Y, Muslin AJ. The role of the Grb2-p38 MAPK signaling pathway in cardiac hypertrophy and fibrosis. J Clin Invest. 2003;111:833–841. doi: 10.1172/JCI16290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YY, Wang SQ, Zhu WZ, Chruscinski A, Kobilka BK, Ziman B, Wang S, Lakatta EG, Cheng H, Xiao RP. Culture and adenoviral infection of adult mouse cardiac myocytes: Methods for cellular genetic physiology. Am J Physiol Heart Circ Physiol. 2000;279:H429–H436. doi: 10.1152/ajpheart.2000.279.1.H429. [DOI] [PubMed] [Google Scholar]