

Abstract
Recent advances in whole genome sequencing have brought the vision of personal genomics and genomic medicine closer to reality. However, current methods lack clinical accuracy and the ability to describe the context (haplotypes) in which genome variants co-occur in a cost-effective manner. Here we describe a low-cost DNA sequencing and haplotyping process, Long Fragment Read (LFR) technology, similar to sequencing long single DNA molecules without cloning or separation of metaphase chromosomes. In this study, ten LFR libraries were made using only ~100 pg of human DNA per sample. Up to 97% of the heterozygous single nucleotide variants (SNVs) were assembled into long haplotype contigs. Removal of false positive SNVs not phased by multiple LFR haplotypes resulted in a final genome error rate of 1 in 10 Mb. Cost-effective and accurate genome sequencing and haplotyping from 10-20 human cells, as demonstrated here, will enable comprehensive genetic studies and diverse clinical applications.
The extraordinary advancements made in DNA sequencing technologies over the past few years have led to the elucidation of ~10,0001-13 individual human genomes (30x or greater base coverage) from different ethnicities and using different technologies2-13 and at a fraction of the cost10 of sequencing the original human reference genome14,15. While this is a monumental achievement, the vast majority of these genomes have excluded a very important element of human genetics. Individual human genomes are diploid in nature, with half of the homologous chromosomes being derived from each parent. The context in which variations occur on each individual chromosome can have profound effects on the expression and regulation of genes and other transcribed regions of the genome16. Further, determining if two potentially detrimental mutations occur within one or both alleles of a gene is of paramount clinical importance.
Almost all recent human genome sequencing has been performed on short read-length (<200 bp), highly parallelized systems starting with hundreds of nanograms of DNA. These technologies are excellent at generating large volumes of data quickly and economically. Unfortunately short reads, often paired with small mate-gap sizes (500b-10kb), eliminate most SNP phase information beyond a few kilobases8. Population based genotype data has been used successfully to assemble short read data into long haplotype blocks3, however these methods suffer from higher error rates and have difficulty phasing rare variants17. Using pedigree information18 or combining it with population data provides additional phasing power, however no combination of these methods are able to resolve de-novo mutations17.
Currently four personal genomes, J. Craig Venter19, a Gujarati Indian (HapMap sample NA20847)11, and two Europeans (Max Planck One (MP1)13 and HapMap Sample NA1287820) which have been sequenced and assembled as diploid. All have involved cloning long DNA fragments in a process similar to that used for the construction of the human reference genome14,15. While these processes generate long phased contigs (N50s of 350 kb19, 386 kb11, 1 Mb13, and full-chromosome haplotypes in combination with parental genotypes20) they require a large amount of initial DNA, extensive library processing, and are currently too expensive11 to use in a routine clinical environment. Additionally, several reports have recently demonstrated whole chromosome haplotyping through direct isolation of metaphase chromosomes21-24. These methods have yet to be used for whole genome sequencing and require preparation and isolation of whole metaphase chromosomes, which can be challenging for some clinical samples. In this paper, we introduce Long Fragment Read (LFR) technology, a process that enables genome sequencing and haplotyping at a clinically relevant cost, quality and scale.
Long Fragment Read technology
The LFR approach can generate long-range phased SNPs because it is conceptually similar to single molecule sequencing of fragments 10-1000 kb25 in length. This is achieved by the stochastic separation of corresponding long parental DNA fragments into physically distinct pools followed by subsequent fragmentation to generate shorter sequencing templates (Fig. 1a). The same principles are used in aliquoting fosmid clones11,13. As the fraction of the genome in each pool decreases to less than a haploid genome, the statistical likelihood of having a corresponding fragment from both parental chromosomes in the same pool dramatically diminishes25. For example, 0.1 genome equivalents (300 Mb) per well yields an approximately 10% chance that two fragments will overlap and a 50% chance those fragments will be derived from separate parental chromosomes. The end result is an approximately 5% overall chance that a particular well will be uninformative for a given fragment. Likewise, the more individual pools interrogated the greater number of times a fragment from the maternal and paternal homologs will be analysed in separate pools. The current version of LFR uses a 384-well plate with 10-20% of a haploid genome in each well, yielding a theoretical 19-38x physical coverage of both the maternal and paternal alleles of each fragment (see Supplementary Materials and Supplementary Table 1 for an explanation of how this amount of material was selected). This high initial DNA redundancy of 19-38x versus recently described strategies using fosmid pools of 3x11 or 6x13 ensures complete genome coverage and higher variant calling and phasing accuracy.
Figure 1. Overview of the Long Fragment Read (LFR) technology and controlled random enzymatic (CoRE) fragmenting.
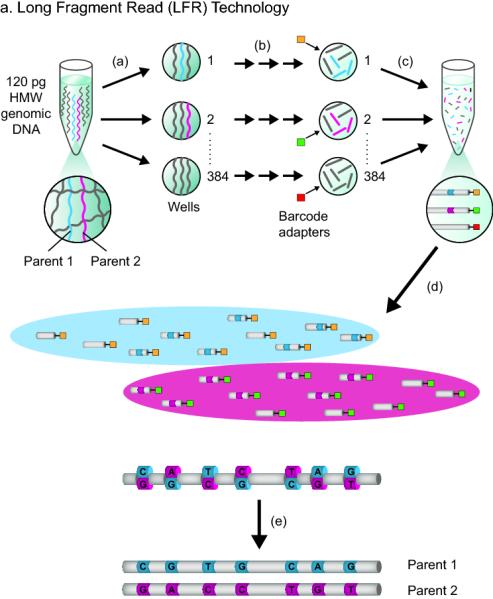
(a) 100-130 pg of high molecular weight DNA is physically separated into 384 distinct wells, (b) through several steps, all within the same well without intervening purifications, the genomic DNA is amplified, fragmented, and ligated to unique barcode adapters, (c) all 384 wells are combined, purified, and introduced into Complete Genomics’ sequencing platform10, (d) mate-paired reads are mapped to the genome using a custom alignment program and barcode sequences are used to group tags into haplotype contigs, (e) the final result is a diploid genome sequence.
To prepare LFR libraries in a high-throughput manner we developed an automated process that performs all LFR-specific steps in the same 384-well plate. First, a highly uniform amplification using a modified, phi29 polymerase-based, multiple displacement amplification (MDA)26 is performed to replicate each fragment about 10,000 times. Next, through a process of five enzymatic steps within each well, without intervening purification steps, DNA is fragmented and ligated with barcode adapters. Briefly, long DNA molecules are processed to blunt ended 300-1,500 bp fragments through the novel process of Controlled Random Enzymatic fragmenting (CoRE) (Supplementary Methods and Supplementary Figures 2 and 3). Unique 10-base Reed-Solomon27 error correcting barcode adapters (Supplementary Figure 4) are then ligated to fragmented DNA in each well using a high yield, low chimera formation protocol10. Lastly, all 384 wells are combined and an unsaturated polymerase chain reaction using primers common to the ligated adapters is employed to generate sufficient template for massively parallel short read sequencing platforms (see Supplementary Methods). The addition of the LFR pre-processing steps to the standard library process currently adds about $100 to the reagent cost of our genome sequencing (Supplementary Table 2).
LFR libraries from 100pg of isolated DNA or directly from 10 cells
As a demonstration of the power of LFR to determine an accurate diploid genome sequence we generated three libraries of Yoruban female HapMap sample NA19240, six libraries from European HapMap pedigree 1463 (Supplementary Figure 5), and a single library from Personal Genome Project (PGP) sample NA20431. Pedigree 1463 and NA19240 have been extensively studied in the HapMap Project28,29, the 1,000 Genomes Project30, and our own efforts31. As a result, highly accurate haplotype information can be generated for these samples based on the redundant sequence data from familial samples. One NA19240 LFR library was made from 10 cells of the corresponding immortalized B-cell line, all other libraries were made from an estimated 100-130 pg (equivalent to 15-20 cells) of denatured high molecular weight genomic DNA (Supplementary Figure 6 and Supplementary Methods). Libraries were analysed using Complete Genomics’ sequencing platform10. 35-base mate-paired reads were mapped to the reference genome using a custom alignment algorithm10,32 yielding on average more than 250 Gbs of mapped data with an average genomic coverage >80x (Table 1 and Supplementary Table 3). Analysis of the mapped LFR data shows two distinct characteristics attributable to MDA: slight underrepresentation of GC rich sequences (Supplementary Figure 7) and an increase in chimeric sequences (Supplementary Table 3). Additionally, coverage normalized across 100 kb windows was more variable (Supplementary Figure 8). Nevertheless, almost all genomic regions were covered with sufficient reads (5 or more) demonstrating that 10,000 fold MDA amplification by our optimized protocol can be used for comprehensive genome sequencing. Barcodes were used to group mapped reads based on their physical well location within each library resulting in sparse regions of coverage interspersed between long spans with almost no read coverage (Supplementary Figure 9). Each of these discrete regions of coverage represents a physical DNA fragment. On average, each well contained 10-20% of a haploid genome (300-600 Mb) in fragments ranging from 10 kb to over 300 kb in length with N50s of ~60 kb (Table 1). Initial fragment coverage was very uniform between chromosomes (Supplementary Figure 10). As estimated from all detected fragments, the total amounts of DNA used to make the two NA19240 libraries from extracted DNA were ~62 pg and 84 pg (equivalent to 9.4 and 12.7 cells). This is less than the expected 100-130 pg indicating some lost or undetected DNA or imprecision in DNA quantification. Interestingly, the 10-cell library appeared to be made from ~90 pg (13.6 cells) of DNA, most likely due to some of the cells being in S phase during isolation (Table 1).
Table 1. Comparison of haplotyping performance between different genome assemblies.
Variant calls for standard and LFR assembled libraries were combined and used as loci for phasing except where specified. Two samples were run with Complete Genomics’ pipeline 2.0 algorithms which are expected to call more heterozygous SNPs, the remaining samples were analysed with previous versions (1.7-1.8) of Complete Genomics’ algorithms. The LFR phasing rate was based on a calculation of parental phased heterozygous SNPs (Supplementary Table 4).
| Sample | Ethnicity | Number of Heterozygous Phased SNPs |
LFR Phasing Rate |
Haploid Fragment coverage (cells) |
Fragment Size for N50 DNA (kb) |
Fragment Size for N25 DNA (kb) |
DNA Bases Sequenced (Gb), LFR + STD |
N50 Contig Length (kb) |
|---|---|---|---|---|---|---|---|---|
| NA19240-Replicate 1 | Yoruban | 2,386,741 | 91% | 38 (9.4) | 64 | 84 | 237+176 | 1,210 |
| NA19240-Replicate 2 | Yoruban | 2,433,621 | 91% | 51 (12.7) | 66 | 96 | 313+176 | 1,010 |
| NA19240-10 cell pipeline 2.0 | Yoruban | 2,369,433 | 89% | 54.3 (13.6)# | 80 | 120 | 308+176 | 943 |
| NA19240-Replicate 1 High Coverage | Yoruban | 2,578,903 | 96% | 48 (11.9) | 82 | 116 | 509+176 | 1,429 |
| NA19240-Replicates 1&2 combined | Yoruban | 2,646,352 | 97% | 89 (22.1) | 65 | 90 | 550+176 | 1,577 |
| NA19240-Replicate 1 LFR only pipeline 2.0 | Yoruban | 2,031,514 | 91% | 38 (9.4) | 64 | 84 | 237 | 1,036 |
| NA19240-Replicate 1 High Coverage LFR only | Yoruban | 2,274,696 | 95% | 48 (11.9) | 82 | 116 | 509 | 1,282 |
| NA12877-Replicate 1 | European | 1,831,032 | 93% | 65 (16.3) | 74 | 104 | 258+218 | 530 |
| NA12877-Replicate 2 | European | 1,810,540 | 92% | 51 (12.7) | 76 | 106 | 238+218 | 535 |
| NA12877-Replicates 1&2 combined | European | 1,946,089 | 97% | 116 (29) | 75 | 105 | 496+218 | 600 |
| NA12885 | European | 1,850,409 | 92% | 46 (11.6) | 72 | 98 | 272+221 | 528 |
| NA12886 | European | 1,854,360 | 93% | 44 (11) | 66 | 88 | 293+216 | 535 |
| NA12891 | European | 1,825,427 | 90%* | 46 (11.6) | 80 | 112 | 280+246 | 545 |
| NA12892 | European | 1,917,442 | 93%* | 93 (23.3) | 94 | 138 | 285+213 | 553 |
| NA12892 LFR only | European | 1,720,750 | 97%* | 93 (23.3) | 94 | 138 | 285 | 525 |
| NA20431 High Coverage | European | 1,703,047 | 84%* | 30 (7.4) | 94 | 142 | 514+189 | 411 |
For those individuals without parental genome data (NA12891, NA12892, and NA20431) the phasing rate was calculated by dividing the number of phased heterozygous SNPs by the number of heterozygous SNPs expected to be real (number of attempted to be phased SNPs – 50,000 expected errors). N50 calculations are based on the total assembled length of all contigs to the NCBI build 36 (build 37 in the case of NA19240 10 cell and high coverage and NA20431 high coverage) human reference genome. Haploid fragment coverage is four times greater than the number of cells as a result of all DNA being denatured to single stranded prior to being dispersed across a 384 well plate. The insufficient amount of starting DNA explains lower phasing efficiency in the NA20431 genome.
The 10 cell sample was measured by individual well coverage to contain more than 10 cells, this is likely the result of these cells being in various stages of the cell cycle during collection.
LFR haplotyping results
To ensure complete representation of the genome we maximized the input of DNA fragments for a given read coverage and number of aliquots (Supplementary Materials and Supplementary Table 1). Unlike other experimental approaches11,13,20, this resulted in low-coverage read data (<2x) for each fragment in each of the ~40 wells a fragment is found in. This type of data is not useful for defining haplotypes for each initial fragment and required the development of a novel phasing algorithm that statistically combines reads from related fragments found in separate aliquots (Supplementary Methods and Fig. 2). Application of our algorithm to the LFR libraries resulted in the placement of on average 92% of the phasable heterozygous SNPs into long contigs with N50s of ~1 Mb and ~ 500 kb for NA19240 and European samples, respectively (Table1 and Supplementary Table 4). The large reduction in the N50 contig size for European samples can be explained by many more regions of low heterozygosity (RLHs, Supplementary Tables 5-7, Supplementary Figure 11, and Supplementary Materials) found in these genomes. Doubling the number of reads to ~160x coverage or combining replicate samples (a total of 768 independent wells), each with ~80x coverage, pushed the phasing rate to ~96% (Table 1). Using only the SNP loci called in the LFR library for phasing resulted mostly in a reduction in the total number of phased SNPs by 5-15% (Table 1 and Supplementary Materials). Importantly, the 1.72 million heterozygous SNPs called and phased by the NA12892 LFR library alone was slightly higher than the number of SNPs phased for a comparable sample using a fosmid approach13,20 (Table 1). For NA19240, the 10-cell library phased over 98% of variants phased by the two libraries made from isolated DNA demonstrating that LFR can be successful starting from a small number of cells.
Figure 2. LFR haplotyping algorithm.
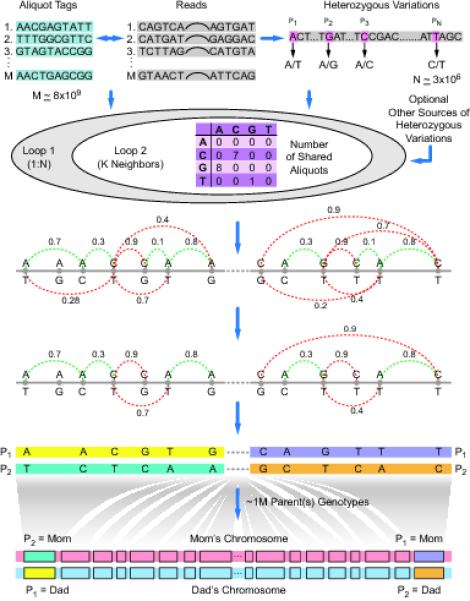
(a) Variation extraction: Variations are extracted from the aliquot tagged reads. The 10-base Reed-Solomon codes enable tag recovery via error correction. (b) Heterozygous SNP-pair connectivity evaluation: The matrix of shared aliquots is computed for each heterozygous SNP-pair within a certain neighbourhood. Loop1 is over all the heterozygous SNPs on one chromosome. Loop2 is over all the heterozygous SNPs on the chromosome which are in the neighbourhood of the heterozygous SNPs in Loop1. This neighbourhood is constrained by the expected number of heterozygous SNPs and the expected fragment lengths. (c) Graph generation: An undirected graph is made, with nodes corresponding to the heterozygous SNPs and the connections corresponding to the orientation and the strength of the best hypothesis for the relationship between those SNPs. The orientation is binary and is shown in the figure with a colour. Red and green depict a flipped and unflipped relationship between heterozygous SNP pairs, respectively. The strength is defined by employing fuzzy logic operations on the elements of the shared aliquot matrix. (d) Graph optimization: The graph is optimized via a minimum spanning tree operation. (e) Contig generation: Each sub-tree is reduced to a contig by keeping the first heterozygous SNP unchanged, and flipping or not flipping the other heterozygous SNPs on the sub-tree, based on their paths to the first heterozygous SNP. The designation of Parent 1 (P1) and Parent 2 (P2) to each contig is arbitrary. The gaps in the chromosome-wide tree define the boundaries for different sub-trees/contigs on that chromosome. (f) Optional mapping of LFR contigs to parental chromosomes: Using parental information, a Mom or Dad label is placed on the P1 and P2 haplotypes of each contig.
LFR reproducibility and phasing error rate analysis
To test LFR reproducibility we compared haplotype data between the two NA19240 replicate libraries. In general, the libraries were very concordant, with only 64 differences per library in ~2.2 million heterozygous SNPs phased by both libraries (Supplementary Table 8) or 1 of this error type in 44 Mb. LFR was also highly accurate when compared to the conservative but accurate whole chromosome phasing generated from the parental genomes NA19238 and NA19239 previously sequenced by multiple methods28-31 (Supplementary Table 4). Only ~60 instances in 1.57 million comparable individual loci were found in which LFR phased a variant inconsistent with that of the parental haplotyping (false phasing rate of 0.002% if half of discordances are due to sequencing errors in parental genomes). The LFR data also contained ~135 contigs per library (2.2%) with one or more flipped haplotype blocks (Supplementary Table 8). Extending these analyses to the European replicate libraries of sample NA12877 (Supplementary Table 8) and comparing them with a recent high quality family-based analysis18 yielded similar results assuming each method contributes half of the observed discordance (Supplementary Table 9). In both NA19240 and NA12877 libraries several contigs had dozens of flipped segments. The majority of these contigs were located in regions of low heterozygosity, low read coverage regions, or repetitive regions observed in an unexpectedly large number of wells (e.g., subtelomeric or centromeric regions). Most of these errors can be corrected by forcing the LFR phasing algorithms to end contigs in these regions. Alternatively, these errors can be removed with the simple, low cost addition of standard high density array genotype data (~1 million or greater SNPs) from at least one parent to the LFR assembly. We found that parental genotypes can connect 98% of LFR phased heterozygous SNPs in full chromosome haplotypes. Additionally, this data allows haplotypes to be assigned to maternal and paternal lineages; information that is critical for incorporating parental imprinting in genetic diagnoses in any experimental haplotyping approach. If parental data is unavailable population genotype data could also be used to connect many of these LFR contigs, although at the cost of increased phasing errors17.
Phasing de-novo mutations
As a demonstration of the completeness and accuracy of our diploid genome sequencing we assessed phasing of 35 de-novo mutations recently reported in the genome of NA1924033. 34 of these mutations were called in either the standard genome or one of the LFR libraries. Of those, 32 de-novo mutations were phased (16 coming from each parent) in at least one of the two replicate LFR libraries (Supplementary Table 10). Not surprisingly, the two non-phased variants reside in RLHs. Of these 32 variants, 21 were phased by Conrad et al.33 and 18 were consistent with LFR phasing results (Matthew Hurles personal communication). The three discordances are likely due to errors in the previous study (Matthew Hurles personal communication) confirming LFR accuracy, but not affecting the substantive conclusions of the report.
Error reduction enabled by LFR for accurate genome sequencing from 10 cells
Substantial error rates (~1 SNV in 100-1,000 called kilobases) are a common attribute of all current massively parallelized sequencing technologies2-10,18. These rates are most likely too high for diagnostic use and complicate many studies searching for novel mutations. The vast majority of errors are no more likely to occur on the maternal or paternal chromosome. This lack of consistent phasing or presence in only a few aliquots can be exploited by LFR to eliminate these errors from the final assembled haplotypes. To demonstrate this we defined a set of heterozygous SNPs in NA19240 and NA12877 LFR libraries that were reported with high confidence in each of the individual’s parents as matching the human reference genome at both alleles (>85% of all heterozygous SNPs). There were about 44,000 of these heterozygous SNPs in NA19240 and 30,000 in NA12877 that met this criterion. By virtue of their nonexistence in the parental genomes these variations are de novo mutations, cell line specific somatic mutations, or false positive variants. Approximately 1,000-1,500 of these variants were reproducibly phased in each of the two replicate LFR libraries from samples NA19240 and NA12877 (Supplementary Table 11). These numbers are similar to those previously reported for de-novo and cell line specific mutations in NA1924033. The remaining variants are likely to be initial false positives of which only about 500 are phased per library. This represents a 60 fold reduction of the false positive rate in those variations that are phased. Only ~2,400 of these false variants are present in the standard libraries, of which only ~260 are phased (<1 false positive SNV in 20 Mb; 5700 haploid Mb / 260 errors). Each LFR library exhibits a 15 fold increase, compared to a genome sequenced by the standard process, in library specific false positive calls before phasing. The majority of these false positive SNVs are likely to have been introduced by MDA; sampling of rare cell-line variants may be responsible for a smaller percentage. Despite making LFR libraries from 100 pg of DNA and introducing a large number of errors through MDA amplification, applying the LFR phasing algorithm described above reduces the overall sequencing error rate to 99.99999% (~600 false heterozygous SNVs/6 Gb), approximately 10 fold lower than the previous published error rates using the same ligation-based sequencing chemistry18. These accurate haplotypes allow detection of highly diverged human sequences (Supplementary Materials and Supplementary Table 13) and many other applications.
Many genes have putative inactivating variations in both alleles
To demonstrate how LFR could be used in a diagnostic/prognostic environment we analysed the coding SNP data of all libraries for two or more nonsense, splicesite, or PolyPhen234 predicted detrimental missense variations that co-occur in the same gene. Of these, approximately 40 genes were found in each individual that contained a least one detrimental variation in each allele (Table 2). Extending this analysis to variants which disrupt transcription factor binding sites (TFBS) introduces an additional ~100 genes per individual (additional analyses of the effects of TFBS disruption on allele specific expression can be found in Supplementary Materials and Supplementary Table 12). Due to the high accuracy of LFR it is unlikely that these variants are a result of sequencing errors and many could have been introduced in the propagation of these cell lines. Further, some of these variants are likely to have little to no effect on the function of these gene products35 and much more work is required to understand how changes in TFBS sites effect transcription. A few of these variants were found in unrelated individuals suggesting that they could be improperly annotated or the result of a systematic mapping or reference error. The genome of NA19240 contained an additional ~10 genes predicted to have complete loss of function; this is most likely due to biases introduced by using a European reference genome to annotate an African genome. Nonetheless, these numbers are similar to those found in several recent studies on phased individual genomes13,35,36 and suggest that most generally healthy individuals probably have a small number of genes, not absolutely required for normal life, which encode ineffective protein products. Additional studies are required to understand the meaning of these types of changes. Importantly, we have demonstrated that LFR is able to identify genes in which two detrimental variants are found in different alleles without the need for costly verification35. This information is critical for effective clinical interpretation of patient genomes.
Table 2. Number of genes with multiple detrimental variations in each analysed sample.
All phased SNPs were analysed by PolyPhen234 and a custom splice site detection algorithm (Supplementary Methods) to find variants with a high probability of coding for non-functional proteins. Only variants that were contained within the same contig for each gene were examined. Because LFR contigs are very long (N50>500 kb) very few variants were excluded based on this criteria. In each gene 5 kb of the regulatory region upstream of the transcription start site and 1 kb downstream were scanned for SNVs that significantly altered over 300 transcription factor binding sites (TFBS)38,39. These potentially detrimental variations in TFBSs were also phased with coding SNPs to create a more comprehensive list of genes whose function and/or expression might be altered in these individuals (Supplementary Methods).
| Sample | Ethnicity | Coding Only | Coding and TFBS | ||
|---|---|---|---|---|---|
| Both Alleles | One Allele | Both Alleles | One Allele | ||
| NA19240 Replicate 1 | Yoruban | 47 | 79 | 182 | 162 |
| NA19240 Replicate 2 | Yoruban | 55 | 85 | 207 | 174 |
| NA19240-10 cell pipeline 2.0 | Yoruban | 62 | 86 | 197 | 156 |
| NA19240-Replicate 1 High Coverage | Yoruban | 65 | 95 | 235 | 185 |
| NA19240-Replicates 1&2 combined | Yoruban | 65 | 99 | 241 | 197 |
| NA12877 Replicate 1 | European | 45 | 78 | 144 | 144 |
| NA12877 Replicate 2 | European | 44 | 82 | 146 | 141 |
| NA12877-Replicates 1&2 combined | European | 49 | 96 | 167 | 168 |
| NA12885 | European | 34 | 79 | 143 | 141 |
| NA12886 | European | 32 | 101 | 140 | 168 |
| NA12891 | European | 36 | 69 | 130 | 140 |
| NA12892 | European | 37 | 65 | 125 | 136 |
| NA20431 High Coverage | European | 36 | 70 | 115 | 127 |
Discussion
In this study we have demonstrated the efficiency of LFR to accurately phase up to 97% of all detected heterozygous SNPs in a genome into long contiguous stretches of DNA (N50s 400-1500 kb in length). Even LFR libraries phased without candidate heterozygous SNPs from standard libraries, and thus using only 10-20 human cells, are able to phase >90% of the available SNP. In several instances, the LFR libraries used in this paper had less than optimal starting input DNA (NA20431, Table1). Phasing rate improvements seen by combining two replicate libraries or starting with more DNA (NA12892, Table 1) agree with this conclusion. Additionally, underrepresentation of GC rich sequences resulted in less of the genome being called (Supplementary Table 3). Improvements to the MDA process, removal of amplification steps as future single molecule sequencing processes improve, or modifications to how we perform base and variant calling in LFR libraries will help increase the coverage in these regions (see Supplementary Materials and Supplementary Figure 12 for a demonstration of how LFR can make calls in low coverage regions). Moreover, as the cost of whole genome sequencing continues to fall, higher coverage libraries, demonstrated in this paper to dramatically improve call rates and phasing, will become more affordable.
A consensus haploid sequence is sufficient for many applications; however it lacks two very important pieces of data for detecting disease causing variants in personal genomes: phased heterozygous variants and identification of false positive and negative variant calls. By providing sequence data from both the maternal and paternal chromosomes independently, LFR is able to detect regions in the genome assembly where only one allele has been covered. Likewise, false positive calls are avoided because LFR independently, in separate aliquots, sequences both the maternal and paternal chromosomes 10-20 times. The result is a statistically low probability that random sequencing or DNA amplification errors would repeatedly occur in several aliquots at the same base position on one parental allele. Thus, LFR allows, for the first time, both accurate and cost-effective sequencing of a genome from a few human cells in spite of the required extensive DNA amplification. Further, by phasing SNPs over hundreds of kilobases (or over entire chromosomes by integrating LFR with routine genotyping of at least one parent), LFR is able to more accurately predict the effects of compound regulatory variants and parental imprinting on allele specific gene expression and function in various tissue types. Additionally, separation of mate-pair reads by haplotype may also help in detecting expanded tri-nucleotide repeats in diseases like Huntingtons, even though LFR does not provide direct length measure of these or similar repeats. Taken together this provides a highly accurate report about the potential genomic changes that could cause gain or loss of protein function. This kind of information, obtained inexpensively for every patient, will be critical for clinical use of genomic data. Moreover, successful and affordable diploid sequencing of a human genome starting from ten cells opens the possibility for comprehensive and accurate genetic screening of micro biopsies from diverse tissue sources such as circulating tumor cells or pre-implantation embryos generated through in vitro fertilization.
Methods Summary
High molecular weight DNA was purified from cell lines GM12877, GM12878, GM12885, GM12886, GM12891, GM12892 GM19240, and GM20431 (Coriell Institute for Medical Research, Camden, NJ) using a RecoverEase DNA isolation kit (Agilent, La Jolla, CA) following the manufacturer’s protocol. Individual cells of NA19240 were isolated under 200x magnification with a micromanipulator (Eppendorf, Hamburg, Germany) and deposited into a 1.5 ml microtube with 10 ul of dH2O. LFR libraries were made as outlined in the main text; a more detailed description can be found in the Supplementary Methods. LFR libraries were sequenced, mapped, and assembled using the Complete Genomics’ sequencing pipeline. Phasing was performed using custom haplotyping algorithms as described in Figure 2 and in further detail in the Supplementary Methods. Variations adversely affecting protein function or expression were found using several methods. Misssense variations were analyzed using Polyphen234. For this study both “possibly damaging” and “probably damaging” were considered to be detrimental to protein function as were all nonsense mutations. Variations determined to adversely affect mRNA splicing were found with a custom algorithm based on consensus splice position models from Steve Mount’s database37. JASPAR models38,39 were used to extract potential TFBSs from the reference genome with mast40. Variations falling with these regions were compared to the models to determine what affect they had upon transcription factor binding. Genes found to have two or more detrimental mutations were only further analyzed if all mutations were found within the same haplotype contig. More detailed descriptions of all methods used in this paper can be found in the online Supplementary Methods.
Supplementary Material
Acknowledgments
We would like to acknowledge the on-going contributions and support of all Complete Genomics employees, in particular M. McElwain, D. Bailey, D. Kruse, and J. Turcotte for their help with preparing the manuscript. We also wish to thank W. Chao for his help with Figures 1 and 2. Some of this work was supported by the National Institute of Standards and Technology grant 70NANB7H7027 and National Institutes of Health grant P50HG005550. Employees of Complete Genomics have stock options in the company. Complete Genomics has filed several patents on this work.
Footnotes
Author Contributions B.A.P., B.G.K., A.B.S., and R.D. conceived the study. B.A.P., B.G.K., R.D., O.A., Y.T.T., J.H., J.E., J.B., A.L.H. and G.B.N. performed analyses. B.A.P., A.B.S., P.H., A.A., Y.J., F.D., J.E.P., H.P., G.Y., J.L., and L.C. developed the lab processes and generated the LFR libraries. K.K., M.T.S., and K.P.P. developed the basecaller and parts of the analysis pipeline. M.I.K. formatted, managed, and uploaded data to the public archives. K.R., A.W.Z., J.H.L., M.P.B., and G.C. generated and analysed the RNA sequencing data. B.A.P., B.G.K., and R.D. coordinated the study and wrote the paper. All authors contributed to revision and review of the manuscript.
Author Information Tagged read data has been deposited with the NCBI short read archive (accession number SRP012316.1). All sequence data and haplotype information for LFR libraries generated in this study are also available at www.completegenomics.com/LFR. Correspondence and requests for materials should be addressed to B.A.P. (bpeters@completegenomics.com) or R.D. (rdrmanac@completegenomics.com).
References
- 1.Human genome: Genomes by the thousand. Nature. 2010;467:1026–1027. doi: 10.1038/4671026a. [DOI] [PubMed] [Google Scholar]
- 2.Wheeler DA, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- 3.Wang J, et al. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–65. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ley TJ, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bentley DR, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahn SM, et al. The first Korean genome sequence and analysis: full genome sequencing for a socio-ethnic group. Genome Res. 2009;19:1622–1629. doi: 10.1101/gr.092197.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim JI, et al. A highly annotated whole-genome sequence of a Korean individual. Nature. 2009;460:1011–1015. doi: 10.1038/nature08211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKernan KJ, et al. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Res. 2009;19:1527–1541. doi: 10.1101/gr.091868.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pushkarev D, Neff NF, Quake SR. Single-molecule sequencing of an individual human genome. Nat Biotechnol. 2009;27:847–852. doi: 10.1038/nbt.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drmanac R, et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science. 2010;327:78–81. doi: 10.1126/science.1181498. [DOI] [PubMed] [Google Scholar]
- 11.Kitzman JO, et al. Haplotype-resolved genome sequencing of a Gujarati Indian individual. Nat Biotechnol. 2011;29:59–63. doi: 10.1038/nbt.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rothberg JM, et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature. 2011;475:348–352. doi: 10.1038/nature10242. [DOI] [PubMed] [Google Scholar]
- 13.Suk EK, et al. A comprehensively molecular haplotype-resolved genome of a European individual. Genome Res. 2011;21:1672–1685. doi: 10.1101/gr.125047.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Venter JC, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 15.Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 16.Tewhey R, Bansal V, Torkamani A, Topol EJ, Schork NJ. The importance of phase information for human genomics. Nat Rev Genet. 2011;12:215–223. doi: 10.1038/nrg2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Browning SR, Browning BL. Haplotype phasing: existing methods and new developments. Nat Rev Genet. 2011;12:703–714. doi: 10.1038/nrg3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roach JC, et al. Chromosomal Haplotypes by Genetic Phasing of Human Families. Am J Hum Genet. 2011;89:382–397. doi: 10.1016/j.ajhg.2011.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levy S, et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duitama J, et al. Fosmid-based whole genome haplotyping of a HapMap trio child: evaluation of Single Individual Haplotyping techniques. Nucleic Acids Res. 2012;40:2041–2053. doi: 10.1093/nar/gkr1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang K, et al. Long-range polony haplotyping of individual human chromosome molecules. Nat Genet. 2006;38:382–387. doi: 10.1038/ng1741. [DOI] [PubMed] [Google Scholar]
- 22.Ma L, et al. Direct determination of molecular haplotypes by chromosome microdissection. Nat Methods. 2010;7:299–301. doi: 10.1038/nmeth.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan HC, Wang J, Potanina A, Quake SR. Whole-genome molecular haplotyping of single cells. Nat Biotechnol. 2011;29:51–57. doi: 10.1038/nbt.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang H, Chen X, Wong WH. Completely phased genome sequencing through chromosome sorting. Proc Natl Acad Sci U S A. 2011;108:12–17. doi: 10.1073/pnas.1016725108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Drmanac R. Nucleic Acid Analysis by Random Mixtures of Non-Overlapping Fragments. 2006. WO 2006/138284 A2.
- 26.Dean FB, et al. Comprehensive human genome amplification using multiple displacement amplification. Proc Natl Acad Sci U S A. 2002;99:5261–5266. doi: 10.1073/pnas.082089499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kermani BG, Shannon KW. Method and apparatus for quantification of dna sequencing quality and construction of a characterizable model system using reed-solomon codes. 2010. PCT/US2010/023083.
- 28.Consortium IH. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frazer KA, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. http://www.completegenomics.com/sequence-data/download-data/
- 32.Carnevali P, et al. Computational Techniques for Human Genome Resequencing Using Mated Gapped Reads. Journal of Computational Biology. 2011;19 doi: 10.1089/cmb.2011.0201. [DOI] [PubMed] [Google Scholar]
- 33.Conrad DF, et al. Variation in genome-wide mutation rates within and between human families. Nat Genet. 2011;43:712–714. doi: 10.1038/ng.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.MacArthur DG, et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335:823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lohmueller KE, et al. Proportionally more deleterious genetic variation in European than in African populations. Nature. 2008;451:994–997. doi: 10.1038/nature06611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. http://www.life.umd.edu/labs/mount/RNAinfo.
- 38.Sandelin A, Alkema W, Engstrom P, Wasserman WW, Lenhard B. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004;32:D91–94. doi: 10.1093/nar/gkh012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bryne JC, et al. JASPAR, the open access database of transcription factor-binding profiles: new content and tools in the 2008 update. Nucleic Acids Res. 2008;36:D102–106. doi: 10.1093/nar/gkm955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. http://meme.sdsc.edu/meme/mast-intro.html.
- 41.Rozowsky J, et al. AlleleSeq: analysis of allele-specific expression and binding in a network framework. Mol Syst Biol. 2011;7:522. doi: 10.1038/msb.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gibson J, Morton NE, Collins A. Extended tracts of homozygosity in outbred human populations. Hum Mol Genet. 2006;15:789–795. doi: 10.1093/hmg/ddi493. [DOI] [PubMed] [Google Scholar]
- 43.Roach JC, et al. Analysis of Genetic Inheritance in a Family Quartet by Whole-Genome Sequencing. Science. 2010;328:636–639. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li H, Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475:493–496. doi: 10.1038/nature10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cook PR. A general method for preparing intact nuclear DNA. EMBO J. 1984;3:1837–1842. doi: 10.1002/j.1460-2075.1984.tb02056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hobolth A, Dutheil JY, Hawks J, Schierup MH, Mailund T. Incomplete lineage sorting patterns among human, chimpanzee, and orangutan suggest recent orangutan speciation and widespread selection. Genome Res. 2011;21:349–356. doi: 10.1101/gr.114751.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hammer MF, Woerner AE, Mendez FL, Watkins JC, Wall JD. Genetic evidence for archaic admixture in Africa. Proc Natl Acad Sci U S A. 2011;108:15123–15128. doi: 10.1073/pnas.1109300108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Green RE, et al. A draft sequence of the Neandertal genome. Science. 2010;328:710–722. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.