

Abstract
We designed, synthesized and evaluated thirteen novel tricyclic indeno[2,1-d]pyrimidines as RTK inhibitors. These analogues were synthesized via a Dieckmann condensation of 1,2-phenylenediacetonitrile followed by cyclocondensation with guanidine carbonate to afford the 2-amino-3,9-dihydro-indeno[2,1-d]pyrimidin-4-one. Sulfonation of the 4-position followed by displacement with appropriately substituted anilines afforded the target compounds. These compounds were potent inhibitors of platelet-derived growth factor receptor β (PDGFRβ) and inhibited angiogenesis in the chicken embryo chorioallantonic membrane (CAM) assay compared to standards. In addition, compound 7 had a two digit nanomolar GI50 against nine tumor cell lines, a submicromolar GI50 against twenty nine of other tumor cell lines in the preclinical NCI 60 tumor cell line panel. Compound 7 also demonstrated significant in vivo inhibition of tumor growth and angiogenesis in a B16-F10 syngeneic mouse melanoma model.
Introduction
Angiogenesis is the formation of new blood vessels from pre-existing vasculature.1 Based on Folkman’s seminal observation,2 in order to grow beyond a few millimeters in diameter, solid tumors depend on angiogenesis for the transport of nutrients and removal of metabolite waste from tumor cells. It is now well established that angiogenesis plays a key role in the growth of solid tumors, tumor invasion and metastasis. 3 Angiogenesis is primarily a receptor-mediated process by growth factors that cause signal transduction, for the most part, via receptor tyrosine kinases (RTK). These RTK, including platelet-derived growth factor receptor (PDGFR), fibroblast growth factor receptor (FGFR), vascular endothelial growth factor receptor (VEGFR), insulin-like growth factor receptor (IGFR) and epidermal growth factor receptor (EGFR) among several others. 4 The catalytic tyrosine kinase domain of RTKs contains binding sites for both ATP and substrates, allowing for autophosphorylation, which is critical for signal transduction and angiogenesis. 5 Dysfunctional, hyperactive growth factor RTKs have been associated with several tumors and play a pivotal role in tumor angiogenesis. 5,6 Abrogation of angiogenesis via RTK inhibition provides a viable approach for the treatment of cancer. 7
In the early stage of RTK inhibitor development, the majority of the effort was focused on targeting a single RTK by small molecules. Examples of such clinically used agents include gefitinib (specific EGFR inhibitor; approved for limited use for the treatment of non small cell lung cancer) 8 and erlotinib (specific EGFR inhibitor; approved for the treatment of non small cell lung cancer) (Figure 1). 9 Since there are redundant signaling pathways for angiogenesis, tumors often survive through alternative signaling pathways and develop resistance to agents that target single RTK. Currently, the paradigm for RTK inhibitors in cancer chemotherapy is the inhibition of multiple, rather than single, RTKs to block potential ‘escape routes’ from single RTK inhibition.10 Clinical studies have recently shown that the inhibition of multiple kinases either by single-agents or with combinations of two or more agents have the potential to increase antitumor activity. Sunitinib (SU11248, inhibiting PDGFR, VEGFR, Kit, Fms-related tyrosine kinase 3) (Figure 1) was the first multitargeted RTK inhibitor approved by FDA for the treatment of renal cell carcinoma (RCC) and imatinib resistant gastrointestinal stromal tumor (GIST).11 Sorafenib (Figure 1) is another multi-targeted inhibitor of PDGF, VEGFR-2 and -3 kinases and is approved by the FDA for the treatment of advanced hepatocellular carcinoma (primary liver cancer) and renal cell carcinoma (primary kidney cancer).12 Since RTK inhibitors are generally cytostatic against tumors.13–18 Thus the combination of RTK inhibitors with standard cytotoxic chemotherapeutic agents is the subject of several clinical trials to improve long-term survival in cancer patients. We19 reasoned that the combination of RTK inhibition along with cytotoxic activity in single molecules would provide single agent(s) with ‘combination chemotherapeutic potential.’ Such agents would have both cytotoxic and antiangiogenic activity. These single agents would be tumoricidal and may have much lower cytotoxic activity and hence toxicity than a chemotherapeutic agent used in combination with a RTK inhibitor. In addition, such single agents could circumvent or delay the emergence of resistance and simplify the pharmacokinetics and toxicity issues compared to two or more separate agents.19
Figure 1.

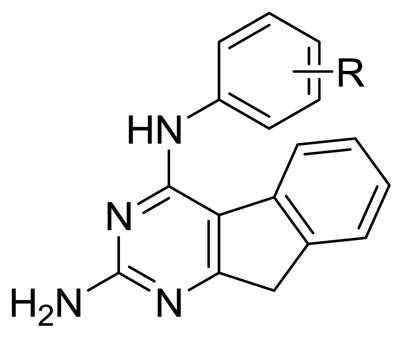
In 1999, Showalter et al.20 reported that tricyclics with a third ring fused to the 6, 7- positions of general bicyclic RTK inhibitors could moderately enhance RTK inhibitory activity over the parent compounds. We have shown that similar tricyclic molecules could bind to VEGFR-2 as well as to PDGFR-β.19 We hypothesized that the introduction of a 2-NH2 group on the pyrimidine ring could form additional hydrogen-bonds in the Hinge region 19, 21 with the backbone carbonyl of the ATP binding sites to perhaps afford an increase in inhibitory activities over those reported by Showalter et al.20 We23 have shown that inclusion of the 2-NH2 moiety does, in some cases, result in better inhibitory activity for RTK. In this study we report novel tricyclic indeno[2, 1-d]pyrimidines 1-13 (Figure 2) with different anilino substitutions at the 4-position of the pyrimidine ring. The anilino ring of the molecule is expected to be involved in RTK binding at the Hydrophobic Region 121 and influence inhibition, selectivity as well as the antitumor activity. Thus, the variations in the anilino ring include electron donating and withdrawing groups.
Figure 2.

Target compounds.
Chemistry
The synthesis of the target compounds was accomplished as shown in Scheme 1. 2-Oxo-indan-1-carboxylic acid ethyl ester 15 was obtained by hydrolysis of 1,2-phenylenediacetonitrile 14 with conc. H2SO4 followed by Dieckmann condensation (42% yield for two steps). Reaction of 15 with guanidine in the presence of potassium t-butoxide at 150 °C in a microwave reactor gave 16 (43% yield). The usual synthetic methodology for similar 4-anilino substituted compounds is to chlorinate the 4-oxo moiety of 16 to the corresponding 4-chloro followed by displacement with appropriate anilines.21 However, several attempts at chlorination of 16 with POCl3 with variation in time and temperature, even at reflux, were unsuccessful. This failure of chlorination could be attributed to the lower aromaticity of the tricyclic ring system of 16. Thus, the 4-oxo was first converted to the 4-sulfonate which is a better leaving group for SNAr reactions. Treatment of 16 with 4-nitrobenzensulfonyl chloride afforded 17 (36% yield). Nucleophilic displacement of 17 with the appropriate anilines at reflux for 12–18 h afforded target compounds 1-13 (yields 18% to 59%).
Scheme 1a.

a Reagents: (a) 1. Conc. H2SO4, EtOH, reflux; 2. toluene, Na, reflux, 3 h, ( 42% for 2 steps); (b) guanidine carbonate, t-butanol, potassium t-butoxide, μW, 4 h (34%); (c) 4-nitrobenzenesulfonylchloride, DMAP, Et3N, dichloromethane, rt.(36%); (d) 1,4-dioxane, substituted aniline, reflux, 12-18 h (18-59%).
Biological Evaluation and Discussion
The RTK inhibitory activities of the compounds were determined using human tumor cells known to express high levels of EGFR, VEGFR-2 and PDGFRβ, respectively using ELISA assay. Cytotoxicity studies against the growth of A431 cells, which overexpress EGFR, in culture, were also carried out for these compounds. In addition, the effect of 1-13 on blood vessel formation (angiogenesis) was assessed using the chicken embryo chorioallantonic membrane (CAM) assay, a standard test for angiogenesis. Since the IC50 values of compounds vary under different assay conditions, we used a standard (control) compound in each of the evaluations. Compound 18 along with erlotinib (Figure 1) were used as standards for EGFR inhibition. (Z)-3-[4-(dimethylamino)benzylidenyl]indolin-2-one (SU4312, Figure 1,) 19 sunitinib and erlotinib were used as standards for PDGFRβ inhibition. Semaxanib, sunitinib and erlotinib (Figure 1) were used as standards for VEGFR-2 inhibition, and semaxanib for the CAM assay, and cisplatin (Figure 1) was used as a standard for the A431 cytotoxicity assay. The results are reported in Table 1. We have used whole cell assays in preference to isolated enzyme assays since whole cell assay results are usually more amenable to translation to in vivo studies that isolated enzyme results.
Table 1.
IC50 values (μM) of kinase inhibition, A431 cytotoxicity and inhibition of the CAM assay
| R | EGFR kinase | VEGFR-2 kinase | PDGFRβ kinase | A431 cytotoxicity | CAM angiogenesis | |
|---|---|---|---|---|---|---|
| 1 | H | >200 | 171.2±18.0 | 43.1±4.7 | 4.9±0.7 | 50.8±6.7 |
| 2 | 2-CH(CH3)2 | >200 | >200 | 21.7±2.6 | 126.4±18.0 | 2.12±0.18 |
| 3 | 4-CH(CH3)2 | 42.6±5.8 | 11.0±1.8 | 7.8±0.9 | 126.4±13.9 | 14.8±1.6 |
| 4 | 3-F | >300 | 55.7±7.0 | 7.1±1.0 | 9.8±1.3 | 78.3±8.3 |
| 5 | 4-F | 276.1±30.2 | 89.3±9.2 | 5.9±0.7 | 50.3±7.0 | 46.7±5.9 |
| 6 | 3-Cl | 19.6±2.7 | 133.9±16.8 | >500 | 235.0±29.1 | ND |
| 7 | 4-Cl | 26.0±3.2 | >200 | 0.8±0.08 | 9.7±1.1 | 0.82±0.09 |
| 8 | 3-Br | 24.1±3.1 | >200 | 126.3±15.0 | >500 | 104.3±14.2 |
| 9 | 4-Br | >300 | 30.1±4.8 | 38.9±4.0 | 103.1±14.8 | 19.1±2.0 |
| 10 | 4-CF3 | >300 | >200 | 23.1±0.38 | 306.3±40.1 | 8.1±1.4 |
| 11 | 3-OCH3 | >200 | 43.0±5.9 | 75.5±9.1 | 13.9±1.6 | 36.4±3.9 |
| 12 | 4-F,3-Cl | >200 | 72.9±0.9 | 6.4±0.7 | 158.1±17.9 | ND |
| 13 | 2-F, 4-Cl | 11.7±2.1 | 197.1±2.6 | 3.6±0.6 | 9.7±0.8 | 3.67±0.4 |
| 18 | 0.23±0.03 | |||||
| 19 | 3.75±0.06 | |||||
| semaxanib | 12.9±2.7 | 0.04±0.009 | ||||
| sunitinib | 172.1±19.4 | 18.9±2.7 | 83.1±10.1 | |||
| erlotinib | 1.2±0.2 | 124.7±18.2 | 12.2±1.9 | |||
| cisplatin | 10.6±2.9 |
ND. Not determined.
In the EGFR assay (Table 1), all analogues had comparatively lower or no inhibitory activity compared to the standard 18. Among these analogues, the most potent compound was the 2-F, 4-Cl analogue 13 (IC50 = 11.7 μM) and it was about 50-fold less potent than the standard 18. The 4-Cl analogue 7 was comparable to 13 and both analogs were the most potent of the series in the A431 assay.
Against VEGFR-2 the most potent compound was the 4-isopropyl analogue 3, and it was equipotent with the standard, semaxanib and better than sunitinib and erlotinib. The smaller 3-fluoro (electron-withdrawing group), 4 was 4.5-fold less potent than semaxanib and also less potent than sunitinib but more potent than erlotinib. Larger halogen atoms such as chlorine or bromine at the 3-position resulted in a lower, or loss of, inhibitory activity against VEGFR-2. Electron-donating substituent (3-OCH3) in 11 was 4-fold less potent than semaxanib, about 2-fold less potent than sunitinib but 3-fold better than erlotinib. Large hydrophobic groups at the 4-postion (3 and 9) were most conducive to VEGFR-2 inhibition.
Six of the analogues showed excellent activity against PDGFRβ. The 4-chloro analogue 7 was the most potent compound and was about 100-fold more potent than sunitinib, 15-fold more potent than erlotinib and 4.5-fold more potent than the standard PDGFRβ inhibitor, 19. The 2-fluoro, 4-chloro analogue 13 was 23-fold more potent than sunitinib, 4-fold more potent than erlotinib and equipotent with the standard 19. Compounds 3-5 and 12 were about 10-times more potent than sunitinib, 1.5-fold more potent than erlotinib and one half as potent as the standard 19. In general, these modifications indicate that an electron-withdrawing group at the 4-position (except 4-CF3) is preferred for PDGFRβ activity and an electron-donating group is detrimental for inhibitory activity. The 4-position is the optimum position for substitution for PDGFRβ inhibition (5,7 and 13).
Cytotoxicity studies against the growth of A431 cells in culture provided interesting results. The standard compound, cisplatin, is a cytotoxic chemotherapeutic agent and not a kinase inhibitor. In the current study, the most potent analogue 1 was 2-fold more potent than the standard, cisplatin. Compounds 4, 7, 11 and 13 were equipotent against A431 compared with cisplatin. In keeping with the fact that EGFR is overexpressed in A431 cells, compounds 7 and 13 were also potent against EGFR kinase, however 4 and 11 do not show potent inhibition of EGFR kinase. That the IC50 values for A431 cytotoxicity are lower than for EGFR kinase suggests that there could be other mechanism(s) which could account for the potent inhibitory activity of these compounds against A431 cells in culture.11
In the CAM angiogenesis assay, 7 was the most potent compound with an IC50 value of 0.8 μM. The next most potent compounds were 2 and 13. These three analogues 2, 7 and 13 had no inhibitory activity against VEGFR-2, the principle mediator of angiogenesis, however 7 and 13 were the most potent analogues against PDGFRβ which has also shown importance in angiogenesis.22
Some of the analogues demonstrated dual RTK inhibitory activity. Compounds 3-5 all had good VEGFR-2 and PDGFRβ activity.
Compound 7 was chosen by the National Cancer Institute24 for evaluation in its preclinical 60 tumor cell line panel. Compound 7 had a two digit nanomolar GI50 against nine tumor cell lines, a submicromolar GI50 against twenty nine of the other tumor cell lines and micromolar GI50 against 16 tumor cell lines (Table 2). These results indicate selectivity for tumors within a class. For example, compound 7 was 8- to 10-fold more potent against CCRF-CEM and MOLT-4 leukemia cells than against other leukemia cells in culture (Table 2)
Table 2.
Tumor cell line inhibitory activity GI50 (nM) of 7(NCI).
| Panel/ Cell line | GI50 (nM) |
|---|---|
| Leukemia | |
| CCRF-CEM | 31.2 |
| HL-60(TB) | 150 |
| K-562 | 366 |
| MOLT-4 | 66.1 |
| RPMI-8226 | 489 |
| SR | 484 |
| NSCLC | |
| A549/ATCC | 43.4 |
| EKVX | 2710 |
| HOP-62 | 654 |
| HOP-92 | 18100 |
| NCI-H226 | 2140 |
| NCI-H23 | 565 |
| NCI-H322M | 1350 |
| NCI-H460 | 19.3 |
| NCI-H522 | 622 |
| Colon Cancer | |
| COLO 205 | 638 |
| HCT-116 | 38.1 |
| HCT-15 | 434 |
| KM12 | 330 |
| SW-620 | 63.0 |
| CNS Cancer | |
| SF-268 | 86.2 |
| SF-295 | 547 |
| SF-539 | 381 |
| SNB-19 | 113 |
| SNB-75 | 5030 |
| U251 | 184 |
| Prostate Cancer | |
| PC-3 | 715 |
| DU-145 | 212 |
| Melanoma | |
| LOX IMVI | 308 |
| MALME-3M | >10000 |
| M14 | 431 |
| SK-MEL-2 | 558 |
| SK-MEL-28 | 1510 |
| SK-MEL-5 | 513 |
| UACC-257 | 84.5 |
| UACC-62 | 641 |
| Ovarian cancer | |
| IGROVI | 287 |
| OVCAR-3 | 216 |
| OVCAR-4 | 2090 |
| OVCAR-8 | 216 |
| SK-OV-3 | 2090 |
| Renal Cancer | |
| 786 - 0 | 514 |
| A498 | 2490 |
| ACHN | 3420 |
| CAKI-1 | 1080 |
| RXF 393 | 2910 |
| SN12C | 189 |
| TK10 | 2150 |
| UO-31 | 790 |
| Breast Cancer | |
| MCF7 | 73.4 |
| NCI/ADR-RES | 787 |
| MDA-MB-231/ATCC | 641 |
| HS 578T | 2220 |
| MDA-MB-435 | 554 |
| BT-549 | 2830 |
| T-47D | 2900 |
Since RTK inhibition in most instances is only a cytostatic effect, it is interesting that compound 7 showed potent cytotoxic activity against the NCI 60 tumor cell lines. The mechanism of action of the potent tumor cytotoxicity of 7 must be due to mechanism(s) in addition to its RTK inhibitory activity, since most tumor cells in culture are not angiogenic. Thus, the NCI COMPARE analysis25 was performed for 7 to elucidate possible mechanism(s) of action of 7 by the similarity responses of the 60 cell lines to known compounds. The five compounds whose cell type selectivity profile showed the highest Pearson correlation coefficients (PCC)26 (Table 3) with 7 were all well known dihydrofolate reductase (DHFR) or dihydroorotate dehydrogenase (DHODH) inhibitors. Thus it was necessary to evaluate 7 and selected analogues for inhibitory activities against DHFR and DHODH.
Table 3.
COMPARE analysis data for compound 7.
| Drug | Correlation coefficient |
|---|---|
| Biphenquinate (DHODH inhibitor) | 0.745 |
| Trimetrexate (DHFR inhibitor) | 0.716 |
| Pyrazofurin (OMP decarboxylase inhibitor) | 0.682 |
| Acivicin (γ-glutamyl transpeptidase inhibitor) | 0.645 |
| Triazinate (DHFR inhibitor) | 0.635 |
Compounds 3, 7, 8, 12 and 13 were evaluated against DHFR27 and thymidylate synthase (TS)28 from human, Eshcerichia coli (E. coli) and Toxoplasma gondii (T. gondii) and the results are included in Table 4. None of these compounds showed activity against either DHFR or TS. The IC50 values of these compounds were all greater than 10 μM. Compounds 1, 4, 7, 8, 11 and 13 were also evaluated as inhibitors of DHODH from human (h) and Plasmodium falciparum (Pf).29 None of the analogues inhibited hDHODH or PfDHODH at IC50 less than 200 μM compared to the standard compound N-(4-trifluoromethylphenyl)-2-cyano-3-hydroxycrotonamide-2-cyano-3-hydroxy-N-[4-(trifluoromethyl)phenyl]-2-butenamide (A77-1726, Figure 1) 20 (data not shown). Thus the cytotoxic mechanism of action of 7 is currently under investigation.
Table 4.
IC50 value (M) of DHFR and TS inhibition for selected compounds.
| DHFR (IC50M) | TS (IC50 M) | |||||
|---|---|---|---|---|---|---|
| Human | E. coli | T. gondii | Human | E. coli | T. gondii | |
| 3 | >1.6×10−5 | >1.6×10−5 | >1.6×10−5 | >1.4×10−5 | >1.4×10−5 | >1.4×10−5 |
| 7 | >1.6×10−5 | >1.6×10−5 | >1.6×10−5 | >1.4×10−5 | >1.4×10−5 | >1.4×10−5 |
| 8 | >1.4×10−5 | >1.4×10−5 | >1.4×10−5 | >1.2×10−5 | >1.2×10−5 | >1.2×10−5 |
| 12 | >1.6×10−5 | >1.6×10−5 | >1.6×10−5 | >1.3×10−5 | >1.3×10−5 | >1.3×10−5 |
| 13 | >1.6×10−5 | >1.6×10−5 | >1.6×10−5 | >1.3×10−5 | >1.3×10−5 | >1.3×10−5 |
| MTX | 2.0×10−8 | 8.8×10−9 | 3.3×10−8 | |||
| Trimethoprim | >3.4×10−4 | 1.0×10−8 | 6.8×10−6 | |||
| Pyrimethamine | 6.0×10−6 | 6.6×10−6 | 2.0×10−7 | |||
| PDDF | 8.5×10−8 | 1.9×10−8 | 4.3×10−7 | |||
| Raltitrexed | 2.9×10−7 | 2.3×10−6 | 4.8×10−7 | |||
| Pemetrexed | 2.9×10−5 | 1.5×10−5 | 1.4×10−5 | |||
Compound 7 with its promising in vitro PDGFRβ inhibitory activity, CAM assay results and results from the preclinical NCI 60 tumor cell line panel, was evaluated in vivo for the tumor growth inhibition, antiangiogenic effects and metastasis of primary B16-F10 mouse melanoma tumor cells in athymic mice at a dose of 35 mg/kg 3 × weekly (M, W, F) for two weeks. (Z)-3-(2,4-Dimethyl-5-(2-oxo-1,2-dihydro-indol-3-ylidenemethyl)-1H-pyrrol-3-yl)-propionic acid 21 (Figure 1), a multi-targeted RTK inhibitor, was used as a standard. Compound 7 was equipotent with the standard 21 both in decreasing the growth rate of B16-F10 tumor cells (Figure 2, A) and inhibiting tumor angiogenesis (Figure 2, B). However, unlike standard 21, compound 7 did not decrease tumor metastasis activity compared to control (Figure 2, C).
Figure 2.

Growth rate of primary B16-F10 mouse melanoma tumor cells in athymic mice (A) and inhibition of tumor angiogenesis (B) and metastasis (C) in response to 21 and 7 given at 35 mg/kg 3 × weekly (M, W, F)
*** = P<0.001, **P<0.05 by one-way ANOVA and Neuman-Keuls post-test. N= 7–10. Average metastasis in untreated group- 2.5/lobe.
In summary a novel series of 4-anilino substituted tricyclic indeno[2,1-d]pyrimidines 1-13 were synthesized and biologically evaluated. Most of the analogues showed potent inhibitory activity against PDGFRβ compared to a standard 19. Compound 7 was about 4.5-fold more potent against PDGFRβ than 19. Compound 3 was equipotent with the standard semaxanib against VEGFR-2. Compound 3 was also a dual inhibitor of VEGFR-2 and PDGFRβ. Some analogues (1, 4, 7, 11, and 13) exhibited potent A431 cytotoxicity as well, compared to the standard, cisplatin. In addition, compound 7 inhibited most of the NCI 60 tumor cell lines with GI50s at nanomolar to micromolar levels. Thus, compound 7 possess both antiangiogenic and cytotoxic activity and could be used alone or in combination in cancer chemotherapy. The cytotoxic mechanism(s) of action of 7 is currently under investigation. The in vivo results showed that 7 at 35 mg/kg decreased tumor growth rate and inhibited angiogenesis similar to the standard compound 21 and much more than in the untreated control animals.
Experimental
General Methods for Synthesis
All evaporations were carried out in vacuo with a rotary evaporator. Analytical samples were dried in vacuo (0.2 mm Hg) in an Abderhalden drying apparatus over P2O5. Thin-layer chromatography (TLC) was performed on silica gel plates with fluorescent indicator. Spots were visualized by UV light (254 and 365 nm). All analytical samples were homogeneous on TLC in at least two different solvent systems. Purification by column and flash chromatography was carried out using Merck silica gel 60 (200–400 mesh). The amount (weight) of silica gel for column chromatography was in the range of 50-100 times the amount (weight) of the crude compounds being separated. Columns were dry packed unless specified otherwise. Solvent systems are reported as volume percent mixture. Melting points were determined on a Mel-Temp II melting point apparatus and are uncorrected. Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker WH-300 (300MHz) spectrometer. The chemical shift (δ) values are reported as parts per million (ppm) relative to tetramethylsilane as internal standard; s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, bs = broad singlet, exch = protons exchangeable by addition of D2O. Elemental analyses were performed by Atlantic Microlab, Inc., Norcross, GA. Elemental compositions were within ± 0.4% of the calculated values. Fractional moles of water or organic solvents frequently found in some analytic samples could not be removed despite 24 h of drying in vacuo and were confirmed, where possible, by their presence in the 1H NMR spectrum. The purity of some final compounds was determined by UPLC/UV/ELSD/MS (see supporting information for UPLC/UV/ELSD/MS method). Average purity of UV and ELSD was >95%.30 All solvents and chemicals were purchased from Aldrich Chemical Co. and Fisher Scientific and were used as received except anhydrous solvents which were freshly dried in the laboratory.
Ethyl 2-oxo-2,3-dihydro-1H-indene-1-carboxylate (15)
1,2-Phenylenediacetonitrile (3.0 g, 2 mmol) was dissolved in ethyl alcohol (5 mL) and conc. sulfuric acid (2 mL) in a 25 mL round bottom flask. The mixture was stirred and heated to reflux for 6 h. After neutralization with ammonium hydroxide in the cold, the reaction solution was extracted with ethyl acetate (3 × 50 mL). The organic phase was combined and dried with anhydrous Na2SO4. Evaporation of ethyl acetate afforded a yellow liquid. Without further purification, the yellow liquid was diluted in toluene (100 mL) in a 250 mL round bottom flask and then to the solution was added sodium metal (0.46 g, 2 mmol). The mixture was heated to reflux for 4 h. After the reaction solution was neutralized with dilute hydrochloric acid, the resulting solution was extracted with ethyl acetate (3 × 50 mL). The organic phase was combined and dried with anhydrous Na2SO4. Concentration of ethyl acetate afforded a brown solid. To this residue was added silica gel (3 g) and ethyl acetate (30 mL), and the solvent was evaporated to afford a plug. The silica gel plug obtained was load onto a silica gel column and eluted with 10:1 hexane/acetyl acetate. Fractions containing the product (TLC) were pooled, and the solvent was evaporated to afford 1.64 g (42% over two steps) of 15 as a white solid: TLC Rf 0.72 (Hexane/EtOAc, 5:1); mp 59–61 °C; 1H NMR (DMSO-d6) δ 3.64 (s, 2 H, Ar-CH2CO), 4.43 (q, 2 H, OCH2), 7.60 (m, 4 H, Ar-H), 11.0 (bs, 1 H, OH, exch).
2-Amino-3H-dihydro-indeno[2,1-d]pyrimidin-4(9H)-one (16)
Compound 15 (0.1 g, 0.49 mmol), guanidine hydrochloride (0.05 g, 0.52 mmol) and potassium t-butoxide (0.12 g, 1.1 mmol) were dissolved in t-butanol (5 mL). The condition of the microwave reaction was 150 °C for 4 h. The solid was filtered and washed with methanol. To the filtrate was added silica gel (300 mg), and the solvent was evaporated to afford a plug. The silica gel plug obtained was load onto a silica gel column and eluted with 10:1 chloroform/ methanol. Fractions containing the product (TLC) were pooled, and the solvent was evaporated to afford 23 mg (34%) of 16 as a light yellow solid: TLC Rf 0.56 (CHCl3/CH3OH, 5:1); mp: ~330 °C (dec.); 1H NMR (DMSO-d6) δ 3.65 (s, 2 H, CH2), 6.71 (s, 2 H, NH2, exch), 7.0-7.67 (m, 4 H, Ar-H), 10.93 (bs, 1 H, NH, exch). Anal. (C11H9N3O.0.1H2O): C, H, N.
2-Amino-9H-indeno[2,1-d]pyrimidin-4-yl 4-nitrobenzenesulfonate (17)
A solution of 16 (0.3 g, 1.5 mmol), triethylamine (0.42 mL, 3 mmol), DMAP (20 mg) and 4-nitrobenzenesulfonyl chloride (0.67 g, 3 mmol) in dichloromethane (40 mL) was stirred at room temperature for 4 h. To this solution was added silica gel (1.5 g), and the solvent was evaporated to afford a plug. The silica gel plug obtained was load onto a silica gel column and eluted with 2:1 hexane/chloroform. Fractions containing the product (TLC) were pooled, and the solvent was evaporated to afford 0.21 g (36%) of 17 as a yellow solid: TLC Rf 0.47 (CHCl3/CH3OH, 10:1); mp: ~189.6 °C (dec); 1H NMR (DMSO-d6) δ 3.92 (s, 2 H, CH2), 7.18 (bs, 2 H, NH2, exch), 7.15-7.64 (m, 4 H, Ph-H), 8.40-8.60 (m, 4 H, 4-NO2-Ph-H). HRMS (EI) cacd for C17H13N4O5S 385.0607; found, 385.0584.
General procedure for the synthesis of compounds 1-13
To a 50 mL round-bottom flask was added 17, the appropriate substituted aniline and anhydrous 1,4-dioxane (10 mL), the mixture was heated to reflux for 12–18 h. To the resulting solution was added silica gel, and the solvent was evaporated to afford a plug. The silica gel plug obtained was load onto a silica gel column and eluted with 2:1 hexane/chloroform. Fractions containing the product (TLC) were pooled, and the solvent was evaporated to afford pure compound.
N4-Phenyl-9H-indeno[2,1-d]pyrimidine-2,4-diamine (1)
Compound 1 was synthesized from 17 (0.05 g, 0.13 mmol) and aniline (24 mg, 0.26 mmol) using the general procedure described above to afford after purification 13.8 mg (37%) as a light brown solid: TLC Rf 0.37 (CHCl3/CH3OH, 10:1); mp: 215.3-216.9 °C; 1H NMR (DMSO-d6) δ 3.72 (s, 2 H, CH2), 6.41 (bs, 2 H, NH2, exch), 7.03-7.89 (m, 9 H, Ph-H), 8.22 (s, H, NH, exch). HRMS (EI) calcd. for C17H14N4 274.1218, found, 274.1218.
N4-(2-Isopropylphenyl)-9H-indeno[2,1-d]pyrimidine-2,4-diamine (2)
Compound 2 was synthesized from 17 (0.1 g, 0.26 mmol) and 2-isopropyl aniline (70 mg, 0.52 mmol) using the general procedure described above to afford after purification 15 mg (18%) as a light brown solid: TLC Rf 0.43 (Et3N/EtOAc/Hex, 1:3:5); mp 193.4-195.3 °C; 1H NMR (DMSO-d6) δ 1.14-1.16 (d, J = 6 Hz, 6 H, 2CH3), 3.10-3.24 (m, H, CH), 3.70 (s, 2 H, CH2), 6.13 (bs, 2 H, NH2, exch), 7.11-7.93 (m, 8 H, Ph-H), 7.91 (s, H, NH, exch). HRMS (EI) calcd. for C20H21N4 317.1766; found, 317.1751.
N4-(4-Isopropylphenyl)-9H-indeno[2,1-d]pyrimidine-2,4-diamine (3)
Compound 3 was synthesized from 17 (75 mg, 0.2 mmol) and 4-isopropyl aniline (54 mg, 0.4 mmol) using the general procedure described above to afford after purification 36.4 mg (59%) as a light brown solid: TLC Rf 0.30 (CHCl3/CH3OH, 10:1); mp 178.5-180.4 °C; 1H NMR (DMSO-d6) δ 1.16-1.18 (d, J = 6 Hz, 6 H, 2CH3), 2.64-2.87 (m, H, CH), 3.72 (s, 2 H, CH2), 6.30 (bs, 2 H, NH2, exch), 7.16-7.84 (m, 8 H, Ph-H), 8.10 (s, H, NH, exch). HRMS (EI) calcd. for C20H20N4 316.1688; found, 316.1704.
N4-(3-Fluorophenyl)-9H-indeno[2,1-d]pyrimidine-2,4-diamine (4)
Compound 4 was synthesized from 17 (0.15 g, 0.39 mmol) and 3-fluoroaniline (87 mg, 0.78 mmol) using the general procedure described above to afford after purification 15 mg (13%) as a light brown solid: TLC Rf 0.42 (CHCl3/CH3OH, 10:1); mp 198.7-200.1 °C; 1H NMR (DMSO-d6) δ 3.78 (s, 2 H, CH2), 6.56 (bs, 2 H, NH2, exch), 7.11-7.89 (m, 8 H, Ph-H), 8.37 (s, H, NH, exch). HRMS (EI) calcd. for C17H13FN4 292.1124; found, 292.1123.
N4-(4-Fluorophenyl)-9H-indeno[2,1-d]pyrimidine-2,4-diamine (5)
Compound 5 was synthesized from 17 (0.12 g, 0.3 mmol) and 4-fluoroaniline (67 mg, 0.6 mmol) using the general procedure described above to afford after purification 46 mg (50%) as a light brown solid: TLC Rf 0.54 (CHCl3/CH3OH, 10:1); mp 204.7-205.8 °C; 1H NMR (DMSO-d6) δ 3.72 (s, 2 H, CH2), 6.38 (bs, 2 H, NH2, exch), 7.11-7.89 (m, 8 H, Ph-H), δ 8.19 (s, H, NH, exch). Anal (C17H13FN4. 0.4H2O): C, H, N, F.
N4-(3-Chlorophenyl)-9H-indeno[2,1-d] pyrimidine-2,4- diamine (6)
Compound 6 was synthesized from 17 (0.1 g, 0.26 mmol) and 3-chloroaniline (66 mg, 0.52 mmol) using the general procedure described above to afford after purification 23 mg (35%) as a light brown solid: TLC Rf 0.65 (CHCl3/CH3OH, 10:1); mp 224.1-225.2 °C; 1H NMR (DMSO-d6) δ 3.73 (s, 2 H, CH2), 6.48 (bs, 2 H, NH2, exch), 7.06-7.84 (m, 8 H, Ph-H), 8.32 (s, H, NH, exch). HRMS (EI) calcd. for C17H13ClN4 308.0829; found, 308.0838.
N4-(4-Chlorophenyl)-9H-indeno[2,1-d]pyrimidine-2,4-diamine (7)
Compound 7 was synthesized from 17 (0.10 g, 0.26 mmol) and 4-chloroaniline (0.07 g, 0.78 mmol) using the general procedure described above to afford after purification 30 mg (38%) as a light brown solid: TLC Rf 0.33 (CHCl3/CH3OH, 10:1); mp 215.9-216.8 °C; 1H NMR (DMSO-d6) δ 3.76 (s, 2 H, CH2), 6.50 (bs, 2 H, NH2, exch), 7.14-7.92 (m, 8 H, Ph-H), 8.33 (s, H, NH, exch). Anal (C17H13ClN4): C, H, N, Cl.
N4-(3-Bromophenyl)-9H-indeno[2,1-d]pyrimidine-2,4-diamine (8)
Compound 8 was synthesized from 17 (0.1 g, 0.26 mmol) and 3-bromoaniline (89 mg, 0.52 mmol) using the general procedure described above to afford after purification 46 mg (50%) as a light brown solid: TLC Rf 0.47 (Et3N/EtOAc/Hex, 1:3:5); mp 233–236 °C; 1H NMR (DMSO-d6) δ 3.76 (s, 2 H, CH2), 6.54 (bs, 2 H, NH2, exch), 7.15-7.94 (m, 8 H, Ph-H), 8.35 (s, H, NH, exch). HRMS (EI) calcd. for C17H14BrN4 353.0402; found, 353.0387.
N4-(4-Bromophenyl)-9H-indeno[2,1-d]pyrimidine-2,4-diamine (9)
Compound 9 was synthesized from 17 (0.13 g, 0.34 mmol) and 4-bromoaniline (117 mg, 0.68 mmol) using the general procedure described above to afford after purification 72 mg (61%) as a white-off solid: TLC Rf 0.44 (CHCl3/CH3OH, 10:1); mp 213.6-214.8 °C; 1H NMR (DMSO-d6) δ 3.75 (s, 2 H, CH2), 6.45 (bs, 2 H, NH2, exch), 7.14-7.89 (m, 8 H, Ph-H), 8.31 (s, H, NH, exch). Anal (C17H13BrN4. 0.6CH3OH): C, H, N, Br.
N4-(4-(Trifluoromethyl)phenyl)-9H-indeno[2,1-d]pyrimidine-2,4-diamine (10)
Compound 10 was synthesized from 17 (0.22 g, 0.57 mmol) and 4-trifluoroaniline (184 mg, 1.04 mmol) using the general procedure described above to afford after purification 44 mg (27%) as a light brown solid: TLC Rf 0.48 (CHCl3/CH3OH, 10:1); mp 229.2-230.8 °C; 1H NMR (DMSO-d6) δ 3.78 (s, 2 H, CH2), 6.56 (bs, 2 H, NH2, exch), 7.18-8.02 (m, 8 H, Ph-H), 8.68 (s, H, NH, exch). Anal (C18H13F3N4): C, H, N, F.
N4-(3-Methoxyphenyl)-9H-indeno[2,1-d]pyrimidine-2,4-diamine (11)
Compound 11 was synthesized from 17 (75 mg, 0.76 mmol) and m-anisidine (0.1 mL) using the general procedure described above to afford after purification 27 mg (45%) as a light brown solid: TLC Rf 0.47 (CHCl3/CH3OH, 10:1); mp186.6- 187.4 °C; 1H NMR (DMSO-d6) δ 3.74 (s, 2 H, CH2), 3.82 (s, 3 H, CH3), 6.44 (bs, 2 H, NH2, exch), 6.62-7.84 (m, 8 H, Ph-H), 8.08 (s, H, NH, exch). HRMS (EI) calcd. for C18H16N4O 304.1324, found, 304.1390.
N4-(3-Chloro-4-Fluorophenyl)-9H-indeno[2,1-d]pyrimidine-2,4-diamine (12)
Compound 12 was synthesized from 17 (0.12 g, 0.31 mmol) and 3-chloro-4-fluoroaniline (0.09 g, 0.62 mmol) using the general procedure described above to afford after purification 53 mg (52%) as a light brown solid: TLC Rf 0.43 (CHCl3/CH3OH, 10:1); mp 231.7-232.8 °C; 1HNMR (DMSO-d6) δ 3.74 (s, 2 H, CH2), 6.47 (bs, 2 H, NH2, exch), 7.14-8.02 (m, 7 H, Ph-H), 8.28 (s, H, NH, exch). HRMS (EI) calcd. for C17H12ClFN4 326.0735, found, 326.0744.
N4-(4-Chloro-2-fluorophenyl)-9H-indeno[2,1-d]pyrimidine-2,4-diamine (13)
Compound 13 was synthesized from 17 (50 mg, 0.59 mmol) and 4-chloro-2-fluoroaniline (0.06 mL) using the general procedure described above to afford after purification 15 mg (18%) as a light brown solid: TLC Rf 0.58 (Et3N/EtOAc/Hex, 1:3:5); mp 234.7-235.7 °C; 1H NMR (DMSO-d6) δ 3.98 (s, 2 H, CH2), 6.37 (bs, 2 H, NH2, exch), 7.15-7.86 (m, 7 H, Ph-H), 8.16 (s, H, NH, exch). Anal. (C17H12ClFN4): C, H, N, Cl, F.
General Methods for Biological Evaluations
Receptor tyrosine kinase evaluations
Cells
All cells were maintained at 37 ° C in a humidified environment containing 5% CO2 using media from Mediatech (Hemden, NJ). A-431 cells were from the American Type Tissue Collection (Manassas, VA).
Chemicals
All growth factors (VEGF, EGF, and PDGF-BB) were purchased from Peprotech (Rocky Hill, NJ). Compound 18 and semaxanib were purchased from Calbiochem (San Diego, CA). The CYQUANT cells proliferation assay was from Molecular Probes (Eugene, OR). All other chemicals were from Sigma Chemical unless otherwise noted.
Antibodies
The PY-HRP antibody was from BD Transduction Laboratories (Franklin Lakes, NJ). Antibodies against EGFR, PDGFR-β, and VEGFR-2 were purchased from Upstate Biotech (Framingham, MA).
Phosphotyrosine ELISA
Cells used were tumor cell lines naturally expressing high levels of EGFR (A431), VEGFR-2 (U251), and PDGFR-β (SF-539). Expression levels at the RNA level were derived from the NCI Developmental Therapeutics Program (NCI-DTP) web site public molecular target information (http://www.dtp.nci.nih.gov.authenticate.library.duq.edu/mtargets/mt_index.htmlhttp://www.dtp.nci.nih.gov.authenticate.library.duq.edu/mtargets/mt_index.html). Briefly, cells at 60–75% confluence are placed in serum-free medium for 18 h to reduce the background of phosphorylation. Cells were always >98% viable by Trypan blue exclusion. Cells are then pretreated for 60 min with 10, 3.33, 1.11, 0.37, and 0.12 μM compound followed by 100 ng/mL EGF, VEGF, or PDGF-BB for 10 min. The reaction is stopped and cells permeabilized by quickly removing the media from the cells and adding ice-cold Tris-buffered saline (TBS) containing 0.05% Triton X-100, protease inhibitor cocktail and tyrosine phosphatase inhibitor cocktail. The TBS solution is then removed and cells fixed to the plate for 30 min at 60 °C and further incubation in 70% ethanol for an additional 30 min. Cells are further exposed to block (TBS with 1% BSA) for 1 h, washed, and then a horseradish peroxidase (HRP)-conjugated phosphotyrosine (PY) antibody added overnight. The antibody is removed, cells are washed again in TBS, exposed to an enhanced luminol ELISA substrate (Pierce Chemical, Rockford, IL) and light emission measured using a UV products (Upland, CA) BioChemi digital darkroom. The known RTK-specific kinase inhibitor 18 was used as a positive control compound for EGFR kinase inhibition; semaxanib for VEGFR-2 kinase inhibition; 19 for PDGFR-β kinase inhibition. Data were graphed as a percent of cells receiving growth factor alone and IC50 values were calculated from two to three separate experiments (n = 8–24) using non-linear regression dose-response relation analysis.
CYQUANT cell proliferation assay
As a measure of cell proliferation, the CYQUANT cell counting/proliferation assay was used as previously described. 29 Briefly, cells are first treated with compounds for 12 h and then allowed to grow for an additional 36 h. The cells are then lysed and the CYQUANT dye, which intercalates into the DNA of cells, is added and after 5 min the fluorescence of each well measured using an UV products BioChemi digital darkroom. A positive control used for cytotoxicity in each experiment was cisplatin, with an apparent average IC50 value about 8.2 ± 0.65 μM. Data are graphed as a percent of cells receiving growth factor alone and IC50 values calculated from two to three separate experiments (n = 6–15) using nonlinear regression dose-response relation analysis.
Chorioallantoic membrane assay of angiogenesis
The CAM assay is a standard assay for testing antiangiogenic agents.32 The CAM assay used in these studies was modified from a procedure by Sheu33 and Brooks34 and as published previously.35 Briefly, fertile leghorn chicken eggs (CBT Farms, Chestertown, MD) are allowed to grow until 10 days of incubation. The proangiogenic factors, human VEGF-165 and bFGF (100 ng each) are then added saturation to a 6 mm microbial testing disk (BBL, Cockeysville, MD) and placed onto the CAM by breaking a small hole in the superior surface of the egg. Antiangiogenic compounds are added 8 h after the VEGF/bFGF at saturation to the same microbial testing disk and embryos allowed to incubate for an additional 40 h. After 48 h, the CAMs are perfused with 2% paraformaldehyde/3% glutaraldehyde containing 0.025% Triton X-100 for 20 s, excised around the area of treatment, fixed again in 2% paraformaldehyde/3% glutaraldehyde for 30 min, placed on petri dishes, and a digitized image taken using a dissecting microscope (Wild M400; Bannockburn, IL) at 7.5X and SPOT enhanced digital imaging system (Diagnostic Instruments, Sterling Heights, MI). A grid is then added to the digital CAM images and the average number of vessels within 5–7 grids counted as a measure of vascularity. AGM-1470 (a kind gift of the NIH Developmental Therapeutics Program) and semaxanib are used as a positive control for antiangiogenic activity. Data are graphed as a percent of CAMs receiving bFGF/VEGF and IC50 values calculated from two to three separate experiments (n = 5–11) using non-linear regression dose-response relation analysis.
Statistics
All analysis was done using Prism 4.0. (GraphPad Software, San Diego, CA). Tumor growth rates were assessed during the linear growth period and statistical significance of tumor growth between groups was calculated using two-way repeated measures ANOVA with treatments and days after implantation as independent variables with Dunnett’s post-test with the null hypothesis rejected when P < 0.05. Tumor metastases were compared using one-way ANOVA with Bonferonni’s multiple comparison post-test with the null hypothesis rejected when P < 0.05. IC50 values were calculated using a non-linear dose-response relation algorithm and IC50 values compared to one another using one-way ANOVA with Neuman-Keuls post test with the null hypothesis rejected when P < 0.05.
B16-F10 syngeneic mouse melanoma model
50,000 B16-F10 (lung colonizing) mouse melanoma cells were injected orthotopically SQ just behind the ear of athymic NCr nu/nu mal mice, 8 week in age. First, a dose-finding study was done with 5, 10, 15, 20, 25, 35 and 50 mg/kg compound 7 given three times weekly for 4 weeks to 3 animals per treatment group. The dose of 35 mg/kg compound 7 was found to result in no apparent toxicity and no significant loss in weight over the 4 week period. Two experiments were done starting with 5-6 animals/group. Animals were monitored every other day for the presence of tumors. At the time in which most tumors were measurable by callipers (day 9 for this experiment), animals with tumors were randomly sorted into treatment groups and treatment with drugs begun. DMSO stocks (30 mM ) of drugs were further dissolved into sterile water for injection and 35 mg/kg injected intraperitoneally (IP) every Monday (AM), Wednesday (noon) and Friday (PM). Sham treated animals received water only Monday, Wednesday and Friday. The length (long side), width (short side) and depth of the tumors were measured using digital Vernier Caliers each Monday, Wednesday, and Friday. Tumor volume was calculated using the formula length × width × depth. Tumor growth rate was calculated using a linear regression analysis algorithm using the software GraphPad Prism 4.0.c. at the experiment’s end, animals were humanely euthanized using carbon dioxide, tumors and lungs excised, fixed in 20% neutral buffered formalin for 8-10 h, embedded into paraffin, and haematoxylin-eosin (H&E) stain of three separate tissue sections completed to span the tumor/lung. Together with the OUHSC Department of Pathology core, metastases per lung lobe counted using the H&E stained sections. The metastases can be seen as purple clusters of disorganized cells on the highly organized largely pink lung. Together with the OUHSC Department of Pathology core, blood vessels per unit area were counted in 5 fields at 100× magnification and averaged. Tumor growth rates were compared statistically using two-way ANOVA with a Repeated Measures post-test and tumor vascularity and metastases were compared using one-way ANOVA and a Neuman-Keuls post test with the null hypothesis rejected when P<0.05.
Supplementary Material
Acknowledgments
We thank Dr. Margaret Phillips (Department of Pharmacology, University of Texas Southwestern Medical Center, Dallas, Texas) for carrying out the DHODH assays. This work was supported, in part, by the National Institutes of Health, National Cancer Institute grant CA 098850 (AG) and the Duquesne University Adrian Van Kaam Chair in Scholarly Excellence (AG).
aAbbreviations
- RTK
receptor tyrosine kinase
- PDGFRβ
platelet-derived growth factor receptor β
- FGFR
fibroblast growth factor receptor
- VEGFR
vascular epidermal growth factor receptor
- IGFR
insulin growth factor receptor
- EGFR
epidermal growth factor receptor
- CAM
chicken embryo chorioallantonic membrane
- PCC
Pearson correlation coefficients
- DHFR
dihydrofolate reductase
- DHODH
dihydroorotate dehydrogenase
Footnotes
Supporting Information Available: Results from elemental analysis, high resolution mass spectra and UPLC/UV/ELSD/MS method and data is available free of charge.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Carmeliet P. Nat Med. 2003;9:653. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 2.Folkman J. Nat Rev Drug Discov. 2007;6:273. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- 3.Naumov GN, Akslen LA, Folkman J. Cell Cycle. 2006;5:1779. doi: 10.4161/cc.5.16.3018. [DOI] [PubMed] [Google Scholar]
- 4.Herbst RS, Johnson DH, Mininberg E, Carbone DP, Henderson T, Kim ES, Blumenschein G, Jr, Lee JJ, Liu DD, Truong MT, Hong WK, Tran H, Tsao A, Xie D, Ramies DA, Mass R, Seshagiri S, Eberhard DA, Kelley SK, Sandler A. J Clin Oncol. 2005;23:2544. doi: 10.1200/JCO.2005.02.477. [DOI] [PubMed] [Google Scholar]
- 5.Pluda JM. Semin Oncol. 1997;24:203. [PubMed] [Google Scholar]
- 6.Sun L, McMahon G. Drug Discovery Today. 2000;8:344. doi: 10.1016/s1359-6446(00)01534-8. [DOI] [PubMed] [Google Scholar]
- 7.Eskens F. Br J Cancer. 2004;90:1. doi: 10.1038/sj.bjc.6601401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li J, Kleeff J, Giese N, Büchler MW, Korc M, Friess H. Int J Oncol. 2004;25:203. [PubMed] [Google Scholar]
- 9.Comis RL. Oncologist. 2005;5:1280. doi: 10.1634/theoncologist.10-7-467. [DOI] [PubMed] [Google Scholar]
- 10.Gild ML, Bullock M, Robinson BG, Clifton-Bligh R. Nature Rev Endocrinol. 2011;7:617–624. doi: 10.1038/nrendo.2011.141. [DOI] [PubMed] [Google Scholar]
- 11.a) US Food and Drug Administration. FDA approves new treatment for gastrointestinal and kidney cancer. Available at: http://www.fda.gov/bbs/topics/news/2006/NEW01302.html.; b) Mandel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, Schreck RE, Abrams TJ, Ngai TJ, Lee LB, Murray LJ, Carver J, Chan E, Moss KG, Haznedar JO, Sukbuntherng J, Blake RA, Sun L, Tang C, Miller T, Shirazian S, McMahon G, Cherrington JM. Clin Cancer Res. 2003;9:327. [PubMed] [Google Scholar]
- 12.Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, Cao Y, Shujath J, Gawlak S, Eveleigh D, Rowley B, Liu L, Adnane L, Lynch M, Auclair D, Taylor I, Gedrich R, Voznesensky A, Riedl B, Post LE, Bollag G, Trail PA. Cancer Res. 2004;64:7099. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 13.Traxler P, Furet P. Pharmacol Ther. 1999;82:195. doi: 10.1016/s0163-7258(98)00044-8. [DOI] [PubMed] [Google Scholar]
- 14.Hudes G. J Clin Oncol. 1999;17:1093. doi: 10.1200/JCO.1999.17.4.1093. [DOI] [PubMed] [Google Scholar]
- 15.Shaheen RM, Tseng WW, Davis DW, Liu W, Reinmuth N, Vellagas R, Wieczorek AA, Ogura Y, McConkey DJ, Drazan KE, Bucana CD, McMahon G, Ellis LM. Cancer Res. 2001;61:1464. [PubMed] [Google Scholar]
- 16.Levitt ML, Koty PP. Invest New Drugs. 1999;17:213. doi: 10.1023/a:1006372102543. [DOI] [PubMed] [Google Scholar]
- 17.Rusnak DW, Lackey K, Affleck K, Wood ER, Alligood KJ, Rhodes N, Keith BR, Murray DM, Knight WB, Mullin RJ, Gilmer TM. Mol Cancer Therap. 2001;1:86. [PubMed] [Google Scholar]
- 18.Traxler P, Bold G, Buchdunger E, Caravatti G, Furet P, Manley P, O’Reilly T, Wood J, Zimmermann J. Med Res Rev. 2001;21:499. doi: 10.1002/med.1022. [DOI] [PubMed] [Google Scholar]
- 19.Gangjee A, Zaware N, Raghavan S, Ihnat M, Shenoy S, Kisliuk RL. J Med Chem. 2010;53:1563. doi: 10.1021/jm9011142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Showalter HD, Hollis Bridges, Alexander J, Hairong Zhou, Sercel Anthony D, McMichael Amy, Fry David W. J Med Chem. 1999;42:5464. doi: 10.1021/jm9903949. [DOI] [PubMed] [Google Scholar]
- 21.Gangjee A, Zhao Y, Raghavan S, Ihnat MA, Disch BC. Bioorg Med Chem. 2010;18:5261. doi: 10.1016/j.bmc.2010.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blume-Jensen P, Hunter T. Nature. 2001;411:355. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 23.Gangjee A, Namjoshi OA, Ihnat MA, Buchanan A. Bioorg Med Chem Ltrs. 2010;20:3177–3181. doi: 10.1016/j.bmcl.2010.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.We thank the National Cancer Institute for performing the in vitro antitumor evaluation in their 60 tumor preclinical screening program.
- 25.Boyd MR. In: Cancer: Principles and Practice of Oncology. DeVita VT Jr, Hellman S, Resenberg SA, editors. Vol. 3. Lippincott; Philadelphia, PA: 1989. pp. 1–12. [Google Scholar]; b.) Boyd MR, Paull KD, Rubinstein LR. In: Cytotoxic Anticancer Drugs: Models and Concepts for Drug Discovery and Development. Vleriote FA, Corbett TH, Baker LH, editors. Kluwer Academic; Hingham, MA: 1992. pp. 11–34. [Google Scholar]
- 26.a) Bai R, Paull KD, Hearld CL, Pettit GR, Hamel E. Halichondrin B and Homohalichondrin B. J Biol Chem. 1991;266:15882. [PubMed] [Google Scholar]; b) Paull KD, Lin CM, Malspeis L, Hamel E. Cancer Res. 1992;52:3892. [PubMed] [Google Scholar]
- 27.Kisliuk RL, Strumpf D, Gaumont Y, Leary RP, Plante L. J Med Chem. 1977;20:1531. doi: 10.1021/jm00221a038. [DOI] [PubMed] [Google Scholar]
- 28.Wahba AJ, Friedkin M. J Biol Chem. 1962;237:3794. [PubMed] [Google Scholar]
- 29.a) Baldwin J, Farajallah AM, Malmquist NA, Rathod PK, Phillips MA. J Biol Chem. 2002;277:41827. doi: 10.1074/jbc.M206854200. [DOI] [PubMed] [Google Scholar]; b) Baldwin J, Michnoff CH, Malmquist NA, White J, Roth MG, Rathod PK, Phillips MA. J Biol Chem. 2005;280:21847. doi: 10.1074/jbc.M501100200. [DOI] [PubMed] [Google Scholar]
- 30.Lemoff A, Yan B. J Comb Chem. 2008;10:746–751. doi: 10.1021/cc800100g. [DOI] [PubMed] [Google Scholar]
- 31.Wilson SM, Barsoum MJ, Wilson BW, Pappone PA. Cell Prolif. 1999;32:131. doi: 10.1046/j.1365-2184.1999.32230131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vu MT, Smith CF, Burger PC, Klintworth GK. Lab Invest. 1985;53:499. [PubMed] [Google Scholar]
- 33.Sheu JR, Fu CC, Tsai ML, Chung WJ. Anticancer Res. 1998;18:4435. [PubMed] [Google Scholar]
- 34.Brooks PC, Montgomery AM, Cheresh DA. Methods Mol Biol. 1999;129:257. doi: 10.1385/1-59259-249-X:257. [DOI] [PubMed] [Google Scholar]
- 35.Marks MG, Shi J, Fry MS, Xiao Z, Trzyna M, Pokala V, Ihnat MA, Li PK. Biol Pharm Bull. 2002;25:597. doi: 10.1248/bpb.25.597. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.