

Abstract
Oxidative stress plays a crucial role in disruption of neovascularization by alterations in thioredoxin-1 (Trx1) expression and its interaction with other proteins after myocardial infarction (MI). We previously showed that Trx1 has angiogenic properties, but the possible therapeutic significance of overexpressing Trx1 in chronic MI has not been elucidated. Therefore, we explored the angiogenic and cardioprotective potential of Trx1 in an in vivo MI model using transgenic mice overexpressing Trx1. Wild type (W) and Trx1 transgenic (Trx1Tg/+) mice were randomized into W Sham (WS), Trx1Tg/+ Sham (TS), WMI and TMI. MI was induced by permanent occlusion of LAD coronary artery. Hearts from mice overexpressing Trx1 exhibited reduced fibrosis and oxidative stress, and attenuated cardiomyocyte apoptosis along with increased vessel formation compared to WMI. We found significant inhibition of Trx1 regulating proteins, TXNIP and AKAP 12, and increased p-Akt, p-eNOS and p-GSK-3β, HIF-1α, β-catenin, VEGF, Bcl-2 and survivin expression in TMI compared to WMI. Echocardiography performed 30 days after MI revealed significant improvement in myocardial functions in TMI compared to WMI. Our study identifies a potential role for Trx1 overexpression and its association with its regulatory proteins TXNIP, AKAP12 and subsequent activation of Akt/GSK-3β/β-catenin/HIF-1α-mediated VEGF and eNOS expression in inducing angiogenesis and reduced ventricular remodeling. Hence, Trx1 and other proteins identified in our study may prove to be potential therapeutic targets in the treatment of ischemic heart disease.
Keywords: Apoptosis, Myocardial infarction, Neovascularization, Oxidative stress, Thioredoxin1 and Ventricular remodeling
INTRODUCTION
Myocardial infarction (MI) remains as a major cause of morbidity and mortality worldwide. Although, MI triggers a spontaneous angiogenic response which aims at reestablishing myocardial blood flow, this protective response is usually not sufficient to restore physiological levels of coronary perfusion. Hence, therapeutic angiogenesis by using biological factors such as vascular endothelial growth factor (VEGF) or angiopoietin-1 have emerged as an attractive treatment option to restore blood flow and thus the supply of oxygen and nutrients to the ischemic regions of the heart after MI [1, 2]. Accumulating evidence supports the notion that thioredoxin1 (Trx1), with its antioxidant, growth stimulatory, anti-inflammatory and pro-angiogenic properties could play a role in modulating the pathological processes involved in cardiovascular disease progression and therefore might represent a promising therapeutic target for the prevention and treatment of the disease [3, 4].
Trx1 is a cytosolic 12 kDa redox protein with important antioxidative and cell signaling functions that undergoes reversible oxidation of its two active site cysteine residues at its active site where reduction of which is catalyzed by the NADPH-dependent flavoenzyme thioredoxin reductase [5]. The activity of Trx1 is found to be regulated through its interaction with Thioredoxin Interacting Protein (TXNIP). Thus, the ratio of Trx1 to TXNIP expression is an important factor in redox-sensitive regulation of myocardial angiogenesis [6, 7]. We have previously reported that the Ad-Trx1 gene therapy is cardioprotective in the ischemic myocardium of streptozotocin-induced diabetes rat by reducing oxidative stress and apoptosis with a corresponding improvement in myocardial functions [3]. Furthermore, other studies demonstrated that elevated Trx1 levels attenuate myocardial damage induced by ischemia–reperfusion injury while the overexpression of TXNIP has been shown to increase cardiomyocytes sensitivity to oxidative stress-induced apoptosis [7].
A kinase anchoring proteins (AKAPs), which compose a growing list of diverse but functionally related proteins defined by their ability to bind to the regulatory subunit of protein kinase A. AKAPs perform an integral role in the spatiotemporal modulation of a multitude of cellular signaling pathways [8]. Among all the AKAPs found so far, AKAP12 was found to be critical regulator of angiogenesis [9]. Exposure of endothelial cells to AKAP12 significantly reduced VEGF mRNA and increased anti-angiogenic proteins such as thormbospondin-1 [10]. AKAP12 is also known to cause significant downregulation of hypoxia inducible factor (HIF)- 1α and thereby reduce the hypoxia induced VEGF expression [11]. Furthermore, Trx1 may play a crucial role in regulating Akt signaling [12]. The Akt survival pathway is a central signaling node involved in cell growth, proliferation, differentiation, apoptosis and angiogenesis [13]. Akt also affects cell survival by phosphorylating and inactivating GSK-3β, which in turn results in the release, stabilization, and subsequent accumulation of β-catenin in the cytosol, followed by its translocation into the nucleus [14, 15]. Conversely, the phosphorylation of β-catenin marks this protein for ubiquitination and subsequent degradation by the proteosomal pathway [16]. Therefore, when phosphorylation of β-catenin is blocked, it stabilizes, accumulates, and translocates into the nucleus, where it forms a complex with T-cell transcription factors/lymphoid-enhancer binding factor and is able to activate or repress several important angiogenic target genes, such as c-Myc, cyclin D1, fibronectin, VEGF, eNOS, Bcl-2, and survivin [15, 17].
We hypothesized that myocardial angiogenesis is regulated by a complex interplay between pro-angiogenic signaling and anti-angiogenic signaling under the redox control by Trx1. Therefore, the aim of the present study was to establish the role of Trx1 as therapeutic angiogenic growth factor by investigating the possible role of Trx1 overexpression in attenuating oxidative stress, TXNIP and AKAP12 expression, and inducing Akt/GSK-3β/HIF-1α/β-catenin-mediated VEGF and eNOS expression during myocardial infarction by using Trx1 transgenic mice.
METHODS
Experimental Animals
The present study was performed in accordance with the principles of laboratory animal care formulated by the National Society for Medical Research and the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health (Publication No. 85–23, revised 1985). The experimental protocol was examined and approved by the Institutional Animal Care Committee of the Connecticut Health Center (Farmington, CT, USA). Eight-week-old male TrxTg/+ overexpressing C57BL/6 background mice and respective wild type mice were used for the study. The generation, characterization and maintenance of TrxTg/+ mice have been described in supplemental material (Suppl. Fig. 1). Further, the genotype of each animal was confirmed by polymerase chain reaction analysis on purified ear DNA. Moreover, the overexpression of Trx1 in transgenic mice is also shown by Western blot in Fig. 3A.
Fig. 3. Trx1 overexpression altered the expression of TXNIP, AKAP12, VEGF, Bcl-2 and survivin during ischemic stress.

Representative Western blots show the expression of Trx1, TXNIP, AKAP12, VEGF, Bcl-2 and survivin, and their corresponding loading control-GAPDH. Western blot analysis revealed downregulation of AKAP12 (Fig. 3B) and TXNIP (Fig. 3C), whereas increased VEGF (Fig. 3D), Bcl-2 (Fig. 3E) and survivin (Fig. 3F) expression in TMI compared to the WMI. Numbers below the bands represent the average-fold change compared to WS from 3–5 independent experiments. Trx1 representative blot (Fig. 3A) is given for the validation of overexpression of Trx1 in transgenic animals. Abbreviations are as given in Fig. 1.
Experimental design
Eight-week-old male Trx1Tg/+ and respective wild type mice were randomized into four groups: (1) wild type sham (WS); (2) Trx1Tg/+ Sham (TS); (3) wild type MI (WMI) and 4) Trx1Tg/+ MI (TMI). MI was induced by permanent left anterior descending (LAD) coronary artery ligation. Sham groups underwent the same time matched surgical procedure without ligation. The apoptosis and β-catenin nuclear translocation were measured 24 h after surgical intervention, whereas the protein expression profile for the Trx1, TXNIP, AKAP12, VEGF, Bcl-2 and survivin and oxidative stress index was measured in the left ventricular tissue (risk area) 4 days after MI. The extent of phosphorylation of eNOS, Akt and GSK-3β, β-catenin and HIF-1α DNA binding activity was observed 8 hrs after MI. The immunohistostaining was performed to reflect the degree of angiogenesis by calculating the capillary and arteriolar density after 7 days of surgical intervention. The cardiac function was assessed by echocardiogram 30 days after MI.
Surgical procedures
Mice were anesthetized with ketamine (100 mg/kg, ip) and xylazine (10 mg/kg, ip) dissolved in physiological saline, then orally intubated with a 22G IV catheter, and ventilated with a rodent respirator (Harvard Apparatus, Hilliston, USA). Hearts were then exposed through the left lateral thoracotomy. MI was created by permanent LAD ligation with 8-0 polypropylene suture viewed under a stereo zoom dissection microscope. The lungs were inflated by positive end-expiratory pressure and the chest was closed with 6.0 nylon suture. After surgery, the analgesic buprenorphine (0.1 mg/kg, sc) was given, and the animals were weaned from the respirator and then placed on a heating pad for recovery [18, 19].
Assessment of cardiac fibrosis (Masson’s trichrome staining)
To determine the effect of Trx1 overexpression on cardiac fibrosis, the collagenous fibrotic area of the heart was stained by Masson’s Trichrome staining in paraffin embedded sections (of 4 μm thick) [20]. In brief, the sections were deparaffinized in histoclear and rehydrated using sequential passage through 100 to 70% ethanol for 6 min each followed by washing in distilled water three times. The slides were then stained with Weigert’s iron hematoxylin for 10 min and washed under tap water for 10 min. The sections were washed again in distilled water and then stained with Biebrich scarlet-acid fuschin solution for 15 min, in phosphomolybdic-phosphotungstic acid solution for 15 min and aniline blue solution and stained for 10 min. The sections were rinsed briefly in distilled water and were treated with 1% acetic acid solution for 5 min. After a final wash in distilled water the sections were dehydrated through sequential gradient of 70–100% alcohol followed by histoclear wash and then mounted using Permount. The heart tissue sections were digitally imaged in high pixel resolution on an Epson Scanner.
Assessment of oxidative stress by TBARS estimation
The extent of oxidative stress in the heart was determined by measuring the levels of thiobarbituric acid reactive substances (TBARS) (Niehaus and Samuelsson [21] method). To 0.5 ml of tissue homogenate (known weight of the myocardium tissue from risk area was homogenized at 3000 rpm in 0.5 ml of 0.025M Tris-Hcl, pH 7.5), 1 ml of TBA:TCA:HCl in the ratio of 1:1:1 (0.37% thiobarbituric acid:15% trichloroacetic acid:0.25 N HCl) was mixed and placed in a boiling water bath for 15 minutes, cooled and centrifuged at room temperature for 10 min at 1,000 rpm. A series of standard solutions (tetramethoxy propane, Sigma-Aldrich, St. Louis, MO) in the range of 2–10 nmole were treated in a similar manner. The absorbance of the clear supernatant was measured against a reference blank at 535 nm. Results were expressed as nM/mg tissue.
Cardiomyocyte apoptosis
Formaldehyde-fixed left ventricle was embedded in paraffin, cut into transverse sections (4 μm thick), and deparaffinized with a graded series of histoclear and ethanol solutions. Immunohistochemical detection of apoptotic cells was carried out using TUNEL reaction using an In Situ Cell Death Detection Kit and fluorescein as per the kit protocol (Roche Diagnostics, Mannheim, Germany). In brief, the TUNEL reaction preferentially labels DNA strand breaks generated during apoptosis, which can be identified by labeling free 3′-OH termini with modified nucleotides in an enzymatic reaction catalyzed by terminal deoxynucleotidyltransferase (TdT). Fluorescein labels incorporated in nucleotide polymers are detected by fluorescence microscopy. The sections were washed three times in PBS, blocked with 10% normal goat serum in PBS, and incubated with mouse monoclonal sarcomeric actin (Sigma, St. Louis, MO) followed by staining with Alexa Fluor 555 donkey anti-mouse IgG (1:200 dilution, Invitrogen, Carlsbad, CA). After incubation, the sections were rinsed 3 times in PBS and mounted with Vectashield mounting medium (Vector, Burlingame, CA). The sections were observed, and images were captured using a confocal laser Zeiss LSM 510 microscope. For quantitative purposes, the number of TUNEL-positive cardiomyocytes was counted in 15–20 HPF from border zones of heart sections and normalized to 100 HPF [22].
Capillary and arteriolar density
Capillary density and arteriolar density was performed 7 days after the surgical procedure by immunohistochemistry. Mice were sacrificed, hearts removed, and paraffin-embedded sections prepared. Endothelial cells were labeled using goat polyclonal anti-CD31 (1:100 dilution, Santa Cruz Biotechnology, Santa Cruz, CA) followed by a biotinylated horse anti-goat secondary antibody (1:200 dilution, Vector Laboratories). The reaction product (brown) was visualized with 3,3′-diaminobenzidine substrate using the Vector ABC Vectastain Elite Kit (Vector Laboratories, Burlingame, CA). On separate slides, vascular smooth muscle cells were labeled using rabbit polyclonal anti-alpha smooth muscle actin (1:100, Abcam, Cambridge, MA) followed by Alexa Fluor 488 donkey anti-rabbit secondary antibody (1:200 dilution, Invitrogen, Carlsbad, CA). After incubation, the sections were again washed in PBS three times and mounted with a coverslip using mounting medium (Vector Laboratories). Images were captured and stored in digital Tiff file format for later image analysis. Counts of capillary and arteriolar density per square millimeter were obtained after superimposing a calibrated morphometric grid on each digital image using Adobe Photoshop Software [23].
Western blot analysis
To quantify TXNIP, AKAP12, eNOS, p-Akt, p-GSK-3β, β-catenin, VEGF, Bcl-2 and survivin, standard SDS–PAGE Western blot technique was performed as described previously [24]. Cytosolic and nuclear protein were prepared according to the kit protocol of the CelLytic NuCLEAR Extraction Kit from Sigma, St. Louis, MO and protein concentration was determined using a bicinchoninic acid protein assay kit (Pierce, Rockville, IL). The proteins were run on SDS-polyacrylamide electrophoresis gels typically using 10% for TXNIP (Invitrogen, Carlsbad, CA), p-Akt (Serine 473), p-GSK-3β (Serine 9) (Cell Signaling Technologies, Danvers, MA), β-catenin (Santa Cruz Biotechnology, Santa Cruz, CA), VEGF (R&D systems, Minneapolis, MN), and Bcl-2 (Santa Cruz Biotechnology, Santa Cruz, CA), 12% for survivin (Abcam, Cambridge, MA) and 7% for AKAP12 (Sigma, St. Louis, MO) and p-eNOS (Ser 1177) (Cell Signaling Technologies, Danvers, MA).
Immunohistochemistry for β-catenin nuclear translocation
The nuclear translocation of β-catenin was performed after 24h of surgical intervention in the paraffin-embedded heart sections [16]. Myocardial sections were analyzed for the reactivity with 1:100 diluted monoclonal antibody against β-catenin (produced in rabbit) (Santa Cruz Biotechnology, Santa Cruz, CA). Bound antibody was detected with the Vectastain ABC kit (Vector Laboratories, Burlingame, CA) and visualized with DAB and the nucleus was counter stained with haematoxylin (Sigma, St. Louis, MO) and observed under light microscope.
Electrophoretic Mobility Shift Assay (EMSA) analysis for HIF-1α DNA binding activity
EMSA was performed by using 5 μg of the nuclear extracts and extract was incubated for 20 min at room temperature with 32P end-labeled oligonucleotides containing the putative HIF-1α (5′-TCTGTACGTGACCACACTCACCTC-3′) (Santa Cruz Biotechnology, Santa Cruz, CA) binding site. Reaction products were resolved on 5% nondenaturing polyacrylamide gel. The specificity of the DNA-protein interaction was established by competition experiments using 10 X cold HIF-1α oligonucleotide as the competitor. After electrophoresis, gels were dried and visualized by autoradiography [25].
Echocardiography measurements
After 30 days of surgical intervention, mice were sedated using isoflurane (2%, vol/vol). When adequately sedated, the mice were secured with tape in the supine position on a custom-built mold designed to maintain the mouse’s natural body contours of the mouse. The hair on the chest wall was removed using a commercially available hair remover from Nair (Princeton, NJ) and cotton swab. Ultrasound gel was spread over the precordial region, and ultrasound biomicroscopy (Vevo 770, Visual-Sonics Inc., Toronto, ON, Canada) with a 25-MHz transducer was used to visualize the left ventricle. The left ventricle was analyzed in apical, parasternal long axis, and parasternal short axis views for left ventricular (LV) systolic function, LV cavity diameter, wall thickness, diastolic function, and LV end-systolic and end diastolic volume determination. Two-dimensional directed M-mode images of the LV short axis were taken just below the level of the papillary muscles to analyze ventricular wall thickness and chamber diameter. Ejection fraction and fractional shortening were assessed for LV systolic function. All measurements represent the mean of at least three consecutive cardiac cycles. Throughout the procedure, the ECG, respiratory rate, and heart rate were monitored as described previously [3].
Statistical analysis
Results were expressed as mean ± SEM. One way ANOVA followed by Newman-Keuls multiple comparison test or unpaired “t” test (Graph Pad Prism Software) was carried out to determine differences between the mean values of all groups. The results were considered significant at P≤0.05.
RESULTS
Trx1 overexpression attenuates oxidative stress, cardiomyocyte apoptosis and myocardial fibrosis after myocardial infarction
The extent of oxidative stress, evaluated by TBARS analysis, was significantly decreased in TMI when compared to WMI (62.5±1.6 vs. 99.2±5 nM/mg tissue) (Fig. 1A), suggesting the possible antioxidant role of Trx1 during ischemic stress. The extent of cardiomyocyte apoptosis (Fig. 1B) was detected by using TUNEL staining in conjunction with α-sarcomeric actin. The number of apoptotic cardiomyocytes was significantly decreased in the TMI group when compared to the WMI (351.7±49.6 vs. 622±67 counts/100 HPF) (Fig. 1C). These results demonstrate that Trx1 acts as an anti-apoptotic agent during ischemic stress. After 7 days post MI, there was comparable decrease in myocardial fibrosis, evaluated by Masson’s trichrome staining in the TMI compared to the WMI (Fig. 1D). Further, we found there was no significant difference in myocardial fibrosis and oxidative stress between WS and TS animals.
Fig. 1. Trx1 overexpression attenuated ischemic stress-induced oxidative stress and myocardial fibrosis after MI.

A, Shows the quantitative analysis of TBARS concentration after surgical intervention. Trx1 transgenic animals resulted in significant reduction in oxidative stress after MI compared to WT animals (n=4). B, Representative digital micrographs showing cardiomyocyte apoptosis in the border zone of TMI and WMI groups. C, Quantitative analysis of cardiomyocyte apoptosis after MI from 3–5 animals, in counts/100 high-power field (HPF). The apoptotic cardiomyocytes are significantly reduced in TMI compared to WMI. D, Shows the representative picture of myocardial fibrosis (Masson’s trichrome staining) from different groups after surgical intervention. A significant increase in fibrosis (collagen staining in blue colour and thinner infarct) is evident in WMI compared to WS and TS. Transgenic animals subjected to MI resulted in a thicker infarct and less fibrosis (blue colour) with the islands of viable cardiac tissue with markedly less scar extension (n=3–4). WS indicates wild type sham; TS, Trx1 transgenic sham; WMI, wild type animals subjected to MI; TMI, Trx1 transgenic animals subjected to MI. ♣, P≤0.05 vs. WS; #, P≤0.05 vs. TS; *, P≤0.05 vs. WMI.
Trx1 overexpression promotes neovascularization by increasing capillary and arteriolar density during ischemic stress
The role of Trx1 overexpression on the degree of angiogenesis following ischemic stress was measured by capillary and arteriolar density 7 days after surgical intervention. Increased capillary density was observed in the TMI group when compared to the WMI group (2481±76.9 vs. 3119±108 counts/mm2) (Figs. 2A and 2B) (n=3–4). Similarly, increased arteriolar density was also observed in the TMI group as compared to the WMI group (12.9±1.2 vs. 26.7±1.6 counts/mm2) (n=3–4/group) (Fig. 2C and 2D). There was no significant difference in the capillary and arteriolar density between WS and TS groups.
Fig. 2. Trx1 overexpression enhanced the neovascularization after MI.

A, Representative digital micrographs showing capillary density/CD31 immuno-staining after 7 days of surgical intervention in different experimental groups. B, Quantitative analysis of capillary density in counts/mm2. The values are mean±SEM of 3–4 animals per group. C, Representative digital micrographs showing arteriolar density/α-smooth muscle actin immuno-staining in different experimental groups after 7 days of surgical intervention. D, Quantitative analysis of arteriolar density, in counts/mm2. The values are mean±SEM of 3–4 animals per group. Both capillary and arteriolar density was found to be significantly increased in transgenic animals subjected to MI compared to WT animals. Abbreviations are as in Fig. 1. ♣, P≤0.05 vs. WS; #, P≤0.05 vs. TS; *, P≤0.05 vs. WMI.
Trx1 overexpression modulates the expression profile of TXNIP, AKAP12, VEGF, Bcl-2 and survivin in ischemic myocardium
To evaluate the possible in vivo angiogenic and anti-apoptotic role of Trx1 during ischemic stress, the expression profiles of TXNIP, AKAP12, VEGF, Bcl-2 and survivin was carried out 4 days after post-MI. Protein expression of AKAP12 (Fig. 3B) and TXNIP (Fig. 3C) were significantly decreased, whereas increased angiogenic protein VEGF (Fig. 3D) and anti-apoptotic proteins Bcl-2 (Fig. 3E) and survivin (Fig. 3F) were increased in the TMI group as compared with the WMI group. These results indicating the angiogenic and anti-apoptotic role of Trx1 during ischemic stress is probably through TXNIP/AKAP12/VEGF/Bcl-2 and survivin mediated.
Trx1 overexpression increases the phosphorylation of Akt, GSK-3β and eNOS after MI
To evaluate the possible molecular mechanism of Trx1 in neovascularization during ischemic stress, the expression profiles of p-Akt, p-GSK3β, and p-eNOS was also carried out after 8h of surgical intervention. The protein expression of p-Akt/Akt (Fig. 4A), p-GSK3β/GSK-3β (Fig. 4B) and p-eNOS/eNOS (Fig. 4C) were significantly increased in the TMI group as compared with the WMI group.
Fig. 4. Trx1 overexpression influenced the expression of p-Akt, p-GSK-3β and p-eNOS after MI.

Representative Western blots show the expression of p-Akt (Fig. 4A), p-GSK-3β (Fig. 4B) and p-eNOS (Fig. 4C). Bar graphs in A, B and C represent the quantitative analysis and difference in the expression of p-Akt, p-GSK-3β and p-eNOS in between different groups, after they were normalized with corresponding non-phosphorylated protein controls, respectively in arbitrary units. The values are mean±SEM (n=3–5 from each group). There was a significant increase in the expression of p-Akt, p-GSK-3β and p-eNOS in TMI compared with the WMI. Abbreviations are as given in Fig. 1. ♣, P≤0.05 vs. WS; #, P≤0.05 vs. TS; *, P≤0.05 vs. WMI.
Trx1 overexpression increases the β-catenin nuclear translocation after MI
Immunohistochemical staining of β-catenin (Fig. 5A) showed a significant increase in β-catenin nuclear translocation (arrow indicates translocation) in TMI than WMI as well as WS and TS. Western blot results also showed significant increase in the β-catenin levels in nuclear fraction of TMI compared to WMI, WS and TS (Fig. 5B). These results clearly suggests possible role of Trx1 induced angiogenesis is through nuclear translocation of β-catanin. Further, there were no significant changes in the β-catenin nuclear translocation between WS and TS groups.
Fig. 5. Trx1 overexpression increased the nuclear translocation of β-catenin in ischemic stress.

A, Representative digital micrographs (immunohistochemical analysis by DAB staining and counter nuclear staining by haematoxylin) showing β-catenin nuclear translocation (indicated by arrows) after surgical intervention in different experimental groups; n=3 per group. B, Representative Western blots show the expression of β-catenin in nuclear fractions and its corresponding loading control. There was a significant increase in the translocation of β-catenin into the nucleus, evaluated by both immunohistochemistry and Western blot analysis, in TMI compared to WMI. Numbers below the bands represent the average-fold change compared to WS. The values are mean±SEM of 3–5 animals from each group. Abbreviations are as given in Fig. 1.
Trx1 overexpression increases DNA binding activity of HIF-1α during ischemic stress
The role of Trx1 overexpression on transcription factor HIF-1α during ischemic stress was evaluated by EMSA analysis. The DNA binding activity of HIF-1α was significantly increased in TMI compared to WMI (Fig. 6), suggesting the possible role of Trx1 influencing the HIF-1α expression followed by nuclear translocation and its DNA binding activity during ischemic stress.
Fig. 6. Trx1 overexpression increased the HIF-1α DNA binding activity after MI.
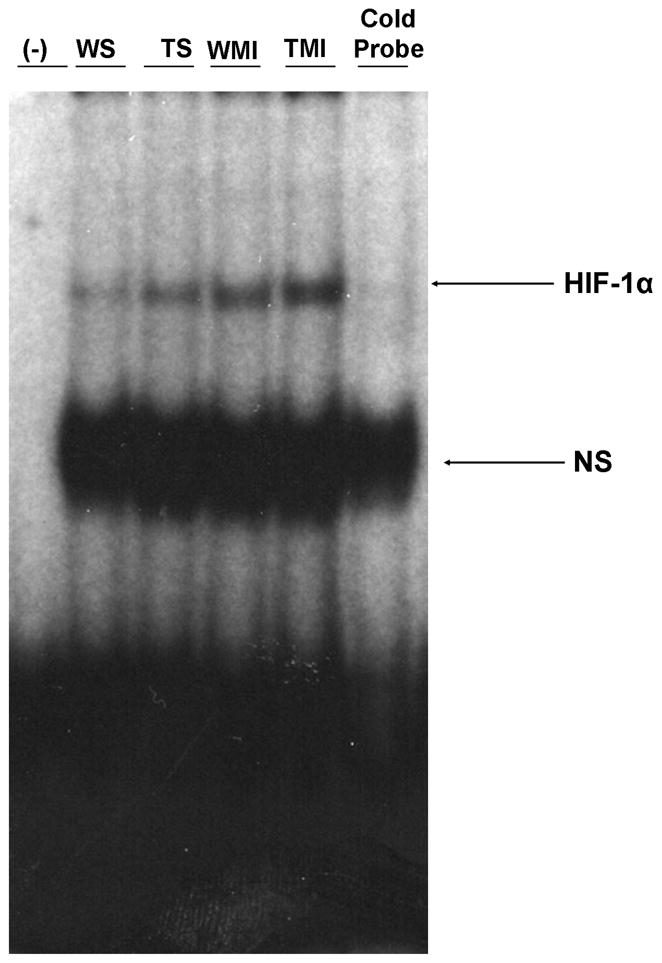
EMSA analysis revealed increased nuclear translocation and DNA binding activity of HIF-1α in TMI compared to WMI (n=3 from each group). The position of the HIF-1α band was confirmed by the disappearance of the corresponding band in the last lane from right side when we used unlabelled probe (Cold probe). WS indicates wild type sham; TS, Trx1 transgenic sham; WMI, wild type animals subjected to MI; TMI, Trx1 transgenic animals subjected to MI; NS, non-specific.
Trx1 overexpression prevents the post-ischemic ventricular remodeling
Left ventricular functional parameters were studied by echocardiography 30 days after surgical procedure. M mode pictures of different groups are given in Fig. 7A. Improvement of left ventricular function was estimated by studying various paramteres, such as ejection fraction (WS vs. WMI : 68±0.6 vs. 39.8±2.4 %) (Fig. 7D) and fractional shortening (WS vs. WMI : 37.1±0.5 vs. 19.5±1.4 %) (Fig. 7E) that was decreased significantly in the WMI group when compared to the WS and TS groups. However, this decrease was found to be preserved in mice overexpressing Trx1 subjected to MI when compared to the WMI group (ejection fraction-TMI vs. WMI : 54.5±1.1 vs. 39.8±2.4 %; fractional shortening- TMI vs. WMI : 27.9±0.7 vs. 19.5±1.3 %). Further, the WMI group exhibited a progressive increase in diastolic left ventricular internal diameter (LVIDd) (Fig. 7B) and systolic LVID (LVIDs) (Fig. 7C) than WS, and TMI {LVIDs (TMI vs. WMI : 2.92±0.07 vs. 3.67±0.1 mm) and LVIDd (TMI vs. WMI : 4.05±0.1 vs. 4.57±0.1 mm)} as compared to WMI group. Therefore, Trx1 overexpression was associated with a progressive, significant increase in LV function as compared to the wild type animals subjected to MI. Further, we have observed 28% increased survival rate in Trx1 transgenic group compared to wild type animals 30 days after MI.
Fig. 7. Trx1 overexpression prevented post-infarcted ventricular remodeling (by echocardiography).

A. Representative echocardiograph pictures of parasternal short-axis M mode images 30 days after surgical intervention from different groups. B, The quantitative data of left ventricular internal diameter in diastole (LVIDd); C, The quantitative data of left ventricular internal diameter in systole (LVIDs); D, ejection fraction; and E, fractional shortening. These results demonstrate that there was significant functional disorder in the WMI compared to WS and TS. Trx1 overexpression significantly improved functional parameters compared to the wild type animals after MI. Values are mean±SEM (n=4–6 per group). WS indicates wild type sham; TS, Trx1 transgenic sham; WMI, wild type animals subjected to MI; TMI, Trx1 transgenic animals subjected to MI. ♣, P≤0.05 vs. WS; #, P≤0.05 vs. TS; *, P≤0.05 vs. WMI.
DISCUSSION
Our present study demonstrates that Trx1 overexpression protects the heart from ischemic stress by reducing apoptosis and increasing neovascularization. Further, Trx1 overexpression significantly reduced the oxidative stress, ventricular remodeling as manifested by reduction in the collagenous fibrotic tissue and improvement in the myocardial mechanical functions.
We have observed decreased expression of AKAP12 and increased expression of eNOS and VEGF along with increased capillary and arteriolar density in hearts from TMI compared to WMI mice. The increased expression of AKAP12 and concomitant impairment in eNOS and VEGF mediated angiogenic signaling might have caused the observed reduction in capillary and arteriolar density in WT infarcted myocardium. Overexpression of TXNIP has been shown to sensitize the cardiomyocytes to oxidative stress induced apoptosis [7]. Additionally, it has been reported that increased oxidative stress is related to the induction of TXNIP, leading to the inhibition of the antioxidant function of Trx1 which reduces its ability to bind to ASK-1 thereby reducing the anti-apoptotic property of Trx1 [26, 27].
Further, increased expression of VEGF and eNOS in TMI could also be correlated with the AKAP12 mediated regulation of HIF-1α, a redox-sensitive transcription factor, which is a critical regulator of VEGF and eNOS expression [11]. Recently, AKAP12, predominant in the endothelial cells has been identified as potential negative regulator of VEGF expression and angiogenesis [28]. Again, AKAP12 has been shown to decrease the stability of HIF-1α by increasing its association with PHD2, one of the isoforms of the prolyl hydroxylase domain enzymes that tags HIF-1α for proteosomal degradation under normoxic conditions [11]. However, HIF-1α protein levels are influenced by Trx1 [29], and Trx1 may cause dissociation of HIF-1α from pVHL, thus blocking HIF-1α degradation [30]. Additionally numerous reports documented that HIF-1α directly influences the expression of VEGF and eNOS [31]. Thus, decreased AKAP 12 in the presence of Trx1 in our study might be related to the stability of HIF-1α. In addition, it is shown that Trx1 maintains cysteine 800 in HIF-1α in a reduced state, which is critical for recruitment of CBP/p300 and thus HIF-1 transactivation [29]. It was also reported that the pro-angiogenic HSPA12B, a member of a new sub-family of HSP70, promotes angiogenesis by suppressing AKAP12 and upregulating VEGF signaling in the myocardium [32, 33]. Our present data demonstrates the decrease of TBARS, an index of oxidative stress, are consistent with other previous reports [34] show that the Trx1 acts as an antioxidant in ischemic heart. Further, our previous study also showed that the Ad-Trx1 gene therapy significantly reduced reactive oxygen species (ROS) in type1 diabetic infarcted myocardium [3]. Taken together, we documented first time that Trx1 overexpression inhibits oxidative stress and promotes angiogenesis through inhibition of TXNIP/AKAP12-that lead to induced significant stabilization of HIF-1α followed by upregulation of VEGF and eNOS.
Moreover, the present study showed that Trx1 overexpression induces Akt-signaling pathway compared to WT mice during ischemic stress. There is growing evidence that ischemia-mediated oxidative stress is known to modulate Akt expression and thus reduce angiogenic factors [35]. Increased Akt expression inhibits the catalytic activity of GSK-3 β, which impedes cytoplasmic β-catenin degradation [13]. In our present study we observed significant increase in p-Akt and p-GSK-3β, along with β-catenin nuclear translocation in hearts from TMI compared with WMI mice. This effect might be due to the decreased oxidative stress by Trx1 overexpression and enhanced Akt expression [34, 35]. Further, our group has previously shown the importance of β-catenin translocation by ischemic preconditioning-mediated cardioprotection using Ad-sh-β-catenin [16]. In that study we showed that the silencing of β-catenin abolished IP-mediated cardioprotection, probably by the inhibition of the survival molecules Bcl-2, survivin, and VEGF, both at the mRNA and protein level. These changes may initiate the death signal that was apparent from significantly increased apoptosis in WMI. Our findings suggest increased neovascularization in TMI is possibly due to the influence of Trx1 on oxidative stress followed by the activation of Akt pathway. Inhibition of oxidative stress and activation of Akt signaling by Trx1 synergistically up regulates the expression of eNOS and VEGF, leading to enhanced angiogenesis and neovascuralization [35, 36]. The resulting improvement in the vascular density in the Trx1 overexpression group might have led to significant improvement in left ventricular myocardial function such as left ventricular dimensions during systole and diastole, fractional shortening and ejection fraction in the TMI group compared with the WMI group. The proposed mechanism of Trx1 overexpression-induced neovascularization and reduced ventricular remodeling is shown in Fig. 8.
Fig. 8.

Schematic diagram shows the possible molecular mechanism of Trx1-induced neovascularization and reduced ventricular remodeling in ischemic myocardium.
In conclusion, the present findings indicate that Trx1 overexpression reduces oxidative stress and apoptosis and thereby induces angiogenesis and neovascularization, subsequently reducing the extent of ventricular remodeling after MI. Our unique and promising preclinical findings therefore support the development of Trx1 as therapeutic myocardial angiogenesis target and call for the initiation of a clinical trial to assess the efficacy of this unique therapeutic strategy in the treatment of human heart failure.
RESEARCH HIGHLIGHTS.
Mice overexpressing Trx1 subjected to MI (TMI) exhibited reduced oxidative stress, cardiomyocyte apoptosis, fibrosis and increased vessel formation compared to WT.
Significant inhibition of Trx1 interacting/regulating proteins TXNIP and AKAP12 and increased p-AKT, p-eNOS and p-GSK-3β/β-catenin was observed in TMI when compared to WMI.
β-Catenin target proteins such as VEGF, Bcl-2 along with survivin were also increased in TMI compared to WMI.
Therefore, Trx1 and other related proteins identified in our study may prove to be potential therapeutic targets in the treatment of ischemic heart disease.
Supplementary Material
Acknowledgments
This study was supported by the grants HL-56803 and HL-85804 to NM.
Non-standard Abbreviations
- AKAP12
A kinase anchoring protein 12
- HIF-1α
Hypoxia inducible factor alpha
- LAD
Left anterior descending coronary artery
- LVIDd
Left ventricular internal diameter at diastole
- LVIDs
Left ventricular internal diameter at systole
- MI
Myocardial infarction
- SEM
Standard error mean
- TBARS
Thiobarbituric acid reactive substances
- TMI
Trx1Tg/+ MI
- Trx1
Thioredoxin-1
- TS
Trx1Tg/+ sham
- TXNIP
Thioredoxin interacting protein
- VEGF
Vascular endothelial growth factor
- WMI
Wild type MI
- WS
Wild type sham
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Molin D, Post MJ. Therapeutic angiogenesis in the heart: protect and serve. Curr Opin Pharmacol. 2007;7:158–63. doi: 10.1016/j.coph.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 2.Losordo DW, Dimmeler S. Therapeutic angiogenesis and vasculogenesis for ischemic disease. Part I: angiogenic cytokines. Circulation. 2004;109:2487–91. doi: 10.1161/01.CIR.0000128595.79378.FA. [DOI] [PubMed] [Google Scholar]
- 3.Samuel SM, Thirunavukkarasu M, Penumathsa SV, Koneru S, Zhan L, Maulik G, et al. Thioredoxin-1 gene therapy enhances angiogenic signaling and reduces ventricular remodeling in infarcted myocardium of diabetic rats. Circulation. 2010;121:1244–55. doi: 10.1161/CIRCULATIONAHA.109.872481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Billiet L, Rouis M. Thioredoxin-1 is a novel and attractive therapeutic approach for various diseases including cardiovascular disorders. Cardiovasc Hematol Disord Drug Targets. 2008;8:293–6. doi: 10.2174/187152908786786179. [DOI] [PubMed] [Google Scholar]
- 5.Ago T, Sadoshima J. Thioredoxin and ventricular remodeling. J Mol Cell Cardiol. 2006;41:762–73. doi: 10.1016/j.yjmcc.2006.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Piao ZH, Yoon SR, Kim MS, Jeon JH, Lee SH, Kim TD, et al. VDUP1 potentiates Ras-mediated angiogenesis via ROS production in endothelial cells. Cell Mol Biol (Noisy-le-grand) 2009;55(Suppl):OL1096–103. [PubMed] [Google Scholar]
- 7.Wang Y, De Keulenaer GW, Lee RT. Vitamin D(3)-up-regulated protein-1 is a stress-responsive gene that regulates cardiomyocyte viability through interaction with thioredoxin. J Biol Chem. 2002;277:26496–500. doi: 10.1074/jbc.M202133200. [DOI] [PubMed] [Google Scholar]
- 8.Mauban JR, O’Donnell M, Warrier S, Manni S, Bond M. AKAP-scaffolding proteins and regulation of cardiac physiology. Physiology (Bethesda) 2009;24:78–87. doi: 10.1152/physiol.00041.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Y, Gao L, Gelman IH. SSeCKS/Gravin/AKAP12 attenuates expression of proliferative and angiogenic genes during suppression of v-Src-induced oncogenesis. BMC Cancer. 2006;6:105. doi: 10.1186/1471-2407-6-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi YK, Kim K-W. AKAP12 in astrocytes induces barrier functions in human endothelial cells through protein kinase C zeta. FEBS Journal. 2008;275:2338–53. doi: 10.1111/j.1742-4658.2008.06387.x. [DOI] [PubMed] [Google Scholar]
- 11.Choi YK, Kim JH, Kim WJ, Lee HY, Park JA, Lee S-W, et al. AKAP12 Regulates Human Blood-Retinal Barrier Formation by Downregulation of Hypoxia-Inducible Factor-1{alpha} J Neurosci. 2007;27:4472–81. doi: 10.1523/JNEUROSCI.5368-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou F, Gomi M, Fujimoto M, Hayase M, Marumo T, Masutani H, et al. Attenuation of neuronal degeneration in thioredoxin-1 overexpressing mice after mild focal ischemia. Brain Res. 2009;1272:62–70. doi: 10.1016/j.brainres.2009.03.023. [DOI] [PubMed] [Google Scholar]
- 13.Dimmeler S, Zeiher AM. Akt takes center stage in angiogenesis signaling. Circ Res. 2000;86:4–5. doi: 10.1161/01.res.86.1.4. [DOI] [PubMed] [Google Scholar]
- 14.Tsai JN, Lee CH, Jeng H, Chi WK, Chang WC. Differential expression of glycogen synthase kinase 3 genes during zebrafish embryogenesis. Mech Dev. 2000;91:387–91. doi: 10.1016/s0925-4773(99)00300-7. [DOI] [PubMed] [Google Scholar]
- 15.Ding VW, Chen RH, McCormick F. Differential regulation of glycogen synthase kinase 3beta by insulin and Wnt signaling. J Biol Chem. 2000;275:32475–81. doi: 10.1074/jbc.M005342200. [DOI] [PubMed] [Google Scholar]
- 16.Thirunavukkarasu M, Han Z, Zhan L, Penumathsa SV, Menon VP, Maulik N. Adeno-sh-beta-catenin abolishes ischemic preconditioning-mediated cardioprotection by downregulation of its target genes VEGF, Bcl-2, and survivin in ischemic rat myocardium. Antioxid Redox Signal. 2008;10:1475–84. doi: 10.1089/ars.2008.2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaga S, Zhan L, Altaf E, Maulik N. Glycogen synthase kinase-3beta/beta-catenin promotes angiogenic and anti-apoptotic signaling through the induction of VEGF, Bcl-2 and survivin expression in rat ischemic preconditioned myocardium. J Mol Cell Cardiol. 2006;40:138–47. doi: 10.1016/j.yjmcc.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 18.Vidavalur R, Swarnakar S, Thirunavukkarasu M, Samuel SM, Maulik N. Ex vivo and in vivo approaches to study mechanisms of cardioprotection targeting ischemia/reperfusion (i/r) injury: useful techniques for cardiovascular drug discovery. Curr Drug Discov Technol. 2008;5:269–78. doi: 10.2174/157016308786733555. [DOI] [PubMed] [Google Scholar]
- 19.Koneru S, Penumathsa SV, Thirunavukkarasu M, Zhan L, Maulik N. Thioredoxin-1 gene delivery induces heme oxygenase-1 mediated myocardial preservation after chronic infarction in hypertensive rats. Am J Hypertens. 2009;22:183–90. doi: 10.1038/ajh.2008.318. [DOI] [PubMed] [Google Scholar]
- 20.Edelberg JM, Lee SH, Kaur M, Tang L, Feirt NM, McCabe S, et al. Platelet-derived growth factor-AB limits the extent of myocardial infarction in a rat model: feasibility of restoring impaired angiogenic capacity in the aging heart. Circulation. 2002;105:608–13. doi: 10.1161/hc0502.103672. [DOI] [PubMed] [Google Scholar]
- 21.Niehaus WG, Jr, Samuelsson B. Formation of malonaldehyde from phospholipid arachidonate during microsomal lipid peroxidation. Eur J Biochem. 1968;6:126–30. doi: 10.1111/j.1432-1033.1968.tb00428.x. [DOI] [PubMed] [Google Scholar]
- 22.Samuel SM, Thirunavukkarasu M, Penumathsa SV, Paul D, Maulik N. Akt/FOXO3a/SIRT1-mediated cardioprotection by n-tyrosol against ischemic stress in rat in vivo model of myocardial infarction: switching gears toward survival and longevity. J Agric Food Chem. 2008;56:9692–8. doi: 10.1021/jf802050h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Penumathsa SV, Koneru S, Samuel SM, Maulik G, Bagchi D, Yet SF, et al. Strategic targets to induce neovascularization by resveratrol in hypercholesterolemic rat myocardium: role of caveolin-1, endothelial nitric oxide synthase, hemeoxygenase-1, and vascular endothelial growth factor. Free Radic Biol Med. 2008;45:1027–34. doi: 10.1016/j.freeradbiomed.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thirunavukkarasu M, Penumathsa SV, Koneru S, Juhasz B, Zhan L, Otani H, et al. Resveratrol alleviates cardiac dysfunction in streptozotocin-induced diabetes: Role of nitric oxide, thioredoxin, and heme oxygenase. Free Radic Biol Med. 2007;43:720–9. doi: 10.1016/j.freeradbiomed.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Samuel SM, Akita Y, Paul D, Thirunavukkarasu M, Zhan L, Sudhakaran PR, et al. Coadministration of adenoviral vascular endothelial growth factor and angiopoietin-1 enhances vascularization and reduces ventricular remodeling in the infarcted myocardium of type 1 diabetic rats. Diabetes. 2010;59:51–60. doi: 10.2337/db09-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamawaki H, Haendeler J, Berk BC. Thioredoxin: a key regulator of cardiovascular homeostasis. Circ Res. 2003;93:1029–33. doi: 10.1161/01.RES.0000102869.39150.23. [DOI] [PubMed] [Google Scholar]
- 27.World CJ, Yamawaki H, Berk BC. Thioredoxin in the cardiovascular system. J Mol Med. 2006;84:997–1003. doi: 10.1007/s00109-006-0109-6. [DOI] [PubMed] [Google Scholar]
- 28.Lee SW, Kim WJ, Choi YK, Song HS, Son MJ, Gelman IH, et al. SSeCKS regulates angiogenesis and tight junction formation in blood-brain barrier. Nat Med. 2003;9:900–6. doi: 10.1038/nm889. [DOI] [PubMed] [Google Scholar]
- 29.Ema M, Hirota K, Mimura J, Abe H, Yodoi J, Sogawa K, et al. Molecular mechanisms of transcription activation by HLF and HIF1alpha in response to hypoxia: their stabilization and redox signal-induced interaction with CBP/p300. EMBO J. 1999;18:1905–14. doi: 10.1093/emboj/18.7.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou J, Damdimopoulos AE, Spyrou G, Brune B. Thioredoxin 1 and thioredoxin 2 have opposed regulatory functions on hypoxia-inducible factor-1alpha. J Biol Chem. 2007;282:7482–90. doi: 10.1074/jbc.M608289200. [DOI] [PubMed] [Google Scholar]
- 31.Hirota K, Semenza GL. Regulation of angiogenesis by hypoxia-inducible factor 1. Crit Rev Oncol Hematol. 2006;59:15–26. doi: 10.1016/j.critrevonc.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 32.Steagall RJ, Hua F, Thirunazukarasu M, Zhan L, Li C, Maulik N, et al. Abstract 3600: HspA12B Promotes Angiogenesis through Suppressing AKAP12 and Up-Regulating VEGF Pathway. Circulation. 2008;118:S_449. [Google Scholar]
- 33.Steagall RJ, Rusinol AE, Truong QA, Han Z. HSPA12B Is Predominantly Expressed in Endothelial Cells and Required for Angiogenesis. Arterioscler Thromb Vasc Biol. 2006;26:2012–8. doi: 10.1161/01.ATV.0000235720.61091.c7. [DOI] [PubMed] [Google Scholar]
- 34.Hamada Y, Miyata S, Nii-Kono T, Kitazawa R, Kitazawa S, Higo S, et al. Overexpression of thioredoxin1 in transgenic mice suppresses development of diabetic nephropathy. Nephrol Dial Transplant. 2007;22:1547–57. doi: 10.1093/ndt/gfm099. [DOI] [PubMed] [Google Scholar]
- 35.Katare R, Andrea C, Emanueli C, Madeddu P. Benfotiamine improves functional recovery of the infarcted heart via activation of pro-survival G6PD/Akt signaling pathway and modulation of neurohormonal response. J Mol Cell Cardiol. 2010 doi: 10.1016/j.yjmcc.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kobayashi H, Yasuda S, Bao N, Iwasa M, Kawamura I, Yamada Y, et al. Postinfarct treatment with oxytocin improves cardiac function and remodeling via activating cell survival signals and angiogenesis. J Cardiovasc Pharmacol. 2009;54:510–9. doi: 10.1097/FJC.0b013e3181bfac02. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.