

Abstract
Synapse formation requires contact between dendrites and axons. Although this process is often viewed as axon mediated, dendritic filopodia may be actively involved in mediating synaptogenic contact. Although the signaling cues underlying dendritic filopodial motility are mostly unknown, brain-derived neurotrophic factor (BDNF) increases the density of dendritic filopodia and conditional deletion of tyrosine receptor kinase B (TrkB) reduces synapse number in vivo. Here, we report that TrkB associates with dendritic growth cones and filopodia, mediates filopodial motility, and does so via the phosphoinositide 3 kinase (PI3K) pathway. We used genetic and pharmacological manipulations of mouse hippocampal neurons to assess signaling downstream of TrkB. Conditional knock-out of two downstream negative regulators of TrkB signaling, Pten (phosphatase with tensin homolog) and Nf1 (neurofibromatosis type 1), enhanced filopodial motility. This effect was PI3K-dependent and correlated with synaptic density. Phosphatidylinositol 3,4,5-trisphosphate (PIP3) was preferentially localized in filopodia and this distribution was enhanced by BDNF application. Thus, intracellular control of filopodial dynamics converged on PI3K activation and PIP3 accumulation, a cellular paradigm conserved for chemotaxis in other cell types. Our results suggest that filopodial movement is not random, but responsive to synaptic guidance molecules.
Keywords: BDNF, TrkB, filopodia, spine, PIP3, PIP2
Introduction
The establishment of appropriate synaptic connections between neurons during development is a prerequisite for normal nervous system function, beginning with contact between presynaptic elements and motile dendritic filopodia (Dailey and Smith, 1996; Ziv and Smith, 1996; Fiala et al., 1998; Niell et al., 2004). Neural activity in developing networks must involve extracellular cues that guide filopodial motility. Activity-dependent release of brain-derived neurotrophic factor (BDNF), a 13.5 kDa neurotrophin, is an attractive candidate for this purpose. In vitro application of BDNF elicits filopodia and spine formation (Dunaevsky et al., 1999; Eom et al., 2003; Ji et al., 2005). Furthermore, conditional ablation of the high affinity BDNF receptor, tyrosine receptor kinase B (TrkB), reduces synaptic density indicating its participation in hippocampal synapse formation in vivo (Luikart et al., 2005). Thus, the interaction of BDNF with TrkB could drive activity-dependent synaptic morphogenesis (Patapoutian and Reichardt, 2001; Poo, 2001; Cohen-Cory, 2002).
TrkB activation drives signaling through the phospholipase Cγ (PLCγ), phosphoinositide 3 kinase (PI3K), and Ras pathways. These signaling pathways are highly regulated and mediate context-dependent cellular responses including differentiation, survival, growth, and synaptic potentiation. For example, the neurofibromatosis type 1 (Nf1) tumor suppressor, functions as a Ras GTPase-activating protein (GAP) that negatively regulates Ras signaling (Zhu et al., 2001). The loss of Nf1 results in neurotrophin-independent survival of neurons (Vogel et al., 1995). The lipid phosphatase, phosphatase with tensin homolog (Pten) opposes PI3K signaling by catalyzing the conversion of phosphatidylinositol 3,4,5-trisphosphate (PIP3) into phosphatidylinositol 4,5-bisphosphate (PIP2). Pten overexpression inhibits neurotrophin-dependent growth and differentiation (Musatov et al., 2004), whereas conditional deletion of Pten in the hippocampus and cortex results in neuronal hypertrophy and behavioral abnormalities reminiscent of human autism (Kwon et al., 2006). These observations and the fact that mutations in BDNF, TrkB, Pten, and Nf1 are associated with impaired cognitive function in humans could indicate a convergence on a common mechanism (North et al., 1997; Yeo et al., 2004; Butler et al., 2005).
Despite extensive knowledge of downstream TrkB signaling, the molecular pathways underlying dendritic filopodial motility and synapse formation are unknown. To identify the signaling pathways involved in TrkB-mediated synapse formation, we imaged dendritic filopodia and synapses in hippocampal neuronal cultures. To alter molecules involved in TrkB signaling, we overexpressed TrkB mutant cDNAs, and used cre-mediated knockdown of TrkB, Pten, or Nf1 in neurons cultured from loxp generated germline alleles. We also used pharmacological inhibitors to complement the genetic studies. Deletion of TrkB, Pten, or Nf1 all caused abnormal dendritic filopodial motility, and altered excitatory synapse formation by a common PI3K-dependent mechanism.
Materials and Methods
Mice.
TrkBflox/flox, NF-1flox/flox, and Ptenflox/flox mice have all been described previously (Zhu et al., 2001; Luikart et al., 2005; Kwon et al., 2006). Mutant mice were generated by crossing parents that were homozygous for the floxp alleles. All mouse procedures used in were approved by institutional animal care and advisory committees at the University of Texas Southwestern Medical Center and Oregon Health & Sciences University.
Constructs.
TrkB cDNAs including point mutations were PCR cloned from previously described adenoviral genomic DNA or plasmids (Atwal et al., 2000) (sense, ACC ATG TCG CCC AGG TGG CAT; TrkB wild type (WT), TrkB Y490F, and TrkB K538N antisense, GTC GAC CCT AGG ATG TCC AGG AA; TrkB Y785F and TrkB Y785/490F antisense, GTC GAC CCT AGG ATG TCC AGG TA). PCR products were cloned into pGEM-T easy (Promega), removed with EcoRI/SalI, and cloned into the same sites of pEYFP-N1 (Clontech).
Primary cell culture and live imaging.
Serum-containing dissociated hippocampal cultures were prepared from postantal day 1 (P1) to P3 mouse pups and transfected at 4–6 d in vitro (DIV) using calcium phosphate as described previously (Luikart et al., 2005). Live imaging at 10–11 DIV was performed using the 63× objective of the LSM510 confocal microscope equipped with a motorized stage, objective heater (temperature control mini, 37.5°C), stage heater (temperature control, 37°C), and humidified climate control chamber (temperature overheat, 1.5°C, CTI controller 3700, 5% CO2). With these settings, the neurons were maintained at an actual temperature of 37°C. All imaging was performed without changing cell-culture media. Images of up to eight cells were captured per imaging session using LSM510 software with the MultiTime Module at a resolution of 640 × 480, an electronic zoom of 2.5×, and a scan speed of 2.56 μm/s. For each neuron, a Z-stack of seven images (Z-step, 0.6 μm) was captured using 1–4% argon laser power with a pinhole of 2.4 μm. Thus, protrusions appeared in at least two images of the stack, and the entire dendritic region remained in the plane of the Z-stack. All equipment was from Zeiss.
Organotypic hippocampal slice cultures.
Slice cultures were prepared from P5 Sprague Dawley rats and transfected using the Helios Gene Gun (Biorad) as described previously. Live imaging was performed using the 63× water-immersion objective with an Axioskop 2 FS microscope (Zeiss) equipped with a motorized stage (Applied Scientific Instrumentation), inline solution heater (Warner Instruments; bath temp, 35°C), spinning disk unit (Solamere Technology), and an XR/Mega10 CCD camera (Stanford Photonics). Imaging was performed in a solution containing (in mm) 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 2.0 CaCl2, 1.0 MgCl2, and 25 d-glucose, bubbled with 95% O2/5% CO2. Images of a single field of view were captured per imaging session using QED In Vivo software. For each neuron, a Z-stack of 20–30 images (Z-step, 0.4 μm) was captured using a 33 ms exposure time and signal averaging of four images. Thus, protrusions appeared in at least two images of the stack and the entire dendritic region remained in the plane of the Z-stack.
Imaging quantification.
We imaged the cultures at a time (10 DIV) when spine formation has not yet occurred and filopodia are the predominant dendritic protrusion. We thus defined filopodia as any dendritic protrusion under 10 μm in length. We quantified filopodial turnover as the average of the sum of new filopodia appearing and the number of filopodia eliminated per dendrite length every 5 min for a total of 30 min. This measurement was performed manually using the manual object counting feature of MetaMorph. Filopodial motility was calculated as the average of the absolute value of the sum of changes in filopodial lengths per dendrite length every 5 min for a total of 30 min. The length of each filopodia was measured by manually tracing using MetaMorph. The relative fluorescence of dendritic versus filopodial pleckstrin homology (PH)-green fluorescent protein (GFP) was quantitated by manually tracing the parent dendrite, and filopodia arising from this dendrite, and using the color profiler plugin for Image J. The fluorescence index was defined as filopodial 488/568 ratio divided by the dendritic 488/568 ratio. Measurements were performed by an investigator blind to experimental conditions.
Immunocytochemistry.
Neurons were prefixed with 4% paraformaldehyde in PBS at a 1:1 ratio with cell culture media (37°, 2 min) followed by a change into 37°C 4% paraformaldehyde for 5 min. Using this protocol, we observed minimal filopodial collapse; however, no fixation protocol completely maintained filopodia as observed in the living cells. After fixation, cells were washed for 5 min with PBS, 5 min with 0.3% Triton X-100 in PBS, and blocked with 6% NGS in PBS for 30 min. Primary antibodies were applied overnight at 4°C in blocking solution. All antibodies were used at the following concentrations: postsynaptic density-95 (PSD-95; Affinity Bioreagents; MA1-046) at 1:400; synaptophysin (Zymed; 18-0130) at 1:800; and anti-TrkB at 1:100 (a gift from David Kaplan, Hospital for Sick Children, Toronto, Ontario, Canada). Secondary Cy3 anti-mouse and Cy5 anti-rabbit antibodies were applied for 30 min (The Jackson Laboratory). Cells were washed 3 × 5 min after each antibody application.
Results
TrkB containing puncta traffic to dendritic growth cones and filopodia
To gain insight into the cellular functions of TrkB during synaptogenesis, we examined TrkB dynamics using an enhanced-yellow fluorescent protein (YFP)-tagged TrkB fusion protein. Furthermore, we reduced the signaling contribution of the endogenous TrkB receptors by cotransfecting TrkB-YFP and cre-dsRed into cultured hippocampal neurons from mice homozygous for the conditional TrkB flox allele. Both dsRed and YFP colocalized in a diffuse pattern throughout the cell. In addition to the diffuse signal, TrkB-YFP was also found concentrated in highly motile puncta. TrkB-YFP puncta within the shafts of primary dendrites of 10–11 DIV neurons were transported in both anterograde and retrograde directions (Fig. 1A,B, supplemental Video 1, available at www.jneurosci.org as supplemental material). We confirmed that the puncta indeed expressed TrkB using immunohistochemistry (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). These puncta were rarely stable, traveling anterograde at 1.008 ± 0.082 μm/s and retrograde at 0.988 ± 0.071 μm/s (mean ± SEM; n = 50 and 51 puncta from 6 cells, respectively). Transport appeared to be saltatory with brief pauses in movement followed by rapid transport. TrkB-YFP puncta traveled in and out of dendritic growth cones and filopodia (Fig. 1C,D, supplemental Video 2, available at www.jneurosci.org as supplemental material) at considerably slower velocities. For secondary dendrites with growth cones and in filopodia TrkB-YFP, puncta moved at 0.015 ± 0.002 μm/s (mean ± SEM; n = 85 puncta from 3 cells). Thus, movement of TrkB was reduced in dendritic growth cones and filopodia compared with primary dendrites suggesting that TrkB is trafficked to and from these dynamic structures. The velocities measured for TrkB motility within the primary dendrite are within the ranges defined as active transport by kinesin and dynein (Hill et al., 2004).
Figure 1.
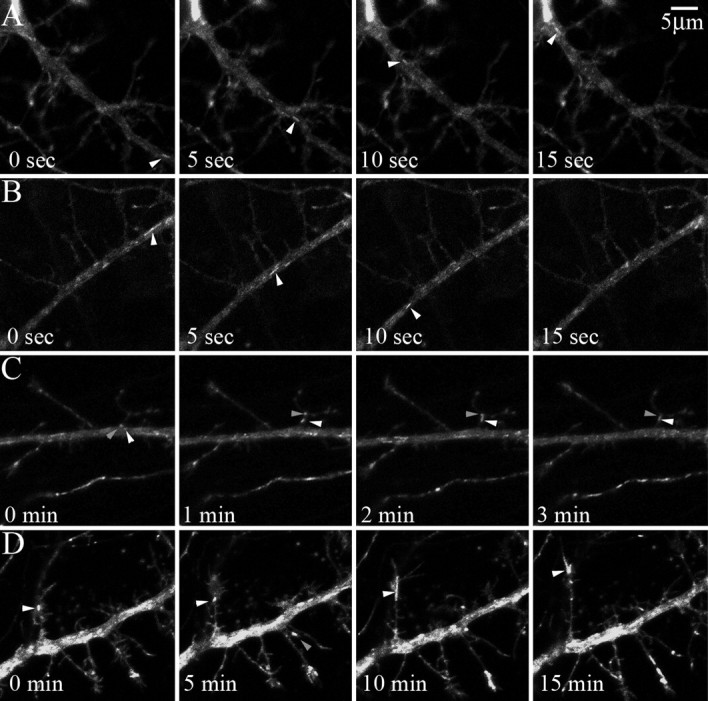
TrkB-YFP puncta are rapidly transported within primary dendrites and stable in dendritic growth cones and filopodia. A, B, We observed TrkB puncta being rapidly transported in both the retrograde (A, arrowheads) and anterograde (B, arrowheads) directions. Within primary dendrites, TrkB puncta traveled at an average velocity of 1.008 ± 0.082 μm/s in the anterograde and 0.988 ± 0.071 μm/s in the retrograde direction (mean ± SEM; n = 50 and 51 puncta from 6 cells, respectively). C, D, At longer time intervals (1 and 5 min), we resolved that TrkB puncta were less mobile in immature dendritic branches, growth cones, and filopodia (arrowheads). In these structures, TrkB puncta moved at an average velocity of 0.015 ± 0.002 μm/s (mean ± SEM; n = 85 puncta from 3 cells).
TrkB signaling mediates filopodial motility
To examine TrkB signaling, we used mutant cDNAs fused in frame with enhanced-YFP that disrupt specific downstream pathways (Atwal et al., 2000). The relative activity of mutant proteins was assessed by transfection of the TrkB-YFP fusion proteins into NG108 cells in the presence of BDNF (50 ng/ml, 10 min) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). As anticipated, TrkB-YFP increased phosphorylation of mitogen-activated protein kinase (MAPK), AKT, and PLC-γ, whereas a kinase dead receptor, incapable of binding ATP (TrkB K538N-YFP), did not support phosphorylation of these downstream effectors. Mutation of the TrkB Shc/FRS-2 binding site (TrkB Y490F-YFP) selectively inhibited phosphorylation of extracellular signal-regulated kinase (ERK) and AKT, whereas mutation of the PLC-γ binding site (TrkB Y785F-YFP) abolished PLC-γ phosphorylation and the double mutant TrkB Y490F/Y785F abolished all BDNF-induced phosphorylation.
To test whether TrkB puncta in dendritic growth cones and filopodia influence structural dynamics, we analyzed filopodial motility (Fig. 2, Table 1), which is enhanced by BDNF (50 ng/ml). Overexpression of wild-type TrkB cDNA (TrkB-YFP) increased filopodial motility, whereas the kinase dead receptor reduced filopodial motility below the level of wild-type cells, consistent with a dominant negative activity (Table 1). The expression of the kinase dead receptor was also characterized by decreased filopodial density (0.31/μm TrkB-YFP vs 0.09/μm TrkB K538N; p < 0.00004) and increased filopodial length (2.66 μm TrkB-YFP vs 5.04 μm TrkB K538N; p < 0.0004. To examine which signaling pathways were responsible for the effect of TrkB on dendritic filopodia, we expressed the TrkB mutants in cre-dsRed-positive TrkBflox/flox hippocampal neurons so that we could blunt background effects of the wild-type receptor (supplemental Videos 3–7, respectively, available at www.jneurosci.org as supplemental material). We found that, whereas introduction of the PLCγ signaling mutant had similar enhancement of filopodial motility compared with TrkB-YFP, overexpression of the Ras/MAPK/PI3K signaling-deficient TrkB cDNA did not support the enhancement of motility (Table 1). These data confirm the TrkB activity in filopodial motility and refine its signaling requirement for the Ras pathway.
Figure 2.

Dendritic filopodial motility quantitation. A, Time-lapse images of dendrites from neurons expressing GFP. B, Filopodial motility was calculated as the average of the absolute value of the sum of changed filopodial lengths per dendrite length every 5 min for a total of 30 min. The colored dots represent the tip of each filopodium and the corresponding line shows the distance the filopodium travels over time. For this example, the dendritic segment is 17.56 μm long. The average distance each filopodium moves every 5 min is 2.91 μm, and the motility is 0.166 (2.91/17.56).
Table 1.
PI3K mediates dendritic filopodial motility
| Motility mean ± variance (n) | p value | % of control | Implicated pathway | |
|---|---|---|---|---|
| GFP | 0.218 ± 0.008 (27) | |||
| GFP + BDNF (50 ng/ml) | 0.386 ± 0.019 (11) | 6.11 × 10−4** | 177% | PLCγ, ERK, PI3K |
| TrkB WT | 0.354 ± 0.015 (8) | 2.76 × 10−2* | 162% | PLCγ, ERK, PI3K |
| TrkB Y490/785F | 0.178 ± 0.017 (7) | 9.75 × 10−1 | 82% | PLCγ, ERK, PI3K |
| TrkB K538N | 0.094 ± 0.004 (7) | 4.93 × 10−2* | 43% | PLCγ, ERK, PI3K |
| TrkB Y490F | 0.215 ± 0.011 (10) | 1.00 | 99% | ERK, PI3K |
| TrkB Y785F | 0.346 ± 0.027 (8) | 4.66 × 10−2* | 158% | Not PLCγ |
| GFP + DMSO | 0.246 ± 0.017 (24) | |||
| TrkB WT + DMSO | 0.468 ± 0.010 (7) | 1.76 × 10−4** | 190% | PLCγ, ERK, PI3K |
| Nf1 KO + DMSO | 0.368 ± 0.012 (12) | 2.87 × 10−2* | 150% | ERK, PI3K |
| Pten KO + DMSO | 0.474 ± 0.018 (14) | 2.32 × 10−6** | 193% | PI3K |
| TrkB WT + PD98059 (10 μm) | 0.409 ± 0.025 (9) | 4.19 × 10−3** | 166% | Not ERK |
| Nf1 KO + PD98059 (10 μm) | 0.356 ± 0.010 (13) | 5.56 × 10−2 | 144% | Not ERK |
| Pten KO + PD98059 (10 μm) | 0.439 ± 0.009 (11) | 1.11 × 10−4** | 178% | Not ERK |
| TrkB WT + LY294002 (10 μm) | 0.154 ± 0.004 (12) | 1.83 × 10−1 | 63% | PI3K |
| Nf1 KO + LY294002 (10 μm) | 0.194 ± 0.025 (13) | 7.97 × 10−1 | 79% | PI3K |
| Pten KO + LY294002 (10 μm) | 0.156 ± 0.004 (15) | 1.39 × 10−1 | 63% | PI3K |
The average filopodial motility values for all genetic and pharmacological manipulations performed on the dissociated hippocampal neurons are shown. Compared with the GFP control, both the application of exogenous BDNF and overexpression of TrkB WT enhanced filopodial motility, implicating the PLCγ, Erk, and PI3K pathways in motility regulation. Expression of TrkB Y785F, but not TrkB Y490F, supported enhanced motility, indicating that the PI3K or ERK pathways (not PLCγ ) are primarily responsible for this effect. The expression of the kinase dead point mutation (TrkB K538N) resulted in a reduction of filopodial motility indicating the dominant negative role of this receptor. Compared with control, the overexpression of TrkB WT, knock-out of Nf1, and knock-out of Pten all supported an increase in filopodial motility. In the presence of the Erk inhibitor PD98059 these genetic manipulations still support an increase in filopodial motility. However, in the presence of the PI3K inhibitor LY294002, this increase was completely abolished. Together, these results indicate that PI3K signaling mediated enhanced dendritic filopodial motility (statistics using a one-way ANOVA and Dunnett's post hoc test against either the GFP or GFP + DMSO control).
*Significant at the 5% level; **significant at the 1% level.
TrkB Y490 has two major bifurcating effector pathways: PI3K and Raf/ERK (Atwal et al., 2000). To distinguish between these downstream pathways, we used specific pharmacological inhibitors (Vlahos et al., 1994; Alessi et al., 1995). The ERK inhibitor [2-(2-amino-3-methyoxyphenyl)-4H-1-benzopyran-4-one (PD98059), 10 μm] had no effect on TrkB-mediated filopodial motility, whereas block of PI3K signaling (LY294002 [2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one], 10 μm) completely prevented TrkB-mediated enhancement of filopodial motility (Table 1). Thus, consistent with the TrkB mutant studies, the Ras pathway is critical, but Raf/ERK signaling is dispensable whereas PI3K signaling is required.
To examine the in vivo relevance of our observations, we turned to genetic systems using mouse conditional knock-outs. The Nf1 tumor suppressor encodes a Ras-GAP that is a known negative regulator of Trk family receptor signaling (Dasgupta and Gutmann, 2003) and the Pten tumor suppressor negatively regulates PI3K signaling (Wu et al., 1998). Thus, elimination of either of these two tumor suppressors results in constitutive activation of the Ras and PI3K pathways, respectively. We reasoned that if reduction of TrkB activity diminished filopodial motility, then genetic activation of the Ras/PI3K pathway should have an apposing effect. We cultured hippocampal neurons from Nf1flox/flox and Ptenflox/flox mice to examine the consequence of genetic ablation on filopodial motility (Backman et al., 2001; Zhu et al., 2001). GFP-cre mediated recombination of either Nf1 or Pten resulted in increased filopodial motility (Table 1). Thus, genetic activation of the PI3K pathway demonstrates its role in filopodial motility downstream of TrkB.
We next examined the activity of pharmacological inhibitors on genetically modified neurons. The PI3K antagonist eliminated the enhanced filopodial motility that resulted from Nf1 or Pten ablation. Conversely, ERK pathway inhibition had no discernible effect when compared with Nf1 or Pten knock-out neurons (Table 1). Thus, both our pharmacologic and genetic data converge to identify the requirement for PI3K activity in mediating neuronal filopodial motility.
Synaptic marker density correlates with filopodial motility
Filopodial motility and turnover can be associated with synapse formation (Ziv and Smith, 1996). We therefore examined whether the observed changes in filopodial motility correlated with synapse number as assessed by immunocytochemical overlap of presynaptic and postsynaptic proteins (synaptophysin and PSD-95). TrkB overexpression increased synaptic density when compared with neurons expressing GFP alone (0.287 ± 0.023 synapses/μm2 vs 0.218 ± 0.021 synapses/μm2, respectively; p < 0.05 using a two-tailed two-sample equal-variance Student's t test). Consistent with our observations using TrkB mutant cDNAs in filopodial motility, the PI3K deficient mutant, but not the PLCγ deficient mutant, resulted in decreased PSD-95/synaptophysin overlap (Fig. 3). Thus, changes in synapse density paralleled changes in filopodial motility in a TrkB-dependent manner. These data were further validated in similar studies using conditional Pten and Nf1 neurons in which synaptic density was enhanced. We next examined synapse formation in vivo. In the CNS dendritic spines are considered to reflect synaptic density. Using the Golgi technique, we examined dendritic spine density in the dentate gyrus of mice with conditional hippocampal ablation of TrkB (Luikart et al., 2005), Pten (Kwon et al., 2006), and Nf1 (Zhuo et al., 2001). We found that TrkB ablation resulted reduced dendritic spines, whereas Pten and Nf1 abated brains exhibited increased numbers of spines. Thus, in vivo, TrkB and downstream effector activity governs normal dendritic spine formation (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
Figure 3.

Synapse density in TrkB, Nf1, and Pten mutant hippocampal neurons. A, Immunohistochemistry for PSD-95 (red) and synaptophysin (blue) in neurons expressing the TrkB-YFP fusion protein (green). Synapses were identified for quantitation as points at which the red, green, and blue signals overlapped (A, arrowheads). Synaptic density was measured for TrkB-YFP (WT; n = 80 neurons), TrkB Y490F-YFP (n = 79), TrkB Y785F-YFP (n = 77), TrkB Y490/785F-YFP (n = 78), and TrkB K538N-YFP (KD, n = 77). The Y490F, Y490/785F, and K538N mutations resulted in a significant decrease in synapse number when compared with TrkB-YFP. B, However, TrkB Y785F-YFP was not significantly different from TrkB-YFP. Synapse density was measured for neurons from Nf1 flox and Pten flox animals transfected with GFP or GFP-cre. C, The deletion of both Nf1 (n = 30 neurons) and Pten (n = 30) resulted in a significant increase in synaptic density when compared with GFP (n = 34; p values calculated using two-sample equal variance t test).
Accumulation of PIP3 in filopodia
PI3K and Pten direct the spatial accumulation of PIP3 and, thus, actin polymerization in Dictyostelium discoideum during chemotaxis (Iijima and Devreotes, 2002). To test whether a similar focal accumulation of PIP3 accompanies filopodial motility, we examined the cellular localization of the PIP3 indicator, PH-GFP (Jin et al., 2000). For these experiments, we used biolistic gene transfer into organotypic hippocampal cultures. We used the ratio of PH-GFP and monomeric Cherry fluorescent protein (mCherry) as a measure of PIP3 accumulation. To address whether this ratio was altered between filopodia and the parent dendrite, we defined the fluorescence index as the filopodial GFP/cherry ratio divided by the dendritic GFP/cherry ratio. We found a significant accumulation of PH-GFP in dendritic filopodia (1.349 ± 0.0261) compared with GFP (1.028 ± 0.0236, mean ± SEM; p < 1.0 × 10−5) (Fig. 4). Treatment of slice cultures with BDNF produced a 1.682 ± 0.0442-fold increase in filopodial PH-GFP (mean ± SEM; p < 1.0 × 10−5, BDNF-treated versus untreated slices) (Fig. 4). Thus, PI3K activity downstream of TrkB increased PIP3 within dendritic filopodia indicating activity of the receptor within this dynamic structure.
Figure 4.
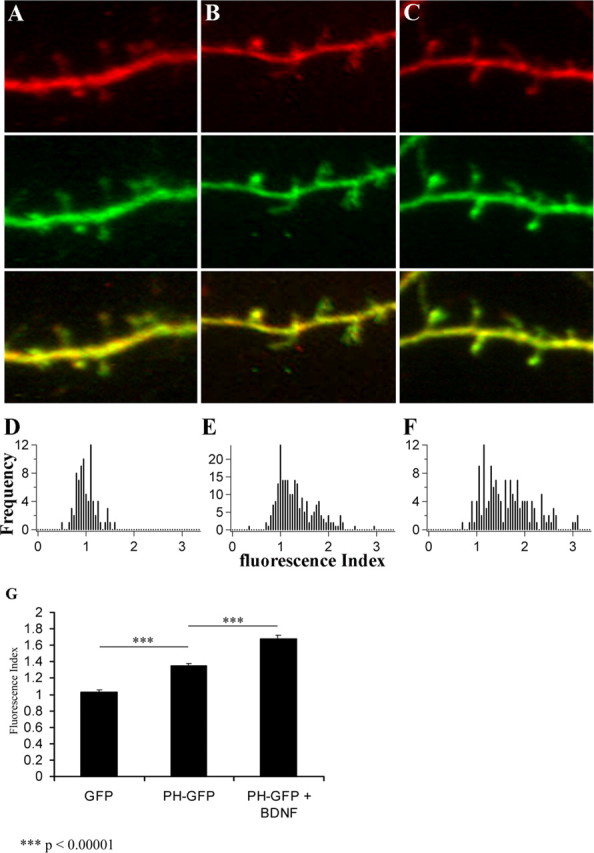
BDNF-dependent accumulation of PH-GFP in dendritic filopodia. A–C, Representative images of dendritic segments expressing GFP and mCherry (A), PH-GFP and mCherry (B), and PH-GFP and mCherry from BDNF-treated (50 ng/ml for 4–6 h) slices (C). The distribution of GFP fluorescence in filopodia relative to dendrites was quantified as the filopodial 488/568 ratio divided by the dendritic 488/568 ratio (fluorescence index). Thus, numbers >1 indicate increased relative GFP fluorescence in the filopodia versus dendrites. D–F, The distribution of these values was plotted for GFP (D; n = 79 filopodia), PH-GFP (E; n = 230 filopodia), and BDNF treated PH-GFP (F; n = 151 filopodia). The equal distribution of GFP and mCherry between dendrites and filopodia is indicated by the distribution of the fluorescence index around 1. The shift in this distribution toward numbers >1 in the PH-GFP condition indicates accumulation of PIP3 in dendritic filopodia. Application of BDNF further increased the filopodial PIP3 levels when compared with untreated cells (p values calculated using the Newman–Keuls test).
Discussion
TrkB localization
TrkB is necessary for the normal development of synaptic connectivity of the hippocampus in vivo (Luikart et al., 2005). We first examined the cellular localization of TrkB-YFP in primary hippocampal cultures during synaptogenesis. In agreement with the study by Gomes et al. (2006), TrkB was trafficked in anterograde and retrograde directions through both dendrites and axons. Furthermore, once in filopodia and dendritic growth cones, TrkB receptors appeared to stabilize. Using antibodies specific to the extracellular domain of TrkB, Gomes et al. (2006) reported that puncta in dendritic growth cones and filopodia are exposed to the extracellular environment. Therefore, it appears that these receptors are poised to transduce signals in response to extracellular BDNF. Our calculated velocity of TrkB puncta movement in dendrites was faster than that observed by Gomes et al. (2006) in axons. This difference is not unexpected as we performed live imaging at 37°C whereas they recorded at room temperature. Our results demonstrate that TrkB is associated with dendritic growth cones and filopodia, and is thus in position to regulate dynamics of those structures.
PI3K signaling and filopodial motility
Our results demonstrate that PI3K is the mediator of TrkB-dependent filopodial motility. The mechanism by which PI3K activity is linked to cytoskeletal dynamics has been studied in cellular chemotaxis (Song and Poo, 2001; Van Haastert and Devreotes, 2004; Sasaki and Firtel, 2006). In response to extracellular guidance molecules, the cellular distribution of PI3K and Pten regulates the spatial distribution of PIP3 in Dictyostelium (Iijima and Devreotes, 2002; Huang et al., 2003). Accumulation of PIP3 results in actin polymerization and forward movement of the cell. In dendritic filopodia, the accumulation of PIP3 and the regulation of motility by Pten and PI3K suggests that filopodial dynamics use an analogous mechanism. Trk receptors form complexes with components of the PI3K signaling cascade (Howe et al., 2001) and associate with dendritic filopodia. BDNF application results in filopodial PIP3 accumulation, increased motility, and increased filopodial number. BDNF is a synaptically released molecule with limited diffusion capability in vivo, suggesting that its dendritic growth promoting properties are likely localized. Furthermore, the molecular machinery underlying BDNF enhancement of filopodial motility has been conserved in chemotaxis indicating that it may elicit directional movements of dendritic filopodia. This chemoattractive aspect of BDNF has been documented for growing axons (Song and Poo, 2001). However, this is the first study to link spatially restricted TrkB signaling to PI3K activation, PIP3 accumulation, and dendritic filopodial motility.
Although it is unclear how PI3K activity and PIP3 accumulation may lead to actin polymerization, it is ultimately the activation of Rac, Rho, and Cdc42 that mediate actin dynamics (Etienne-Manneville and Hall, 2002) and dendritic growth (Threadgill et al., 1997). PI3K is required for Rac activation and critical for the activation of Rac by Ras (Cantrell, 2001; Innocenti et al., 2003). T-cell lymphoma invasion and metastasis 1 (Tiam1), the Rac1-GEF (guanine-nucleotide exchange factor) that mediates activity-dependent dendritic elaboration, is regulated directly by TrkB, PIP3, and calcium through NMDA receptor activation (Innocenti et al., 2003; Fleming et al., 2004; Miyamoto et al., 2006). Thus, an attractive model would place Tiam1 as the nexus for neurotrophin and glutamate regulation of dendritic growth.
Filopodial motility and synapse formation
The enhancement of filopodial motility resulting from the overexpression of TrkB, or reduction of Pten and Nf1, correlated with increased synaptic density. In our studies, enhancement of motility correlates positively with ultimate stabilization of synaptic contacts. BDNF-dependent activation of TrkB promoted dendritic filopodial motility and synapse formation via the activation of PI3K. For synaptogenesis to occur, there must not only be contact between neurons, but also stabilization of contacts and accumulation of synaptic proteins (Akins and Biederer, 2006). Although our data addresses the molecular pathways by which TrkB mediates enhanced filopodial motility and synapse formation, it does not address mechanisms by which TrkB may contribute to the stabilization of synaptic contacts. Nonetheless, it appears that Trk signaling does contribute to stabilization (Vaillant et al., 2002; Hu et al., 2005).
The PI3K pathway has been highly conserved throughout evolution, mediating chemoattractive responses in Dictyostelium, neutrophil chemotaxis, wound healing, and tumor cell metastasis (Merlot and Firtel, 2003; Van Haastert and Devreotes, 2004). This paradigm suggests that BDNF not only enhances filopodial motility, but may use chemoattractive molecular pathways allowing for directed movement of filopodia toward sites of BDNF release. BDNF synthesis and secretion is regulated by neuronal activity (Patterson et al., 1992; Hartmann et al., 2001; Kohara et al., 2001). Thus, filopodial motility may not be random, but influenced by extracellular cues allowing for the activity-dependent sculpting of neuronal connectivity during development and activity-dependent remodeling of synaptic connectivity in the adult (Luikart and Parada, 2006). Indeed, recent evidence suggests that filopodia are not distributed randomly with respect to mature synaptic boutons (Nägerl et al., 2007; Toni et al., 2007). Although BDNF is an attractive candidate to mediate this extracellular targeting, other neuromodulators also influence PI3K-dependent and -independent pathways that are important for dendritic development. For example, glutamate and GABA influence dendritic development through non-PI3K pathways. Thus, a variety of extracellular influences likely converge on intracellular pathways mediating directional growth and movement. The modulation of dendritic development and synapse formation can have global consequences on CNS function. Accordingly, human mutations in the TrkB, Pten, and Nf1 genes cause cognitive impairment (North et al., 1997; Yeo et al., 2004; Butler et al., 2005). Our data provide a putative link between the function of these genes and normal synaptic development.
Footnotes
This work was supported by National Institutes of Health Grant R37NS33199, Department of Defense Grant W81XWH-05-1-0265-01, National Institute of Neurological Disorders and Stroke Center Grant P50 NS 052606, The Simons Foundation, and National Institutes of Health Grants F32MH079548 (B.W.L.) and MH46613 (G.L.W.). L.F.P. is an American Cancer Society Research Professor. We thank Peter Devreotes for the PH-GFP construct and David Kaplan for the TrkB point mutant constructs.
References
- Akins MR, Biederer T. Cell–cell interactions in synaptogenesis. Curr Opin Neurobiol. 2006;16:83–89. doi: 10.1016/j.conb.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Atwal JK, Massie B, Miller FD, Kaplan DR. The TrkB-Shc site signals neuronal survival and local axon growth via MEK and P13-kinase. Neuron. 2000;27:265–277. doi: 10.1016/s0896-6273(00)00035-0. [DOI] [PubMed] [Google Scholar]
- Backman SA, Stambolic V, Suzuki A, Haight J, Elia A, Pretorius J, Tsao MS, Shannon P, Bolon B, Ivy GO, Mak TW. Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat Genet. 2001;29:396–403. doi: 10.1038/ng782. [DOI] [PubMed] [Google Scholar]
- Butler MG, Dasouki MJ, Zhou XP, Talebizadeh Z, Brown M, Takahashi TN, Miles JH, Wang CH, Stratton R, Pilarski R, Eng C. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet. 2005;42:318–321. doi: 10.1136/jmg.2004.024646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantrell DA. Phosphoinositide 3-kinase signalling pathways. J Cell Sci. 2001;114:1439–1445. doi: 10.1242/jcs.114.8.1439. [DOI] [PubMed] [Google Scholar]
- Cohen-Cory S. The developing synapse: construction and modulation of synaptic structures and circuits. Science. 2002;298:770–776. doi: 10.1126/science.1075510. [DOI] [PubMed] [Google Scholar]
- Dailey ME, Smith SJ. The dynamics of dendritic structure in developing hippocampal slices. J Neurosci. 1996;16:2983–2994. doi: 10.1523/JNEUROSCI.16-09-02983.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta B, Gutmann DH. Neurofibromatosis 1: closing the GAP between mice and men. Curr Opin Genet Dev. 2003;13:20–27. doi: 10.1016/s0959-437x(02)00015-1. [DOI] [PubMed] [Google Scholar]
- Dunaevsky A, Tashiro A, Majewska A, Mason C, Yuste R. Developmental regulation of spine motility in the mammalian central nervous system. Proc Natl Acad Sci USA. 1999;96:13438–13443. doi: 10.1073/pnas.96.23.13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eom T, Antar LN, Singer RH, Bassell GJ. Localization of a β-actin messenger ribonucleoprotein complex with zipcode-binding protein modulates the density of dendritic filopodia and filopodial synapses. J Neurosci. 2003;23:10433–10444. doi: 10.1523/JNEUROSCI.23-32-10433.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Feinberg M, Popov V, Harris KM. Synaptogenesis via dendritic filopodia in developing hippocampal area CA1. J Neurosci. 1998;18:8900–8911. doi: 10.1523/JNEUROSCI.18-21-08900.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming IN, Batty IH, Prescott AR, Gray A, Kular GS, Stewart H, Downes CP. Inositol phospholipids regulate the guanine-nucleotide-exchange factor Tiam1 by facilitating its binding to the plasma membrane and regulating GDP/GTP exchange on Rac1. Biochem J. 2004;382:857–865. doi: 10.1042/BJ20040916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes RA, Hampton C, El-Sabeawy F, Sabo SL, McAllister AK. The dynamic distribution of TrkB receptors before, during, and after synapse formation between cortical neurons. J Neurosci. 2006;26:11487–11500. doi: 10.1523/JNEUROSCI.2364-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann M, Heumann R, Lessmann V. Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. EMBO J. 2001;20:5887–5897. doi: 10.1093/emboj/20.21.5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill DB, Plaza MJ, Bonin K, Holzwarth G. Fast vesicle transport in PC12 neurites: velocities and forces. Eur Biophys J. 2004;33:623–632. doi: 10.1007/s00249-004-0403-6. [DOI] [PubMed] [Google Scholar]
- Howe CL, Valletta JS, Rusnak AS, Mobley WC. NGF signaling from clathrin-coated vesicles: evidence that signaling endosomes serve as a platform for the Ras-MAPK pathway. Neuron. 2001;32:801–814. doi: 10.1016/s0896-6273(01)00526-8. [DOI] [PubMed] [Google Scholar]
- Hu B, Nikolakopoulou AM, Cohen-Cory S. BDNF stabilizes synapses and maintains the structural complexity of optic axons in vivo. Development. 2005;132:4285–4298. doi: 10.1242/dev.02017. [DOI] [PubMed] [Google Scholar]
- Huang YE, Iijima M, Parent CA, Funamoto S, Firtel RA, Devreotes P. Receptor-mediated regulation of PI3Ks confines PI(3,4,5)P3 to the leading edge of chemotaxing cells. Mol Biol Cell. 2003;14:1913–1922. doi: 10.1091/mbc.E02-10-0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima M, Devreotes P. Tumor suppressor PTEN mediates sensing of chemoattractant gradients. Cell. 2002;109:599–610. doi: 10.1016/s0092-8674(02)00745-6. [DOI] [PubMed] [Google Scholar]
- Innocenti M, Frittoli E, Ponzanelli I, Falck JR, Brachmann SM, Di Fiore PP, Scita G. Phosphoinositide 3-kinase activates Rac by entering in a complex with Eps8, Abi1, and Sos-1. J Cell Biol. 2003;160:17–23. doi: 10.1083/jcb.200206079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, Pang PT, Feng L, Lu B. Cyclic AMP controls BDNF-induced TrkB phosphorylation and dendritic spine formation in mature hippocampal neurons. Nat Neurosci. 2005;8:164–172. doi: 10.1038/nn1381. [DOI] [PubMed] [Google Scholar]
- Jin T, Zhang N, Long Y, Parent CA, Devreotes PN. Localization of the G protein betagamma complex in living cells during chemotaxis. Science. 2000;287:1034–1036. doi: 10.1126/science.287.5455.1034. [DOI] [PubMed] [Google Scholar]
- Kohara K, Kitamura A, Morishima M, Tsumoto T. Activity-dependent transfer of brain-derived neurotrophic factor to postsynaptic neurons. Science. 2001;291:2419–2423. doi: 10.1126/science.1057415. [DOI] [PubMed] [Google Scholar]
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikart BW, Parada LF. Receptor tyrosine kinase B-mediated excitatory synaptogenesis. Prog Brain Res. 2006;157:15–24. doi: 10.1016/s0079-6123(06)57002-5. [DOI] [PubMed] [Google Scholar]
- Luikart BW, Nef S, Virmani T, Lush ME, Liu Y, Kavalali ET, Parada LF. TrkB has a cell-autonomous role in the establishment of hippocampal Schaffer collateral synapses. J Neurosci. 2005;25:3774–3786. doi: 10.1523/JNEUROSCI.0041-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlot S, Firtel RA. Leading the way: Directional sensing through phosphatidylinositol 3-kinase and other signaling pathways. J Cell Sci. 2003;116:3471–3478. doi: 10.1242/jcs.00703. [DOI] [PubMed] [Google Scholar]
- Miyamoto Y, Yamauchi J, Tanoue A, Wu C, Mobley WC. TrkB binds and tyrosine-phosphorylates Tiam1, leading to activation of Rac1 and induction of changes in cellular morphology. Proc Natl Acad Sci USA. 2006;103:10444–10449. doi: 10.1073/pnas.0603914103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musatov S, Roberts J, Brooks AI, Pena J, Betchen S, Pfaff DW, Kaplitt MG. Inhibition of neuronal phenotype by PTEN in PC12 cells. Proc Natl Acad Sci USA. 2004;101:3627–3631. doi: 10.1073/pnas.0308289101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nägerl UV, Köstinger G, Anderson JC, Martin KA, Bonhoeffer T. Protracted synaptogenesis after activity-dependent spinogenesis in hippocampal neurons. J Neurosci. 2007;27:8149–8156. doi: 10.1523/JNEUROSCI.0511-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niell CM, Meyer MP, Smith SJ. In vivo imaging of synapse formation on a growing dendritic arbor. Nat Neurosci. 2004;7:254–260. doi: 10.1038/nn1191. [DOI] [PubMed] [Google Scholar]
- North KN, Riccardi V, Samango-Sprouse C, Ferner R, Moore B, Legius E, Ratner N, Denckla MB. Cognitive function and academic performance in neurofibromatosis. 1: consensus statement from the NF1 Cognitive Disorders Task Force. Neurology. 1997;48:1121–1127. doi: 10.1212/wnl.48.4.1121. [DOI] [PubMed] [Google Scholar]
- Patapoutian A, Reichardt LF. Trk receptors: mediators of neurotrophin action. Curr Opin Neurobiol. 2001;11:272–280. doi: 10.1016/s0959-4388(00)00208-7. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Grover LM, Schwartzkroin PA, Bothwell M. Neurotrophin expression in rat hippocampal slices: a stimulus paradigm inducing LTP in CA1 evokes increases in BDNF and NT-3 mRNAs. Neuron. 1992;9:1081–1088. doi: 10.1016/0896-6273(92)90067-n. [DOI] [PubMed] [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Sasaki AT, Firtel RA. Regulation of chemotaxis by the orchestrated activation of Ras, PI3K, and TOR. Eur J Cell Biol. 2006;85:873–895. doi: 10.1016/j.ejcb.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Song H, Poo M. The cell biology of neuronal navigation. Nat Cell Biol. 2001;3:E81–88. doi: 10.1038/35060164. [DOI] [PubMed] [Google Scholar]
- Threadgill R, Bobb K, Ghosh A. Regulation of dendritic growth and remodeling by Rho, Rac, and Cdc42. Neuron. 1997;19:625–634. doi: 10.1016/s0896-6273(00)80376-1. [DOI] [PubMed] [Google Scholar]
- Toni N, Teng EM, Bushong EA, Aimone JB, Zhao C, Consiglio A, van Praag H, Martone ME, Ellisman MH, Gage FH. Synapse formation on neurons born in the adult hippocampus. Nat Neurosci. 2007;10:727–734. doi: 10.1038/nn1908. [DOI] [PubMed] [Google Scholar]
- Vaillant AR, Zanassi P, Walsh GS, Aumont A, Alonso A, Miller FD. Signaling mechanisms underlying reversible, activity-dependent dendrite formation. Neuron. 2002;34:985–998. doi: 10.1016/s0896-6273(02)00717-1. [DOI] [PubMed] [Google Scholar]
- Van Haastert PJ, Devreotes PN. Chemotaxis: signalling the way forward. Nat Rev Mol Cell Biol. 2004;5:626–634. doi: 10.1038/nrm1435. [DOI] [PubMed] [Google Scholar]
- Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) J Biol Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- Vogel KS, Brannan CI, Jenkins NA, Copeland NG, Parada LF. Loss of neurofibromin results in neurotrophin-independent survival of embryonic sensory and sympathetic neurons. Cell. 1995;82:733–742. doi: 10.1016/0092-8674(95)90470-0. [DOI] [PubMed] [Google Scholar]
- Wu X, Senechal K, Neshat MS, Whang YE, Sawyers CL. The PTEN/MMAC1 tumor suppressor phosphatase functions as a negative regulator of the phosphoinositide 3-kinase/Akt pathway. Proc Natl Acad Sci USA. 1998;95:15587–15591. doi: 10.1073/pnas.95.26.15587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo GS, Connie Hung CC, Rochford J, Keogh J, Gray J, Sivaramakrishnan S, O'Rahilly S, Farooqi IS. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat Neurosci. 2004;7:1187–1189. doi: 10.1038/nn1336. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Romero MI, Ghosh P, Ye Z, Charnay P, Rushing EJ, Marth JD, Parada LF. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev. 2001;15:859–876. doi: 10.1101/gad.862101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31:85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]
- Ziv NE, Smith SJ. Evidence for a role of dendritic filopodia in synaptogenesis and spine formation. Neuron. 1996;17:91–102. doi: 10.1016/s0896-6273(00)80283-4. [DOI] [PubMed] [Google Scholar]