

Abstract
Mild traumatic brain injury (mTBI) patients do not show clear structural brain defects and, in general, do not require hospitalization, but frequently suffer from long-lasting cognitive, behavioral and emotional difficulties. Although there is no current effective treatment or cure for mTBI, tumor necrosis factor-alpha (TNF-α), a cytokine fundamental in the systemic inflammatory process, represents a potential drug target. TNF-α levels increase after mTBI and may induce or exacerbate secondary damage to brain tissue. The present study evaluated the efficacy of the experimental TNF-α synthesis inhibitor, 3,6'-dithiothalidomide, on recovery of mice from mTBI in a closed head weight-drop model that induces an acute elevation in brain TNF-α and an impairment in cognitive performance, as assessed by the Y-maze, by novel object recognition and by passive avoidance paradigms at 72 hr and 7 days after injury. These impairments were fully ameliorated in mice that received a one time administration of 3,6'-dithiothalidomide at either a low (28 mg/kg) or high (56 mg/kg) dose provided either 1 hr prior to injury, or at 1 or 12 hr post injury. Together, these results implicate TNF-α as a drug target for mTBI and suggests that 3,6'-dithiothalidomide may act as a neuroprotective drug to minimize impairment.
Keywords: Mild traumatic brain injury, tumor necrosis factor-α, thalidomide, infliximab, etanercept, interleukin-1β, head injury
Introduction
Notwithstanding some recent improvements in the management of mild traumatic brain injury (mTBI), as yet no effective treatment is available for those afflicted, and thus morbidity and mortality are significant (Cernak 2010). mTBI-associated brain damage can be segregated into two phases: an initial primary phase induced by the injury, itself, that is immediate, irreversible and only minimized by preventive strategies, followed by an extended second phase, which is initiated at the time of original injury and persists over subsequent days, weeks and, possibly, months. This delayed phase involves an array of molecular, cellular, and physiological reactions focused towards restoring the homeostasis of damaged tissue, which, if not appropriately regulated, can lead to additional injury. The frequent occurrence of secondary brain injury provides an opportunity in which therapeutic strategies with neuroprotective properties can be administered. However, to develop effective treatment strategies for TBI, it is clearly necessary to understand the molecular and biochemical cascades that induce secondary injury (Tweedie et al., 2007a; Stoica & Faden, 2010). In this regard, a fundamental pathway recognized to be triggered in response to mTBI is cellular and humoral inflammation (Cederberg & Siesjo, 2010; Svetlovey al., 2010; Ziebell & Morganti-Kossmann, 2010).
Neuroinflammation within the injured brain is widely considered to exacerbate damage induced by mTBI, as it plays a critical role in the initiating processes of a wide variety of other neurodegenerative disorders, such as Alzheimer’s and Parkinson’s diseases and amyotrophic lateral sclerosis (Frankola et al., 2011). Although inflammation represents a first line of defense against injury and infection, an exaggerated inflammatory response can induce additional injury to neural tissues (Tweedie et al., 2007b; Tansey 2010). Within the central nervous system, glial cells, specifically microglia and astrocytes, represent the primary source of inflammatory reactions. Under normal conditions, these cells provide supportive functions to optimize neuronal activity and maintenance of homeostasis. Under stress, however, glial cells proliferate, become activated and generate the production of neurotoxic molecules, including free radical species and proinflammatory cytokines (Lecca D et al., 2008; Yan et al., 2009; Reale et al., 2009).
Key amongst the proinflammatory cytokines, tumor necrosis factor-α (TNF-α) is central both in initiating and regulating the cytokine cascade during an inflammatory response (Tweedie et al., 2007b; Frank-Canon et al., 2009; Tansey 2010; Frankola et al., 2011). Generated as a membrane-bound 26-kDa precursor molecule, it is cleaved by TNF-α converting enzyme (TACE; ADAM17) to a soluble protein that homotrimerizes to provide the active ligand. TNF-α exerts its pharmacological action via two transmembrane-spanning receptors, TNFR1 and TNFR2, which differ in their expression profiles, ligand affinity and downstream signaling pathway activation (Tansey & Szymkowski 2009; Tansey, 2010). An early transient elevation in the mRNA expression of TNF-α has been reported in rodents following closed head injury, and precedes the appearance of the ensuing cytokine (Shohami et al., 1997; Lu et al., 2009; Yang et al., 2010). Furthermore, numerous studies of TBI models have reported commensurate TNF-α protein upregulation in brain within a few hours of trauma (Taupin et al., 1993; Shohami et al., 1994; Knoblach et al, 1999). Manipulating TNF-α levels may thus substantiate its role in mTBI and define its value as a potential treatment target.
Although a reduction in systemic TNF-α can be effectively achieved by antibobies or fusion proteins capturing and clearing TNF-α prior to its receptor activation, exemplified by the utility of infliximab and etanercept in the treatment of rheumatoid arthritis (Taylor 2010), neither drug significantly enters the brain (Tweedie et al., 2007b, Zhou et al., 2011), which limits use in neurological conditions (Andersson et al., 2006; Tobinick 2010). Consequently, in this report, a novel experimental TNF-α synthesis inhibitor, 3,6'-dithiothalidomide (Zhu et al., 2003; Greig et al., 2004; Tweedie et al., 2009), was time-dependently administered prior to and after mTBI and brain biochemistry and behavioral correlates were then evaluated. This lipophilic compound is an analog of the classic, orally active neurological drug, thalidomide (N-phthaloylglutamimide), that has been shown to lower TNF-α protein levels by post- transcriptional mechanisms (Sampaio et al., 1991; Moreira et al., 1993).
Materials and Methods
Materials
3,6'-Dithiothalidomide and thalidomide
3,6'-dithiothalidomide was synthesized according to a published procedure (Luo, et al., 2008) to greater than 99.8% chemical purity, and was freshly prepared prior to each study. Thalidomide was purchased from Sigma (St Louis, MO). In cell culture studies both agents were prepared freshly in tissue culture grade DMSO (100%), whereas in animal studies, 3,6'-dithiothalidomide was prepared as a suspension in 1% carboxymethyl cellulose to provide a final concentration of 28 or 56 mg/kg (0.1 ml/10 g and 0.1 ml/100 g body weight injection in mice and rats, respectively), and was administered by the intraperitoneal (i.p.) route. These concentrations of 3,6'-dithiothalidomide are equimolar to 25 and 50 mg/kg of thalidomide.
Cellular studies
Validation of 3,6’-dithiothalidomide-induced TNF-α lowering in a RAW 264.7 cell model of inflammation
The ability of 3,6'-dithiothalidomide to ameliorate a lipopolysaccharide (LPS)-induced inflammatory response in cultured RAW 264.7 cells was quantified, using equimolar thalidomide as a positive control (Tweedie et al., 2009, 2011). RAW 264.7 cells were purchased from ATCC (Manassas, VA, USA), grown in DMEM media supplemented with 10% FCS; penicillin 100 U/ml; streptomycin 100 μg/ml and maintained at 37°C and 5% CO2. For all experiments RAW 264.7 cells were seeded at a density of 200x103 cells per well in 24 well plates. Cells were allowed to equilibrate for 24 hr after plating; one to two hr prior to the initiation of any pharmacological study the seeding media was replaced with fresh media and the cells were allowed to equilibrate at 37°C and 5% CO2. To initiate the study, RAW 264.7 cells were pretreated with thalidomide or 3,6’-dithiothalidomide (0, 10 μM or 30 μM, in 100% DMSO (Sigma)). Both drugs were then diluted in media to a 1 in 200 working solution one hr prior to the addition of LPS (Sigma, serotype 055:B5) at 60 ng/ml. At 24 hr following the addition of LPS, conditioned media was harvested and used for further analysis. The levels of TNF-α protein secreted into the media were measured by ELISA (BioLegend, San Diego, CA). Nitrite levels in culture media were measured by use of the Griess Reagent System (Promega, Madison, WI), as per the manufacturer’s guidelines. The optical density of unknown samples were read at 520 nm λ and compared to a sodium nitrite standard curve (1.5 μM to 100 μM). The concentration of nitrite measured in the media was expressed as μM units. As an assessment of drug-induced toxicity, cell viability was assessed by use of the CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega), a reduction in absorbance at 490 nm λ is indicative of cell death.
Animal studies
Rodents for these studies included male ICR mice (6–8 weeks age and 30–40 g weight: initially purchased from HSD Jerusalem, Israel, and thereafter bred and raised within the vivarium of the University of Tel Aviv, Tel Aviv, Israel) and F344 rats (3 month old and 200–225 g weight: Taconic Farms, Germantown, NY). Experimental procedures and housing conditions were approved by the Institutional Animal Care and Use Committees of Tel Aviv University (M-10-006) and the Intramural Research Program, National Institute on Aging (ASP 331-LNS-2012). Animals were housed 2 (rats) and 5 (mice) per cage with ad libitum access to food and water on a 12:12 light/dark cycle at 22±1Co, with lighting during the light phase kept constant. All experimental manipulations were undertaken during the light phase of the cycle. A minimum number of animals were used and all efforts were made to minimize potential suffering. Each animal was used for only one experiment.
Procedures
Validation of systemic and brain TNF-α lowering activity of 3,6’-dithiothalidomide in rodent
To verify TNF-α syntheis lowering action of 3,6’-dithiothalidomide in rodents, the agent was administered to animals to lower systemic and brain TNF-α protein levels elevated by LPS challenge. Specifically, F344 rats were anaesthetized (50 mg/kg sodium pentobarbital, 1 ml/kg vol. i.p., n=6) and the right femoral artery was cannulated to permit blood sampling. 3,6’-Dithiothalidomide (56 mg/kg, i.p.), followed 1 hr later by LPS (1 mg/kg i.p., E. coli 055:B5; Sigma), were administered, and serial blood samples then were collected over a subsequent 4 hr period. At 4 hr the animal was euthanized by excess sodium pentobarbital, and the CNS collected. Plasma was derived immediately by centrifugation (8,000 G × 5 min) and, together with the CNS sample, frozen at −80°C for subsequent analysis of TNF-α by ELISA (BioLegend). For determination of secreted levels of brain TNF-α, the brain was briefly perfused with PBS to clear the vascular system prior to freezing. At a later time, 50 micron brain slices were cut by cryostat and then incubated (2 hr, 37°C) in 250 ul cytoprotectant/PBS solution. This solution was then quantified for TNF-α levels by ELISA.
Close head mTBI injury and treatment
Mice were anesthetized lightly with Isoflurane and then placed under a weight drop device. This device consisted of a 50 g weight, a cylindrical–shaped piece of metal with a slight spherical tip, which was dropped through a vertical metal guide tube (13 mm in diameter and 80 cm long). Each mouse was positioned with the temporal right side of its head, between the corner of the eye and the ear, positioned under the tube whilst the head was manually supported and immobilized by a sponge. The weight then was released from the top of the tube, and the sponge supported the head of the mouse allowing some anterior/posterior motion in the absence of any rotational head movement at the moment of impact (Milman et al., 2005; Pan et al., 2003; Zohar et al., 2003). Sham mice underwent an identical procedure as described for mTBI, but in the absence of dropped weight. Treatment comprised of 3,6’-dithiothalidomide at a low (28 mg/kg) or high (56 mg/kg) dose, or vehicle administered as a single dose at one of three separate times: 1 hr prior to injury or 1 and 12 hrs post injury. Thereafter, mice were assessed in behavioral tests 72 hours or 7 days following the injury. In a separate series of animals, brain TNF-α levels together with other markers of apoptosis were time-dependently quantified from brain slices following mTBI.
Validation of elevated brain TNF-α levels following mTBI
To verify the occurrence of TNF-α elevation in our mTBI model and define its time-dependence, mice were subjected to mTBI and euthanized at specific times thereafter (1 to 18 hr (n=3–5 per time)). Animals were perfused with phosphate buffered saline (PBS: 0.1 mM, pH 7.4) to remove blood, and the brain immediately frozen to −−70°C. Brain sections (50 um) were later cut by cryostat, and individually placed (5 sections, per time point, per animal) into separate wells of a 96 well plate containing PBS. Following incubation (2 hr at 37°C), PBS/cytokine admixture was harvested and quantified for TNF-α by ELISA assay (BioLegend).
Validation of brain entry of 3,6’-dithiothalidomide in mice
To define whether actions of 3,6’-dithiothalidomide were centrally mediated, the brain uptake of the agent was assessed at an early time point (1 min) following administration to avoid potential confounds due to metabolism. Specically, three mice were administered 56 mg/kg 3,6’-dithiothalidomide and brain and plasma samples were collected after exactly 1 min and analysed by HPLC to define a brain/plasma ratio. Samples were quantified for 3,6’-dithiothalidomide on an Agilent 1100 series PHLC (Foster City, CA), equipped with an Agilent Zorbax column (Eclipse xDB-c18, 4.8 × 250 mm, 5 μm particle size) and detected at 297 nm λ, which was determined to be a peak of its UV absorbance. The mobile phase was acetonitrile/methanol/20 mM phosphate buffer (15:42:43), run isocratically at a flow rate of 1.0 ml/min. Briefly, plasma samples (200 ul) were mixed with internal standard (50 ul revlimid, 0.1 mg/ml) and TCA (200 ul) was then added, vortexed, and the sample centrifuged (2500 g × 10 min). The supernatant (400 ul) was removed and 10 ul injected on to the HPLC. Brain (300 mg) was sonicated in 700 ul RIPA buffer, internal standard (revlimid 0.1 mg/ml) was added, and the sample centrifuged (20,000 g × 15 min). To the supernatant (800 ul), 20% TCA (400 ul) was added, and the sample again centrifuged (2500 g × 10 min). Thereafter, 10 ul of the supernatant (1000 ul) was injected on to the HPLC.
Behavioral tests
At 72 hr and 7 days after the mTBI procedure, animals were assessed in 3 behavioral tests: Y-maze, object recognition and passive avoidance. Passive avoidance was designated as the last performed test as, otherwise, its aversive nature could potentially impact performance in the other tests. Tests were undertaken at 24 hr intervals. All studies were undertaken in the same sound-insulated romm that had constant illumination.
Y maze paradigm
The Y maze test was used to assess spatial memory. This task takes advantage of the preference of rodents to explore novel rather than familiar places. Comprising three arms (8 × 30 × 15 cm at an angle of 120° from the others), with each distinguished by the presence of a different visual cue (triangle, square, or circle), the Y maze weas constructed of black Perspex (Dellu et al., 1992). From the three arms, one awas randomly selected as the “start” arm. Each animal was placed twice in the “start” arm. During the initial trial, of 5 min duration, one of the two remaining arms was randomly selected to be closed off whereas on the second trial, of 2 min, both arms were open. These trials were separated by a 2 min interlude, during which the mouse was returned to its home cage and the maze was cleaned (70% ethanol solution (v/v)) and dried.
The time spent in each of the arms was quantified. Discrimination of spatial novelty was assessed by a preference index (Dix & Aggleton, 1999) determined as: (time in the new -time in the old arm) / (time in the new + time in the old arm).
Novel object recognition paradigm
An object recognition task was used to appraise recognition memory, as described by Messier (1997). This task takes advantage of a propensity of rodents to discriminate a familiar from a new object. Mice were initially individually habituated to an open field box (59x59x20 cm) for 5 min, 24 hr before the test. During the acquisition phases, two objects (A and B) of identical material, which were sufficiently heavy and high to ensure that mice could neither move nor climb over them, were placed in a symmetric position within the chamber for 5 min duration. At 24 hr after acquisition phase training, one of these objects (A or B randomly) was substituted by a novel one (C), and exploratory behavior was again evaluated for 5 min. All objects were thoroughly cleansed (70% ethanol) between sessions to preclude odor recognition. Exploration of an object was characterized as rearing on it or sniffing it at a distance of less than 2 cm and/or touching it with the nose. Successful recognition was revealed by preferential exploration of the novel object. Discrimination of visual novelty was assessed by a preference index (Dix and Aggleton, 1999), determined as: (time near the new - time near the old object) / (time near the new + time near the old object).
Passive avoidance paradigm
The passive avoidance task was used to evaluate simple non-spatial memory ability. The passive avoidance apparatus (San Diego Instruments, San Diego, CA) was created from black Perspex (48 × 22 × 22 cm) and possessed two separate chambers, one light-illuminated the other dark, of equal dimensions. These chambers were joined by a door that was shut at the start of each trial and could be opened by the experimenter. The test involved two sessions separated by a 24 hr interval. Animals were positioned within the illuminated chamber and, after 30 sec, the connecting door was opened. On crossing into the dark compartment, the door was closed, and the animal received a mild electric foot shock (1 mA for 5 sec). Following the electric shock, animals remained for further 5 sec within the apparatus and then were returned to their home cage. After a 24 hr interval, animals were evaluated for retention of the passive avoidance response to the shock by again placing them in the illuminated compartment. Memory was operationally defined as a failure to enter the dark compartment within 3 min (Milman et al., 2005). No shock was delivered during the second day.
Data analysis
All results are presented as mean ± SEM and were analyzed either by with SPSS 15 software (Genius Systems, Petah Tikva, Israel) or GraphPad InStat Version 3.05 (GraphPad Software, San Diego, CA). The number (n) of studies performed and/or animals is provided in parentheses. One-way ANOVA’s were performed to compare between all groups, followed by LSD post hoc tests. ANOVA’s were used to analyze the results of the Y maze and the object recognition test. Results of the passive avoidance test were evaluated using the non- parametic Chi square test. Statistical comparisons of TNF-α levels in cell culture and animal studies were undertaken by use of a Students t-test with appropriate Bonferroni corrections for multiple comparisons, as necessary. P values of ≤0.05 are considered to be of statistical significance.
Results
Amelioration of LPS-induced cellular inflammation by 3,6’-dithiothalidomide
3,6’-Dithiothalidomide, but not thalidomide, dose-dependently lowered secreted levels of both TNF-α and nitrite in LPS-challenged RAW 264.7 cells, as assessed at 48 hr incubation post LPS (Fig. 1). In contrast, 10 uM thalidomide elevated TNF-α levels, although higher thalidomide concentrations (100 uM) have been reported to lower TNF-α levels in LPS-challenged cultured human peripheral blood mononuclear cells (Zhu et al., 2003). Cell viability, assessed by MTS assay, was unaltered by either drug.
Figure 1. 3,6’-dithiothalidomide lowers TNF-α protein and nitrite generation in LPS-challenged RAW 264.7 cells without loss of viability.
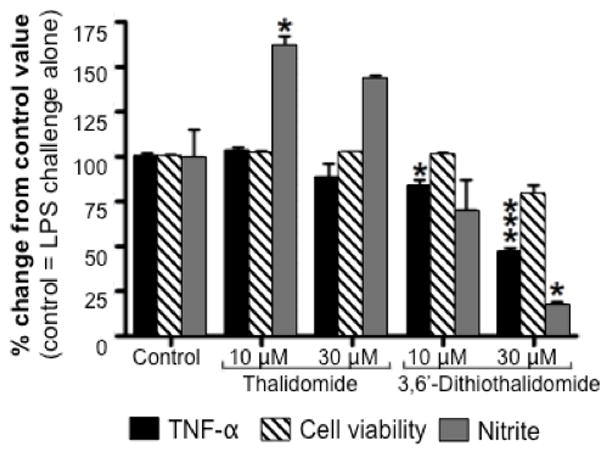
The effects of 3,6’-dithiothalidomide (3,6’-DT) and equimolar thalidomide (TH) on cell viability and LPS-induced elevation of TNF-α protein levels and nitrite generation in RAW 264.7 cells. Cells were incubated for 48 hr after administration of LPS with and without drug. Whereas 3,6’-dithiothalidomide (10 and 30 μM) significantly lowered TNF-α protein and nitrite generation, thalidomide (10 μM) significantly elevated generation of nitrite. Control refers to 100% DMSO.(n=4 observations. *, *** refers to p 0.05 and p 0.001 compared to control, respectively (Dunnett’s t test)).
Amelioration of elevated central and systemic LPS-induced TNF-α levels in rodents by 3,6’dithiothalidomide
3,6’-Dithiothalidomide induced a time-dependent lowering of TNF-α plasma levels in ansethetized rats challenged with LPS to induce increased TNF-α synthesis and secretion (Fig. 2). As assessed at 4 hr, 3,6’-dithiothalidomide likewise lowered CNS TNF-α levels, confirming the compound’s ability to act as a TNF-α synthesis inhibitor in cellular and in vivo models of inflammation.
Figure 2. 3,6’-dithiothalidomide lowers LPS-induced TNF-α levels in plasma and CNS.

(A) Systemically administered LPS induced a rapid, time-dependent 3 log elevation in plasma TNF-α levels that peaked at 90 min. (B) This LPS-induction in plasma TNF-α levels was substantially (up to 80%) inhibited by prior administration of 3,6’-dithiothalidomide (3,6’-DT, 56 mg/kg) – levels are expressed as a percent of those determined at the same time in LPS alone animals. (C) Systemically administered LPS elevated CNS levels of TNF-α by 20% compared to basal levels in pure controls (no LPS). This CNS LPS-induced TNF-α rise was fully blocked by prior adminsitration of 3,6’-dithiothalidomide (56 mg/kg). (n=6. **, *** refers to p 0.01 and p 0.001 compared to LPS alone determined at the same time points, respectively (paired Student’s t test)).
3,6’-Dithithalidomide brain/plasma ratio
With a measured brain/plasma concentration ratio of 1.34 ± 0.63, the neurological actions of 3,6’-dithithalidomide were likely centrally mediated.
mTBI-induced elevation in TNF-α
As illustrated in Supplemental Figure 1, compared to control animals whose brain TNF-α were minimal, mTBI induced an acute rise (up to 4.5-fold) in TNF-α that peaked at 12 hr and returned to resting levels within 18 hr .
mTBI-induced behavioral impairments and their amelioration by 3,6’-dithithalidomide
Y-maze paradigm
72 hr test
As illustrated in Figure 3 (Upper), mTBI mice demonstrated a slight memory deficit, compared with the sham group, that did not reach significance in all three time-dependent studies (1 hr prior to, and 1 and 12 hr post injury, respectively) [F (5,94) =7.09; N.S],[F(5, 53) =6.806; N.S], [F(5, 55) =8.41; N.S.]. This trend was not evident in mTBI mice treated with 3,6’-dithiothalidomide at either the lower or higher dose (28 and 56 mg/kg).
Figure 3. mTBI induces impairment in performance in a Y maze preference index paradigm that is fully ameliorated by 3’6-dithiothalidomide.
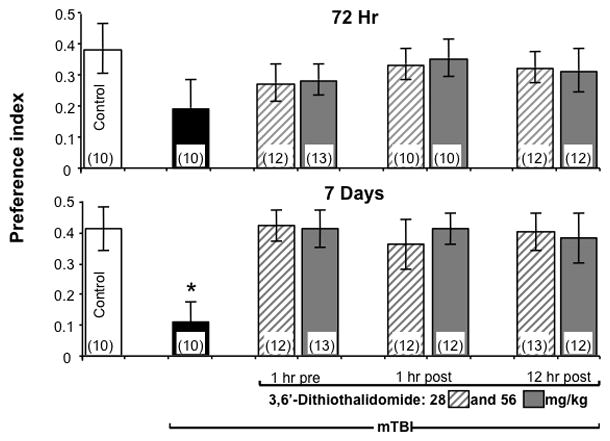
Performace of mice was quantitatively assessed in a Y maze paradigm at 72 hr and 7 days following mTBI as a preference index, calculated as (time new-time old)/(time new+time old). Values are mean ± SEM. (Upper) 72 hr post injury the mTBI group displayed a trend toward lower memory ability that did not reach significance (one-way ANOVA showed no differences; N.S) but this trend was not evident in mTBI drug-treated mice. (Lower) 7 days post injury, one way ANOVA showed that mTBI animals had a deficit in spatial memory performance compared with all the other groups (*p 0.05). No differences were found between any of the other groups, suggesting complete amelioration by 3’6-dithiothalidomide.
7 day test
As illustrated in Figure 3 (Lower), mTBI mice displayed significantly reduced spatial memory compared with all other groups (1 hr prior to, and 1 and 12 hr post injury, respectively) [F (5,84) =4.09; p<0.005] [F(5,51) =4.102; p<0.05], [F(5,59) =3.660; p<0.05].Post-hoc analyses confirmed that the mTBI group was significantly different from all other (in all experiments; p<0.05 compared with all groups). 3,6’-dithiothalidomode at both low and high doses fully reversed this injury-induced effect, whether administered before or after mTBI. Post-hoc analyses revealed that 3,6’-dithiothalidomode abolished mTBI-induced behavioral deficits seen in this Y-maze paradigm.
Novel Object Recognition
72 hr test
As illustrated in Figure 4 (Upper), mTBI mice demonstrated a trend towards a lower visual memory, compared to the sham group, which was not statistically significant in all time-dependent studies (1 hr prior to, and 1 and 12 hr post injury, respectively) [F (5,60) =1.17; N.S;] [F (5,53) =3.17; N.S], [F (5,52) =4.15; N.S.]. This trend was not as evident in 3,6’dithiothalidomide treated mice.
Figure 4. mTBI induces impairment in performance in a novel object recognition preference index paradigm that is fully ameliorated by 3’6-dithiothalidomide.

Performace of mice was quantitatively measured in a novel object recognition paradigm at 72 hr and 7 days post mTBI as a preference index, calculated as (time new-time old)/(time new+time old). Values are mean ± SEM. (Upper) 72 hr post injury the mTBI group displayed a trend toward a lower preference index that did not reach significance (one-way ANOVA showed no differences; N.S) but this trend was not evident in mTBI drug-treated mice. (Lower) 7 days post injury, one way ANOVA showed that mTBI animals demonstrated a deficit in spatial memory performance compared with all the other groups (*p 0.05). No differences were found between any of the other groups, suggesting a complete amelioration by 3’6-dithiothalidomide.
7 day test
Reduced visual memory was clearly evident in mTBI mice compared to all other groups, in all all three time-dependent studies (1 hr prior to, and 1 and 12 hr post injury, respectively) [F (5,73) =7.107; p<0.001], [F (5,52) =4.102; p<0.001], [F (5,57) =4.472; p<0.001] (Fig 4, Lower). Post-hoc analyses showed statistical significance (p<0.05 compared with all groups). Mice administered either dose of 3,6’-dithiothalidomide prior to or post mTBI performed similar to the sham group. Post-hoc analyses showed no differences between all other groups, indicating complete amelioration of this mTBI-induced behavioral deficit by 3,6’dithiothalidomide.
Passive avoidance
72 hr test
As illustrated in Figure 5 (Upper), pair-wise comparisons revealed no differences in memory ability between all groups in all three time-dependent experiments (1 hr prior to, and 1 and 12 hr post injury, respectively) (χ2 (73) = 12.31; N.S), (χ2 (54) = 11.27; N.S), (χ2 (51) = 14.35; N.S).
Figure 5. mTBI induces a trend towards impairment in learning as assessed by passive avoidance that is not evident in mice administered 3’6-dithiothalidomide.

Performace of mice was measured in a passive avoidance paradigm at 72 hr and 7 days post mTBI with learning defined as the percent of animals that did not enter the dark compartment on the second day. Values are in percents. (Upper). 72 hours post injury Chi square tests revealed no differences between all groups. (Lower). 7 days post injury Chi square tests revealed no differences between all groups, although a trend towards decreased learning was evident in mTBI mice that was not seen in animals treated with 3,6’dithiothalidomide prior to or post injury.
7 days
Pair-wise comparisons revealed that the difference between the groups did not approach significance all three experiments (1 hr prior to, and 1 and 12 hr post injury, respectively) (χ2 (79) = 10.31; p=0.06), (χ2 (64) = 9.14; N.S), (χ2 (61) = 11.27; N.S); however, a trend towards reduced memory was evident in mTBI animals that was not noticable in those treated with 3,6’-dithiothalidomide (Fig. 5, Lower).
In summary, across the three behavioral paradigms studied, a general pattern emerged. The mTBI groups presented a consistently (in all the behaviors) inferior performance, as compared to the sham mice. However, this reached statistical significance only at 7 days post injury. 3,6’-dithiothalidomide proved to be well tolerated in mice challenged with mTBI at both low (28 mg/kg) and high (56 mg/kg) doses, and these mice performed at levels that were not statistically different from the sham group in all behaviors. Hence, this compound fully ameliorated mTBI-induced behavioral deficits at both doses.
Discussion
mTBI has become a major and increasing public health concern and represents the most frequent cause of mortality and disability in young adults. Additionally, associated with battlefield injury, blast-TBI is currently of particular concern. At present, no effective pharmaceutical therapies are available for mTBI and existing treatment principally involves optimized intensive care management after injury occurrence (Garner & Brett, 2007; Moppett 2007). The pathology resulting from head injury is becoming better characterized. Mechanical forces induce shearing and compression of neuronal and vascular tissue at the time of impact. A cascade of neurochemical events may then ensue that result in additional damage. Such secondary injury makes the brain vulnerable to further injury but may be amenable to intervention. mTBI has hence been been considered a gateway to the development of neuropsychiatric disorders, such as depression and anxiety (Hesdorffer et al., 2009; Bryant et al., 2010), and also chronic neurodegenerative diseases exemplifed by Alzheimer’s disease and Parkinson’s Disease (Uryu et al., 2003; Kiraly & Kiraly 2007; Johnson et al., 2010).
Our closed head murine injury model attempts to mimic mTBI in humans, and results of the present study support previous work showing a decline in performance in cognitive tests after mTBI (Baratz et al., 2010; Edut et al., 2010; Milman et al., 2005; Zohar et al., 2006) that are also evident in humans (Vakil 2006; Ghajar et al., 2007). Studies of the neurochemical changes induced after injury suggests that mTBI represents a complex neurological condition, encompassing numerous molecular and cellular pathways, including inflammation (Raghupathi 2004; Lu et al., 2009), resulting in the induction of diffuse neuronal dysfuction and apoptosis (Tweedie et al., 2007a; Tashlykov et al., 2009; Rubovitch et al., 2010). The inflammatory cascades triggered by mTBI are mediated by the generation of both pro- and anti-inflammatory cytokines that, although barely detectable in healthy brain, are rapidly upregulated in response to mTBI (Israelsson et al., 2009). In particular, an early transitory rise in TNF-α mRNA expression of up to 30-fold, with similar elevations in IL-1β and IL-6, has been described in rodent closed head injury, and preceded the subsequent arrival of cytokine activity and leukocyte recruitment (Shohami et al., 1997; Yang et al., 2010; Israelsson et al., 2009). Likewise, TNF-α, IL-1β and IL-6 protein upregulation in brain has been described in diverse mTBI models as well as in human CSF within a few hours of trauma (Taupin et al., 1993; Shohami et al., 1994; Knoblach et al., 1999). As evident in Supplemental Figure 1, an acute and rapid rise in brain TNF-α levels was seen in our mTBI murine model, occurring within an hour, and increasing to 4.5-fold above control levels at 12 hr. Such an elevation supports the use of this model to assess agents that lower brain TNF-α. In this respect, prior studies focused on lowering TNF-α with a TNF-α binding protein and the competitive nonselective phosphodiesterase inhibitor, pentoxifylline, that additionally lowers TNF-α transcriptionally, have been reported to ameliorate mTBI over an initial 24 hr (Shohami et al., 1996, 1997), when these agents were administered i.v. immediately following trauma.
The biosynthesis of inflammatory cytokines like TNF-α are tightly regulated post- transcriptionally at the level of mRNA stability via their 3’-untranslated region (3’-UTR) (Moreia et al., 1993), which allows rapid changes in protein expression to adapt to enviromental cues. The occurrence of adenylate-uridylate-rich elements (AREs) within the 3′-UTR of TNF-α mRNA performs a key function in post-transcriptional repression, targeting it for rapid degradation or inhibition of translation (Khera et al., 2010). p38 MAPK has been characterized as a primary signaling cascade impacting TNF-α stability via its 3’-UTR ARE cis elements, mediated through interactions with RNA binding proteins (Patil et al., 2008). Proteins such as HuR have been associated with promoting transcript stabilization. Following export to the cytoplasm, HuR binds and stabilizes ARE-containing transcripts and aids convey them to translational machinery. In contrast, interaction with RNA-binding proteins such as tristetraprolin and alike proteins can accelerate the degradation of bound mRNAs (Stamou & Kontoyiannis, 2010; Khera et al., 2010). Challenges such as LPS extends the half-life of TNF-α mRNA, allowing release of its translational repression. Whereas, administration of agents such as thalidomide have been shown to increase translational blockade and a reduce TNF-α mRNA half-life from 30 min to 17 min (Moreia et al., 1993); thereby lowering its rate of protein synthesis (Sampaio et al.,1991). As assessed by following a luciferase element within the 3’-UTR of TNF-α mRNA, 3,6’-dithiothalidomide appears to likewise regulate mRNA stability (Zhu et al., 2003; Greig et al., 2004).
Although thalidomide is a controversial drug (Melchert & List, 2007), several groups have generated analogs of significant clinical interest (Knight 2005; Aragon-Ching et al., 2007). As illustrated in Figures 1 and 2, the compound 3,6’-dithiothalidomide is a more potent TNF-α synthesis lowering analog than thalidomide in cell culture models. Indeed, a 10 μM thalidomide concentration, which compares favorably with plasma levels observed after a routine 200 mg dose in humans (Teo et al., 2004), was found to mildly elevate TNF-α levels in culture. This finding is in accord with others (Shannon et al., 1996; Tadasse et al., 2004). In contrast, 3,6’-dithiothalidomide elicited a time-dependent decline in TNF-α levels in the plasma and CNS of rodents following a marked, up to 3 log, induction of TNF-α by LPS. As assessed by its brain/plasma ratio of 1.34, which is in accord with its log D value of −0.56 (Zhu et al., 2003; Greig et al., 2004), a measure of its balanced aqueous solubility/lipophilicity, 3,6’-dithiothalidomide appears to readily enter the brain. In light of elevations in TNF-α apparent in our mTBI mouse model, 3,6’-dithiothalidomide was administered as a single dose either an hour before or after mTBI to define its ability to lower TNF-α synthesis prior to and immediately after its induction by mTBI. These are time points when TNF-α levels in brain would be basal and sub-maximally elevated, respectively. In addition, and more related to clinical use, the agent was assessed when administered as a single dose 12 hr post mTBI, coinciding with the peak expression of TNF-α in brain after mTBI (Figure 3). Our chosen doses of 3,6’-dithiothalidomine (28 and 56 mg/kg, equipotent to 25 and 50 mg/kg thalidomide) compare favorably with those of thalidomide used in humans, where doses of up to 1200 mg are administered.
Consistent with prior cognitive loss previously reported in our mTBI model (Zohar et al, 2003, Milman et al, 2005, Baratz et al, 2010), a reduced memory ability was seen following mTBI in both the Y maze and novel object recognition paradigms that was evident at 72 hr and reached significance at 7 days. A trend towards impairment was also apparent in the passive avoidance test. 3,6’-dithiothalidomide, administered in either a single low or high dose prior to or up to 12 hr after injury, fully ameliorated all animal performance deficits. There are few prior studies of thalidomide and analogues in traumatic brain and spinal cord injury (SCI) in humans and animal models. The immediate administration of a combination of thalidomide (100 mg/kg) and the phosphodiesterase 4 inhibitor, rolipram (3 mg/kg), ameliorated functional loss following SCI in rats, with thalidomide alone showing a trend toward improvement (Koopman et al., 2009). In contrast, no behavioral or morphological improvements were evident following either single or repeated doses of thalidomide (100 mg/kg), administered 10 min, 4 and 24 hr after SCI in a study by Reyes-Alva et al., (2009). Moreover, in other animal models of brain injury, such as ischemia, thalidomide has demonstrated efficacy over a narrow dose range when administered prior to injury (Lee et al., 2007; Hyakkoku et al., 2009). By comparison, in the present study, 3,6’-dithiothalidomide compares favorably with the results of past studies of its parent compound, and the full reversal of mTBI-induced cognitive impairments induced by both studied doses, suggest that still lower doses may prove effective and warrant future assessment. A reexamination and reinterpretation of the novel object recognition test has recently been published (McTighe et al., 2010). These investigators suggest, rather than a loss of memory for the familiar object, there is a “false memory” of the novel object as familiar after perirhinal cortex damage.
An evaluation of the behavioral and histopathological outcome of TBI in mice deficient in TNF-α (TNF-α−/−) and their wild-type (wt) littermates has provided insight into the time-dependent roles of TNF-α in brain injury and repair (Scherbel et al., 1999), albeit that the TBI model studied, induced by cortical impact injury, was far more severe than ours. TNF-α −/− mice suffered significantly less behavioral (assessed at a week) and neuromotor deficits (assessed at 2 days) than wt mice. However, whereas the latter demonstrated some recovery over 4 weeks post TBI, the TNF-α−/− lacked improvement, and their cortical brain damage (assessed histologically at 4 weeks) proved greater (Scherbel et al., 1999). Such results suggest that TNF-α has time-dependent actions, playing a detrimental role during the acute phase within the traumatized brain, but providing beneficial actions in the delayed, chronic reparative phase, and are in accord with results of more deleterious effects in TNF-α receptor knockout mice following TBI (Sullivan et al., 1999; Stahel et al., 2000); thereby emphasizing the importance of defining a treatment window.
In summary, the results of the current study support 3,6’-dithiothalidomide as a potent TNF-α synthesis inhibitor and a promising drug in the treatment of experimental mTBI. Administration within the initial 12 hr window following injury resulted in full amelioration of behavioral deficits. Additionally, such protection supports a role for elevated TNF-α levels in the development of mTBI, and its targeting as a treatment strategy (Esposito & Cuzzocrea 2009; Frankola et al., 2011).
Supplementary Material
Brain levels of TNF-α were time-dependently elevated following mTBI, rising acutely within an hour, peaking at 4.5-fold above control values by 12 hr, and returning to basal levels by 18 hr. (n=3–5, *p 0.05 (Dunnett’s t test) compared to control (basal values) (n=5)).
Acknowledgments
This research was supported in part by the Sackler School of Medicine, Tel-Aviv University, and the Intramural Research Programs of both the National Institute on Aging and National Institute on Drug Abuse, National Institutes of Health.
References
- Andersson M, Svenungsson E, Khademi M, Lampa J, Brundin L, Wallstrom E. No signs of immunoactivation in the cerebrospinal fluid during treatment with infliximab. Ann Rheum Dis. 2006;65:1237–1240. doi: 10.1136/ard.2006.055194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragon-Ching JB, Li H, Gardner ER, Figg WD. Thalidomide analogues as anticancer drugs. Recent Pat Anticancer Drug Discov. 2007;2:167–174. doi: 10.2174/157489207780832478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baratz R, Rubovitch V, Frenk H, Pick CG. The influence of alcohol on behavioral recovery after mTBI in mice. J Neurotrauma. 2010;27:555–563. doi: 10.1089/neu.2009.0891. [DOI] [PubMed] [Google Scholar]
- Bryant RA, O'Donnell ML, Creamer M, McFarlane AC, Clark CR, Silove D. The psychiatric sequelae of traumatic injury. Am J Psychiatry. 2010;167:312–320. doi: 10.1176/appi.ajp.2009.09050617. [DOI] [PubMed] [Google Scholar]
- Cernak I, Noble-Haeusslein LJ. Traumatic brain injury: an overview of pathobiology with emphasis on military populations. J Cereb Blood Flow Metab. 2010;30:255–266. doi: 10.1038/jcbfm.2009.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cederberg D, Siesjo P. What has inflammation to do with traumatic brain injury? Childs Nerv Syst. 2010;26:221–226. doi: 10.1007/s00381-009-1029-x. [DOI] [PubMed] [Google Scholar]
- Dellu F, Mayo W, Cherkaoui J, Le Moal M, Simon H. A two-trial memory task with automated recording: study in young and aged rats. Brain Res. 1992;588:132–139. doi: 10.1016/0006-8993(92)91352-f. [DOI] [PubMed] [Google Scholar]
- Dix SL, Aggleton JP. Extending the spontaneous preference test of recognition: Evidence of object-location and object-context recognition. Behav Brain Res. 1999;99:191–200. doi: 10.1016/s0166-4328(98)00079-5. [DOI] [PubMed] [Google Scholar]
- Donkin JJ, Vink R. Mechanisms of cerebral edema in traumatic brain injury: therapeutic developments. Curr Opin Neurol. 2010 doi: 10.1097/WCO.0b013e328337f451. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Edut S, Rubovitch V, Schreiber S, Pick CG. The intriguing effects of ecstasy (MDMA) on cognitive function in mice subjected to a minimal traumatic brain injury (mTBI) Psychopharmacol (Berl) 2010 doi: 10.1007/s00213-010-2098-y. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Esposito E, Cuzzocrea S. TNF-alpha as a therapeutic target in inflammatory diseases, ischemia-reperfusion injury and trauma. Curr Med Chem. 2009;16:3152–3167. doi: 10.2174/092986709788803024. [DOI] [PubMed] [Google Scholar]
- Frank-Cannon TC, Alto LT, McAlpine FE, Tansey MG. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol Neurodegener. 2009;4:47. doi: 10.1186/1750-1326-4-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankola KA, Greig NH, Luo W, Tweedie D. Targeting TNF-alpha to elucidate and ameliorate neuroinflammation in neurodegenerative diseases. CNS & Neurological Disorders - Drug Targets. 2011;10 doi: 10.2174/187152711794653751. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner J, Brett SJ. Mechanisms of injury by explosive devices. Anesthesiol Clin. 2007;25:147–160. doi: 10.1016/j.anclin.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Ghajar J, Ivry RB Cognitive Neurobiological Research Consortium. The predictive brain state: timing deficiency in traumatic brain injury? Neurorehabil Neural Repair. 2008;22:217–227. doi: 10.1177/1545968308315600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig NH, Giordano T, Zhu X, Yu QS, Perry TA, Holloway HW, Brossi A, Rogers JT, Sambamurti K, Lahiri DK. Thalidomide-based TNF-alpha inhibitors for neurodegenerative diseases. Acta Neurobiol Exp. 2004;64:1–9. doi: 10.55782/ane-2004-1486. [DOI] [PubMed] [Google Scholar]
- Hesdorffer DC, Rauch SL, Tamminga CA. Long-term psychiatric outcomes following traumatic brain injury: a review of the literature. J Head Trauma Rehabil. 2009;24:452–459. doi: 10.1097/HTR.0b013e3181c133fd. [DOI] [PubMed] [Google Scholar]
- Hyakkoku K, Nakajima Y, Izuta H, Shimazawa M, Yamamoto T, Shibata N, Hara H. Thalidomide protects against ischemic neuronal damage induced by focal cerebral ischemia in mice. Neuroscience. 2009;159:760–769. doi: 10.1016/j.neuroscience.2008.12.043. [DOI] [PubMed] [Google Scholar]
- Israelsson C, Wang Y, Kylberg A, Pick CG, Hoffer BJ, Ebendal T. Closed head injury in a mouse model results in molecular changes indicating inflammatory responses. J Neurotrauma. 2009;26:1307–1314. doi: 10.1089/neu.2008.0676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH. Traumatic brain injury and amyloid-beta pathology: a link to Alzheimer's disease? Nat Rev Neurosci. 2010 doi: 10.1038/nrn2808. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiraly M, Kiraly SJ. Traumatic brain injury and delayed sequelae: a review--traumatic brain injury and mild traumatic brain injury (concussion) are precursors to later-onset brain disorders, including early-onset dementia. Scientific World Journal. 2007;7:1768–1776. doi: 10.1100/tsw.2007.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khera TK, Dick AD, Nicholson LB. Mechanisms of TNFα regulation in uveitis: focus on RNA-binding proteins. Prog Retin Eye Res. 2010;29:610–25. doi: 10.1016/j.preteyeres.2010.08.003. [DOI] [PubMed] [Google Scholar]
- Knight R. IMiDs: a novel class of immunomodulators. SeminOncol. 2005;32(4 Suppl 5):S24–30. doi: 10.1053/j.seminoncol.2005.06.018. [DOI] [PubMed] [Google Scholar]
- Knoblach SM, Fan L, Faden AI. Early neuronal expression of tumor necrosis factor-α after experimental brain injury contributes to neurological impairment. J Neuroimmunol. 1999;95:115–125. doi: 10.1016/s0165-5728(98)00273-2. [DOI] [PubMed] [Google Scholar]
- Koopmans GC, Deumens R, Buss A, Geoghegan L, Myint AM, Honig WH, Kern N, Joosten EA, Noth J, Brook GA. Acute rolipram/thalidomide treatment improves tissue sparing and locomotion after experimental spinal cord injury. Exp Neurol. 2009;216:490–498. doi: 10.1016/j.expneurol.2009.01.005. [DOI] [PubMed] [Google Scholar]
- Lecca D, Trincavelli ML, Gelosa P, Sironi L, Ciana P, Fumagalli M, Villa G, Verderio C, Grumelli C, Guerrini U, Tremoli E, Rosa P, Cuboni S, Martini C, Buffo A, Cimino M, Abbracchio MP. The recently identified P2Y-like receptor GPR17 is a sensor of brain damage and a new target for brain repair. PLoS One. 2008;3(10):e3579. doi: 10.1371/journal.pone.0003579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CJ, Kim KW, Lee HM, Nahm FS, Lim YJ, Park JH, Kim CS. The effect of thalidomide on spinal cord ischemia/reperfusion injury in a rabbit model. Spinal Cord. 2007;45:149–157. doi: 10.1038/sj.sc.3101931. [DOI] [PubMed] [Google Scholar]
- Lu J, Goh SJ, Tng PY, Deng YY, Ling EA, Moochhala S. Systemic inflammatory response following acute traumatic brain injury. Front Biosci. 2009;14:3795–3813. doi: 10.2741/3489. [DOI] [PubMed] [Google Scholar]
- McTighe SM, Cowell RA, Winters BD, Bussey TJ, Saksida LM. Paradoxical false memory for objects after brain damage. Science. 2010;330:1408–1410. doi: 10.1126/science.1194780. [DOI] [PubMed] [Google Scholar]
- Melchert M, List A. The thalidomide saga. Int J Biochem Cell Biol. 2007;39:1489–1499. doi: 10.1016/j.biocel.2007.01.022. [DOI] [PubMed] [Google Scholar]
- Messier C. Object recognition in mice: improvement of memory by glucose. Neurobiol Learn Mem. 1997;67:172–175. doi: 10.1006/nlme.1996.3755. [DOI] [PubMed] [Google Scholar]
- Milman A, Rosenberg A, Weizman R, Pick CG. Mild traumatic brain injury induces persistent cognitive deficits and behavioral disturbances in mice. J Neurotrauma. 2005;22:1003–1010. doi: 10.1089/neu.2005.22.1003. [DOI] [PubMed] [Google Scholar]
- Moppett IK. Traumatic brain injury: assessment, resuscitation and early management. Br J Anaesth. 2007;99:18–31. doi: 10.1093/bja/aem128. [DOI] [PubMed] [Google Scholar]
- Moreira AL, Sampaio EP, Zmuidzinas A, Frindt P, Smith KA, Kaplan G. Thalidomide exerts its inhibitory action on tumor necrosis factor alpha by enhancing mRNA degradation. J Exp Med. 1993;177:1675–1680. doi: 10.1084/jem.177.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan W, Kastin AJ, Rigai T, McLay R, Pick CG. Increased hippocampal uptake of tumor necrosis factor alpha and behavioral changes in mice. Exp Brain Res. 2003;149:195–199. doi: 10.1007/s00221-002-1355-7. [DOI] [PubMed] [Google Scholar]
- Patil CS, Liu M, Zhao W, Coatney DD, Li F, VanTubergen EA, D'Silva NJ, Kirkwood KL. Targeting mRNA stability arrests inflammatory bone loss. Mol Ther. 2008;16:1657–64. doi: 10.1038/mt.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghupathi R. Cell death mechanisms following traumatic brain injury. Brain Pathol. 2004;14:215–222. doi: 10.1111/j.1750-3639.2004.tb00056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reale M, Greig NH, Kamal MA. Peripheral chemo-cytokine profiles in Alzheimer’s and Parkinson’s diseases. Mini Rev Med Chem. 2009;9:1229–1241. doi: 10.2174/138955709789055199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Alva HJ, Franco-Bourland RE, Martinez-Cruz A, Grijalva I, Fuchs B, Madrazo I, Guizar-Sahagun G. Thalidomide fails to be therapeutic following contusive spinal cord injury in rats. Acta Neurobiol Exp. 2009;69:494–503. doi: 10.55782/ane-2009-1759. [DOI] [PubMed] [Google Scholar]
- Rubovitch V, Edut S, Sarfstein R, Werner H, Pick CG. The intricate involvement of the Insulin-like growth factor receptor signaling in mild traumatic brain injury in mice. Neurobiol Dis. 2010;38:299–303. doi: 10.1016/j.nbd.2010.01.021. [DOI] [PubMed] [Google Scholar]
- Sampaio EP, Sarno EN, Galilly R, Cohn ZA, Kaplan G. Thalidomide selectively inhibits tumor necrosis factor alpha production by stimulated human monocytes. J Exp Med. 1991;173:699–703. doi: 10.1084/jem.173.3.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherbel U, Raghupathi R, Nakamura M, Saatman KE, Trojanowski JQ, Neugebauer E, Marino MW, McIntosh TK. Differential acute and chronic responses of tumor necrosis factor- deficient mice to experimental brain injury. PNAS. 1999;96:8721–8726. doi: 10.1073/pnas.96.15.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon EJ, Sandoval F. Thalidomide can be either agonistic or antagonistic to LPS evoked synthesis of TNF-alpha by mono- nuclear cells. Immunopharmacol Immunotoxicol. 1996;18:59–72. doi: 10.3109/08923979609007110. [DOI] [PubMed] [Google Scholar]
- Shohami E, Bass R, Wallach D, Yamin A, Gallily R. Inhibition of tumor necrosis factor alpha (TNF-alpha) activity in rat brain is associated with cerebroprotection after closed head injury. J Cereb Blood Flow Metab. 1996;16:378–384. doi: 10.1097/00004647-199605000-00004. [DOI] [PubMed] [Google Scholar]
- Shohami E, Gallily R, Mechoulam R, Bass R, Ben-Hur T. Cytokine production in the brain following closed head injury: dexanabinol (HU-211) is a novel TNF-α inhibitor and an effective neuroprotectant. J Neuroimmunol. 1997;72:169–177. doi: 10.1016/s0165-5728(96)00181-6. [DOI] [PubMed] [Google Scholar]
- Shohami E, Novikov M, Bass R, Yamin A, Gallily R. Closed head injury triggers early production of TNF-α and IL-6 by brain tissue. J Cereb Blood Flow Metab. 1994;14:615–619. doi: 10.1038/jcbfm.1994.76. [DOI] [PubMed] [Google Scholar]
- Stahel PF, Shohami E, Younis FM, Kariya K, Otto VI, Lenzlinger PM, Grosjean MB, Eugster HP, Trentz O, Kossmann T, Morganti-Kossmann MC. Experimental closed head injury: analysis of neurological outcome, blood–brain barrier dysfunction, intracranial neutrophil infiltration, and neuronal cell death in mice deficient in genes for pro-inflammatory cytokines. J Cereb Blood Flow Metab. 2000;20:369–380. doi: 10.1097/00004647-200002000-00019. [DOI] [PubMed] [Google Scholar]
- Stamou P, Kontoyiannis DL. Posttranscriptional regulation of TNF mRNA: a paradigm of signal-dependent mRNA utilization and its relevance to pathology. Curr Dir Autoimmun. 2010;11:61–79. doi: 10.1159/000289197. [DOI] [PubMed] [Google Scholar]
- Stoica BA, Faden AI. Cell death mechanisms and modulation in traumatic brain injury. Neurotherapeutics. 2010;7:3–12. doi: 10.1016/j.nurt.2009.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svetlov SI, Larner SF, Kirk DR, Atkinson J, Hayes RL, Wang KK. Biomarkers of blast-induced neurotrauma: profiling molecular and cellular mechanisms of blast brain injury. J Neurotrauma. 2009;26:913–921. doi: 10.1089/neu.2008.0609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PG, Bruce-Keller AJ, Rabchevsky AG, Christakos S, Clair DK, Mattson MP, Scheff SW. Exacerbation of damage and altered NF-κB activation in mice lacking tumor necrosis factor receptors after traumatic brain injury. J Neurosci. 1999;19:6248–6256. doi: 10.1523/JNEUROSCI.19-15-06248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadesse A, Abebe M, Bizuneh E, Mulugeta W, Aseffa A, Shannon EJ. Effect of thalidomide on the expression of TNF-alpha m-RNA and synthesis of TNF-alpha in cells from leprosy patients with reversal reaction. Immunopharmacol Immunotoxicol. 2006;28:431–441. doi: 10.1080/08923970600928023. [DOI] [PubMed] [Google Scholar]
- Tansey MG. Inflammation in neuropsychiatric disease. Neurobiol Dis. 2010;37:491–492. doi: 10.1016/j.nbd.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansey MG, Szymkowski DE. The TNF superfamily in 2009: new pathways new indications new drugs. Drug Discov Today. 2009;14:1082–1088. doi: 10.1016/j.drudis.2009.10.002. [DOI] [PubMed] [Google Scholar]
- Tashlykov V, Katz Y, Volkov A, Gazit V, Schreiber S, Zohar O, Pick CG. Minimal traumatic brain injury induce apoptotic cell death in mice. J Mol Neurosci. 2009;37:16–24. doi: 10.1007/s12031-008-9094-2. [DOI] [PubMed] [Google Scholar]
- Taupin V, Toulmond S, Serrano A, Benavides J, Zavala F. Increase in IL-6, IL-1 and TNF levels in rat brain following traumatic lesion: influence of pre- and post-traumatic treatment with Ro5 4864, a peripheral-type (p site) benzodiazepine ligand. J Neuroimmunol. 1993;42:177–185. doi: 10.1016/0165-5728(93)90008-m. [DOI] [PubMed] [Google Scholar]
- Taylor PC. Pharmacology of TNF blockade in rheumatoid arthritis and other chronic inflammatory diseases. Curr Opin Pharmacol. 2010;10:308–315. doi: 10.1016/j.coph.2010.01.005. [DOI] [PubMed] [Google Scholar]
- Teo SK, Colburn WA, Tracewell WG, Kook KA, Stirling DI, Jaworsky MS, Scheffler MA, Thomas SD, Laskin OL. Clinical pharmacokinetics of thalidomide. Clin Pharmacokinet. 2004;43:311–327. doi: 10.2165/00003088-200443050-00004. [DOI] [PubMed] [Google Scholar]
- Tobinick E. Perispinal etanercept: a new therapeutic paradigm in neurology. Expert Rev Neurother. 2010;10:985–1002. doi: 10.1586/ern.10.52. [DOI] [PubMed] [Google Scholar]
- Tweedie D, Milman A, Holloway HW, Li Y, Harvey BK, Shen H, Pistell PJ, Lahiri DK, Hoffer BJ, Wang Y, Pick CG, Greig NH. Apoptotic and behavioral sequelae of mild brain trauma in mice. J Neurosci Res. 2007a;85:805–815. doi: 10.1002/jnr.21160. [DOI] [PubMed] [Google Scholar]
- Tweedie D, Sambamurti K, Greig NH. TNF-alpha inhibition as a treatment strategy for neurodegenerative disorders: new drug candidates and targets. Curr Alzheimer Res. 2007b;4:378–385. doi: 10.2174/156720507781788873. [DOI] [PubMed] [Google Scholar]
- Tweedie D, Luo W, Short RG, Brossi A, Holloway HW, Li Y, Yu QS, Greig NH. A cellular model of inflammation for identifying TNF-alpha synthesis inhibitors. J Neurosci Methods. 2009;183:182–187. doi: 10.1016/j.jneumeth.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tweedie D, Frankola KA, Luo W, Li Y, Greig NH. Thalidomide analogues suppress lipopolysaccharide-induced synthesis of TNF-α and nitrite, an intermediate of nitric oxide, in a cellular model of inflammation. Open Biochem J. 2011 doi: 10.2174/1874091X01105010037. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uryu K, Giasson BI, Longhi L, Martinez D, Murray I, Conte V, Nakamura M, Saatman K, Talbot K, Horiguchi T, McIntosh T, Lee VM, Trojanowski JQ. Age-dependent synuclein pathology following traumatic brain injury in mice. Exp Neurol. 2003;184:214–224. doi: 10.1016/s0014-4886(03)00245-0. [DOI] [PubMed] [Google Scholar]
- Vakil E. The effect of moderate to severe traumatic brain injury (TBI) on different aspects of memory: a selective review. J Clin Exp Neuropsychol. 2005;27:977–1021. doi: 10.1080/13803390490919245. [DOI] [PubMed] [Google Scholar]
- Yan YP, Lang BT, Vemuganti R, Dempsey RJ. Galectin-3 mediates post-ischemic tissue remodeling. Brain Res. 2009;1288:116–124. doi: 10.1016/j.brainres.2009.06.073. [DOI] [PubMed] [Google Scholar]
- Yang J, You Z, Kim HH, Hwang SK, Khuman J, Guo S, Lo EH, Whalen MJ. Genetic analysis of the role of tumor necrosis factor receptors in functional outcome after traumatic brain injury in mice. J Neurotrauma. 2010;27:1037–1046. doi: 10.1089/neu.2009.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou QH, Boado RJ, Hui EK, Lu JZ, Pardridge WM. Brain-penetrating tumor necrosis factor decoy receptor in the mouse. Drug Metab Dispos. 2011;39:71–6. doi: 10.1124/dmd.110.036012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Giordano T, Yu QS, Holloway HW, Perry TA, Lahiri DK, Brossi A, Greig NH. Thiothalidomides: novel isosteric analogues of thalidomide with enhanced TNF-alpha inhibitory activity. J Med Chem. 2003;46:5222–5229. doi: 10.1021/jm030152f. [DOI] [PubMed] [Google Scholar]
- Ziebell JM, Morganti-Kossmann MC. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics. 2010;7:22–30. doi: 10.1016/j.nurt.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zohar O, Getslev V, Miller AL, Schreiber S, Pick CG. Morphine protects for head trauma induced cognitive deficits in mice. Neurosci Lett. 2006;394:239–242. doi: 10.1016/j.neulet.2005.10.099. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Brain levels of TNF-α were time-dependently elevated following mTBI, rising acutely within an hour, peaking at 4.5-fold above control values by 12 hr, and returning to basal levels by 18 hr. (n=3–5, *p 0.05 (Dunnett’s t test) compared to control (basal values) (n=5)).