

Summary
Gene profiling is an excellent tool to identify the genetic mechanisms, networks, and molecular pathways involved in skeletal muscle development and muscular disorders. Oligonucleotide or cDNA microarray can be the first step to identify the global gene expression in the study of interest. As microarray techniques provide a large set of differentially expressed genes in a given comparison, the expression profile can be narrowed down by taking various parameters into consideration such as fold values, p-values, and their relevance to the study. Every technique has its own limitations. Therefore, further validation of the results with a different technique is always necessary. Quantitative real-time reverse-transcriptase polymerase chain reaction (qRT-PCR) is the most common technique to validate microarray data and to study the relative expression of specific genes in any experimental set-up. Here, we describe, the qRT-PCR technique, in detail, for -successful gene expression studies in skeletal muscle cells and tissues.
Keywords: Skeletal muscle, quantitative real-time PCR, C2C12 myoblasts, RNA isolation, RNA analysis, gene expression
1. Introduction
Skeletal muscle constitutes approximately 40% of human body mass and is required for basic functions such as locomotion, metabolism, and respiration. Myogenesis is a multistep developmental program that generates skeletal muscle in embryos (1, 2). This process is also required for postnatal growth and the regeneration of myofibers after injury (1). Adult skeletal muscle exhibits a very high level of plasticity. Resistance exercise and nutritional uptake leads to skeletal muscle hypertrophy (3). Conversely, skeletal muscle undergoes atrophy or wasting, which is a critical determinant of human morbidity and mortality in a wide variety of disease states and conditions (4-6). Furthermore, mutations in many muscle structural genes lead to muscular dystrophy, a group of fatal genetic disorders involving striated muscle (7).
Skeletal muscle development as well as pathology is governed by coordinated changes in the activity and expression of several genes (1, 8). Different physiological and pathophysiological stimuli modulate the activity of various signaling pathways and transcription factors leading to expression of a specific set of genes in nuclei. A number of gene profiling studies using microarray approaches have been performed leading to the identification of sets of genes involved in skeletal muscle development, plasticity, and muscular disorders such as muscular dystrophy and inflammatory myopathies (9-14). Although microarray is a powerful approach to simultaneously study the expression of a large number of genes, the reliability of microarray results depends on several factors such as array production, RNA extraction, probe labeling, hybridization conditions, and image analysis. Therefore, the genes identified as differentially expressed by microarray approaches must be confirmed by another method. Quantitative real-time PCR (qRT-PCR) is a highly sensitive and rapid technique which requires 1000-fold less RNA compared to conventional assays such as Northern blot for gene expression studies (15). With the advent of multiplexing technology, it has now become possible to study the expression of multiple genes in a single qRT-PCR reaction. Furthermore, the qRT-PCR is an ideal technique to study relative expression of known muscle genes in experimental or clinical settings.
In addition to animal models, the major steps of muscle development and plasticity can be recapitulated using C2C12 myoblasts/myotubes in vitro. Indeed, C2C12 myoblasts are the most commonly used cells for studying skeletal muscle in culture. Here, we provide the detailed methodology for RNA isolation and analysis and qRT-PCR technique for gene expression studies in skeletal muscle cells and tissues. Though most of the procedures given in this chapter are similar across all types of real-time PCR machines, we have provided details of qRT-PCR assays using 7300 Sequence Detection System (Applied Biosystems) and SYBR Green dye.
2. Materials
Prepare all solutions using nuclease-free water and under nuclease-free conditions (see Notes 1, 2 and 3).
2.1 Cell Culture
2.1.1 Cell lines
C2C12 myoblasts (American Type Culture Collection).
2.1.2 Growth Medium
Perform all the following steps aseptically under the hood. All the cell culture reagents are from Invitrogen unless specified.
To 500ml of DMEM high Glucose 1X, add following:
5 ml Hepes from 100X stock
5 ml Penicillin/streptomycin from 100X stock
50 ml heat-inactivated fetal bovine serum d). 0.25% trypsin solution
2.1.3 Differentiation medium
Perform all the following steps aseptically under the hood.
To 500ml of DMEM high Glucose 1X add following:
5 ml Hepes from 100X stock
5 ml Penicillin/streptomycin from 100X stock
10 ml horse serum (Sigma)
2.2. Quantitative Real-Time RT-PCR
2.2.1 Reagents for total RNA isolation
TRIzol Reagent. Store the reagent at 4°C in amber color bottle (see Note 4).
Diethyl Pyrocarbonate (DEPC)-treated water: Add 1ml of 0.1% DEPC to sterile water and mix well. Alternatively, water can be autoclaved after adding DEPC (see Note 5).
75% ethanol: Add 25 ml of DEPC-treated water to 75 ml absolute ethanol. Mix well and refrigerate until further use.
RNeasy kit.
Chlorofom (Molecular Biology grade).
2.2.2 Reagents for cDNA synthesis
-
High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The kit consists of the following components:
10x Reverse transcription (RT) buffer, 100 mM dNTP mix, 10x RT Random Primers, Multiscribe Reverse Transcriptase 50units/μl.
2. DNase/RNase free water.
2.2.3 Reagents for RNA quality analysis using Agilent 2100 Bioanalyzer
RNA samples (up to 12 per chip).
RNA 6000 ladder (Ambion).
-
Bioanalyzer RNA Nano Kit .
Includes chips and reagents.
RNaseZap.
2.2.4 SYBR Green qRT-PCR components
All the components are from Applied Biosystems unless specified.
cDNA made previously (see cDNA synthesis).
DNase/RNase free water (Fisher Scientific).
20 μM Primers, Forward and Reverse from your favorite vendor.
Power SYBR Green PCR Master Mix.
96-well Optical Reaction Plate with barcode 128.
MicroAmp optical Adhesive Film, PCR compatible, DNA/RNA/RNase free.
2.2.5 Primers
We design our primers using Vector NTI software (Invitrogen). Using this software, we get >95% success rate that the primers will work in qRT-PCR assay.
3. Methods
3.1 Culturing C2C12 myoblasts and their differentiation into myotubes
Culture C2C12 myoblasts in growth medium at 37°C and 5% CO2 in an incubator until they reach 70-80% confluency.
Split the myoblasts in 1:3 ratios to maintain the undifferentiated myoblast population.
Differentiate C2C12 myoblast cultures by replacing the growth medium with differentiation medium (DM).
Change differentiation medium every 48h to maintain healthy myotubes.
3.2 Extraction of total RNA from cultured myoblasts/myotubes
Perform all centrifugation steps in an Eppendorf microcentrifuge.
Depending on the study the treated myoblasts/myotubes were harvested by trypsinizing the cells using 0.25% trypsin solution.
Collect the cells in microcentrifuge tubes and pellet them by centrifuging at 8,000 rpm for 2 min at 4°C.
Discard the supernatant and add 1 ml TRIzol Reagent.
Pipette the cell pellet in TRIzol 5-6 times to ensure proper lysis.
To the homogenate, add 200 μl chloroform and mix well by vortexing the tube for 1 min.
Incubate at room temperature for 5 min.
Centrifuge the tubes at 12,000 rpm in refrigerated centrifuge for 15 min.
Carefully transfer the top aqueous phase (approx 600 μl) into a new eppendorf tube, add an equal volume of 75% ethanol, and vortex vigorously for 5 sec.
Add the mixture to the RNeasy spin column from the RNeasy kit.
Centrifuge the tubes at 15,000 rpm for 20 sec and discard the flow-through.
Add 100 μl of buffer RW1 (from RNeasy kit) and spin at 15,000 rpm for 20 sec.
Discard the flow-though and the collection tubes and put columns into new collection tubes.
Add 500 μl of buffer RPE (from RNeasy kit) and centrifuge at 15,000 rpm for 30 sec.
Discard the flow-through and repeat step 13.
Discard the flow-through and centrifuge again at 15,000 rpm for 30 sec without buffer.
Discard the flow-through and collection tubes and put columns into new sterile 1.5 ml eppendorf tubes.
Add 30 μl RNase-free water in the center of the tube.
Incubate at room temperature for 5 min and centrifuge at 15,000 rpm for 1 minute.
19. Measure the concentration of RNA by spectrophotometer or any other standard methods.
3.3 Total RNA Isolation from Skeletal Muscle Tissues
Place approximately 30-40 mg skeletal muscle tissue in a prelabeled microcentrifuge tubes placed on ice.
Chop tissues using a sterile razor blade in a dish (kept on ice) in a drop of TRIzol reagent.
Transfer chopped tissue to a glass mortar and pestle pretreated with DEPC water for 2 h.
Add 1ml TRIzol reagent to it and homogenize well (see Note 6).
Transfer the homogenate into a 1.5 ml eppendorf tube.
For rest of the process, follow steps 5 to 19 as described above for isolation of total RNA from cultured myoblasts.
3.4. Evaluating RNA quality
We use Agilent 2100 Bioanalyzer to check the quality of RNA (see Note 7). The complete protocol for analyzing RNA quality is described as follows:
Clean the electrodes by placing a washing chip filled with 350μl RNAseZap in the Bioanalyzer for 1 minute. Place a washing chip filled with 350μl DEPC water in the Bioanalyzer for 30 sec.
Leave the lid open for 10 sec to allow the electrodes to dry.
Place 400 μl of RNA gel matrix into the top receptacle of a spin filter (included in RNA Nano kit), place the spin filter in a microcentrifuge and spin at 4000 rpm for 10 min. Filtered gel must be used within 4 weeks.
Put 130μl of the filtered RNA gel matrix into an RNAase-free eppendorf tube and add 2μl RNA concentrate provided in the RNA Nano kit.
Vortex vigorously to ensure proper mixing of gel and dye (see Note 8).
Take a new RNA chip out of its sealed bag and place it on the chip priming station.
Take 9.0 μl of the gel-dye mix with a pipette and dispense this in the well marked with a black circle “G”.
Ensure that the plunger is at 1 ml. Then close the chip priming station. Press the plunger until it is held by the syringe clip.
Wait for 30 sec. Slip plunger off clip and wait 5 sec and then slowly pull the plunger back to the 1 ml mark. Open Chip Priming Station.
Pipette 9μl of the gel-dye mix in each of the wells marked with an uncircled “G” (see Notes 9 and 10).
Pipette 5μl of the RNA 6000 Nano Marker and put it in the well marked with the ladder symbol.
Dispense 5 μl of the RNA 6000 Nano Marker into each of the 12 sample wells.
Dilute RNA samples at 1:10 and 1:100 using RNAase-free water. Also put 2μl RNA ladder in a tube.
Heat the tubes at 70°C for 2 min to denature RNA samples followed by snap cooling by putting the tubes on ice.
Pipette 1μl RNA 6000 ladder and put it in the well with the ladder symbol.
Pipette 1μl of each sample and put it in each sample well (see Note 11).
Pipette 1μl of RNA Nano Marker in each unused well (see Note 12).
Place the chip in the Agilent 2100 Bioanalyzer scanner and follow the user manual protocol for operating the machine and extracting the analyzed data.
Finally print results. A typical gel picture and results of one RNA sample of good quality are shown in Figure 1 (see Note 13).
FIGURE 1. Analysis of RNA quality by using Agilent 2100 Bioanalyzer.
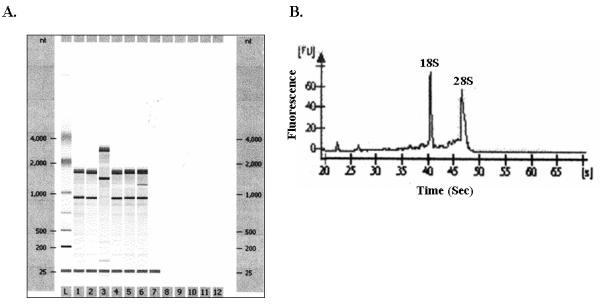
A). A representative gel like picture after running the total RNA samples on Agilent 2100 Bioanalyzer is presented here. Six different C2C12 RNA preparations were analyzed with the Agilent 2100 Bioanalyzer. B). The electropherograms of RNA sample # 1 showing the presence of ribosomal RNA 28S and 18S peaks is shown here.
3.5 cDNA synthesis
Use 2 μg of RNA in a final volume made to 10μl with DNase/RNase-free water in a 0.2 ml PCR tube (Note 14).
Make PCR master-mix using the High Capacity cDNA Reverse Transcription Kit as follows (see Note 15):
Add 10μl of PCR master-mix to RNA sample (i.e. from step 1 above). Total volume is now 20μl.
Briefly vortex and centrifuge the tubes.
Program a PCR machine as follows:
Put the tubes on PCR heating block and run PCR reaction. Store the tubes at −20°C after the PCR reaction is finished.
3.6 Quantitative real-time RT-PCR
The Quantitative real-time RT-PCR consists of 2 steps: 1. Designing Primers, and 2. Setting up the reaction.
3.6.1. Designing Primers
We design primers using Vector NTI software (Note 16). Basic steps for designing good quality primers using Vector NTI software are as follows:
Find the cDNA or mRNA sequence for the gene of interest in PubMed nucleotide database and copy the gene’s unique gene ID number.
Open Vector NTI software followed by clicking on Tools in the main menu, and then click on open link GID.
Paste the gene ID in the dialog box and click OK. The program will download the nucleotide sequence on your computer.
Select a region of 300-400 base pairs (bp) within the cDNA sequence.
Go to Analyses in main menu and click on Primer Design. A dialog box will appear.
-
Make the following entry in only specific fields. The others are default; don’t change the values in them.
Product length: Min:100 bp, Max: 200 bp
Maximum number of output options: 50
Tm (C): ≥55 and ≤ 60
%GC: ≥55 and ≤ 60
Length: ≥20 and ≤ 25
Click “Apply”. A window will open on upper left corner containing sequence of 50 primer sets.
Look at each forward and reverse primer sequence individually. The GC difference should be 0°C. Tm difference should not be more than +1°C between two primers.
Click on the first primer meeting the required parameters, and then select Analyze. A new window will open containing the selected primer information.
Check for palindrome and repeats. Palindrome and Repeats should be 0. If there is any number of Palindrome or Repeats in either forward or reverse primers, do not use this pair. Perform the same on other set of primers.
Next click on “Dimers and Hairpin Loops” icon. A new window will open providing separately the number of hairpin loops and dimers in the selected primer. The ideal situation is that we should have no hairpin loop and dimers. However, it is rare for most of the primers. The following criteria can be used to pick the good primers even with those having hair-pin loops and primer dimers.
Make sure that the primer does not have more than 8 hairpin loops or dimers. Minimum is better but up to 8 are acceptable.
Check the dimer dG and hairpin dG for each dimer and hairpin loop, respectively by clicking >> button on the window.
14. The best value for dG should be 0 kcal/mol. However, dG values between −1.8 to +1.8 kcal/mol can be acceptable. If any of the two primers in the pair has a dG value outside this range, then don’t use this pair and analyze other primer sets (see Note 17).
Once the right primer set is found, copy primer sequence and send for primer synthesis (see Note 18).
Test run qRT-PCR using these primers with a few samples. The primer set which shows a good dissociation curve should be used for qRT-PCR.
Figure 2A shows a typical dissociation curve for a good primer set with no primer dimers. Primer sets having primer dimers are shown in Figure 2B. The dissociation curve of a good qRT-PCR assay should look similar to Figure 2A (see Note 19).
FIGURE 2. Dissociation curves in qRT-PCR assay.
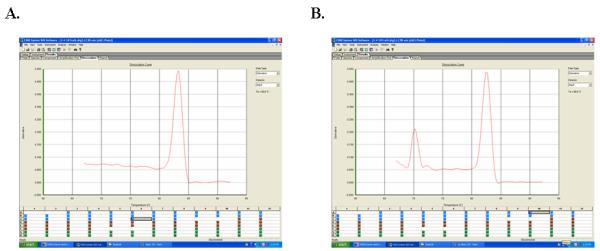
A). A typical dissociation curve with no primer dimer after running a qRT-PCR assay is shown here. B). The left peak represents a primer dimer in this dissociation curve.
3.6.2. Setting up the reaction
After synthesizing the cDNA and obtaining primers, the real time PCR reaction is set up as follows:
Prepare the master-mix with all the ingredients except cDNA.
2. Dispense 1μl of cDNA in the individual wells of 96-well Optical Reaction Plate with barcode 128 and then add 19μl of master mix to each well. All reactions should be carried out in duplicate or triplicate to reduce variation.
Data normalization is accomplished using the endogenous control such Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) or β-actin (see Note 20).
Seal the plate using MicroAmp optical Adhesive Film and spin the plate in PCR plate centrifuge.
Insert the plate into the 7300 Sequence Detection system.
- Set thermal conditions for qRT-PCR using 7300 system SDS software as follows:
- Denaturation at 95 °C for 10 min
- 40 cycles of denaturation at 95°C for 15s, annealing and extension at 60 °C for 1 min
- Finally, a melting curve of 95°C for 15s, 60°C for 15s, and 95°C for 15s.
Click on the 7300 system SDS software icon on the computer attached with 7300 Sequence Detection system. Click on Create New Document tab.
A new window will appear, select ddCt (Relative Quantitation) Plate in the Assay pull down menu.
Click “Next” and enter the name of the primes to be used in left side window. Then select the primer in left window and click Add button. After adding all primer names, click Next tab.
Enter the sample information for each well and save the file as .sds document.
In the same window, click Instrument tab and then click on Add Dissociation Stage tab.
Finally, click on Start tab. This will start the program. Do not disturb the program until the run is finished.
3.7 Analysis of qRT-PCR data
Open 7300 system SDS software on the computer.
Click on Create New Document.
Select ddCt (Relative Quantitation) Study from the pull-down menu of the Assay tab.
Click on Next tab. A new window will appear. Click on Add plates tab.
Select the desired .sds file saved at the time of setting-up the qRT-PCR assay. Click open. The file will appear. Click Finish tab in the dialog box.
A new window will open. Select all the fields in the upper left box and click green arrow in the main menu.
Again select all the fields in upper left window. The corresponding Ct values will appear in bottom left window.
Go in main menu and save file as .sdm (SDS Multi-plate documents).
Click on main menu File tab, and sequentially click on Export, Results, and Both. Save the file as .csv file.
Close the application and proceed for analysis part using .csv file.
Open .csv file using Microsoft Excel program, calculate the averages for the duplicates/triplicates of each sample and normalizing gene. This gives us ΔCT values.
Deduct the ΔCT values of the normalizing gene from the corresponding ΔCT values of the samples.
Calculate the final average by taking the average of all control ΔCT values.
Subtracting the ΔCT values from the final average gives us the ΔΔCt values.
The corresponding fold change is calculated as 2 to the power of ΔΔCt values. This gives us the fold change in the samples as compared to control which can be plotted on a graph.
| Reagents | Volume (μl) |
|---|---|
| 10 × RT Buffer | 2.0 |
| 20 × dNTP mix (100 mM) | 0.8 |
| 10 × RT Random Primers | 2.0 |
| Multiscribe Reverse Transcriptase | 1.0 |
| Nuclease free water | 4.2 |
| Total | 10.0 |
| Conditions | Step 1 | Step 2 | Step 3 | Step 4 |
|---|---|---|---|---|
| Temperature | 25°C | 37°C | 85°C | 4°C |
| Time | 10 min | 120 min | 5 sec | Forever |
| Components | Volume (μl) |
|---|---|
| cDNA | 1 |
| 20 μm Stock Primer 1 | 1 |
| 20 μM Stock primer 2 | 1 |
| RNase-free water | 7 |
| 2X SYBER Green Master Mix |
10 |
| Total | 20 |
ACKNOWLEDGEMENT
This work was supported by National Institute of Health grants (AG029623 and AR059810) to Ashok Kumar.
4. Notes
Prepare and store all cDNA synthesis and qRT-PCR reagents at −20°C (unless indicated otherwise). Cell culture medium should be stored at 4°C.
Diligently follow all waste disposal regulations (especially phenol based reagents used during RNA isolation) according to the material safety data sheet provided by the manufacturer. Store all other reagents at room temperature.
Always use sterile gloves while working with RNA. Avoid contact with skin or All the procedures should be done in RNAse/DNase free environment. Autoclave all tips and eppendorf tubes properly before using. Clean the working area, pipettes and other equipment used for RNA extraction with RNAZap and/or with 70% ethanol to ensure RNase free conditions.
TRIzol reagent may be corrosive and cause irritation. Therefore, avoid contact with directly. Use gloves and lab coats while working with TRIzol reagent (Read MSDS of product for detailed protection and safety).
DEPC is not miscible with water; shake vigorously after adding DEPC to water for proper mixing. Also DEPC should not be stored more than 24 months (See MSDS of the product for full details). Follow the hazardous waste disposal methods for proper disposal of expired DEPC.
For good yield of RNA, muscle tissue should be properly homogenized in TRIzol reagent. When tissue is homogenized, the solution becomes turbid with no major tissue clumps visible.
It is always a good practice to check the quality of RNA before using it for qRT-PCR assay especially when RNA samples were stored at −80°C for several days. RNA can also be stored in small aliquots to avoid repeated freeze thaw.
To protect the Gel-Dye mix from light, cover the tube with foil. Remember to return reagents to the cold room when you are finished.
Loading the Gel-Dye mix could be difficult. If you don’t get the gel distributed evenly and without bubbles throughout the channels, you won’t get good results. Look at the back of the chip for bubbles.
Use the chip within 5 min of preparation to prevent evaporation. Alternatively, cover the chip if it will be left standing for any length of time.
Sample concentrations should ideally be 100-200 ng/μl, though a concentration as low as 50ng/μl can be used.
You must put the nano marker in every sample and the ladder well. Add water or nano marker to unused wells to bring the volume up to 6 μl.
Genomic DNA contamination of RNA samples can produce stray bands or clog the capillary. To check for genomic DNA, treat the samples with DNase. Run a DNase-treated sample next to an untreated sample.
Accurate pipetting is very important. Therefore, use properly calibrated pipettes. Place the tip at the bottom and center of each well when dispensing. Do not try to push pass the first resistance point on the pipette to avoid bubbles. You may pipette up and down gently to mix samples in the wells.
To avoid multiple pipetting, it is better to make a master mix and then add master mix to the tubes containing cDNA.
Having good primer sets is critical for the success of qRT-PCR assay.
It is quite possible that you will not get any good quality primers in a selected region of 300-400 bp. If the primers are not good, move across the sequence (by shifting starting point 200bp downstream) and perform the same search.
If the gene of interest is not giving good primer sets, we order two to three best possible sets of primers and test them in qRT-PCR assays.
After finishing the run, it is a good idea to run the PCR products on agarose gel electrophoresis. This gel should show a single PCR product and no primer dimer.
In pathological conditions, skeletal muscles are infiltrated by other cells such as immunocytes and fibroblasts. Since GAPDH and β-actin are expressed by all cell types, using these genes as normalizing controls, it is not possible to distinguish whether the observed change in expression of a specific gene is because of its altered expression in skeletal muscle or other cell type. To determine the changes in expression level only in skeletal muscle, we suggest using some skeletal muscle specific endogenous gene for normalization purposes. Myosin heavy chain 4 or muscle creatine kinase could serve as good normalizing controls.
5. References
- 1.Charge SB, Rudnicki MA. Cellular and molecular regulation of muscle regeneration. Physiol Rev. 2004;84:209–238. doi: 10.1152/physrev.00019.2003. [DOI] [PubMed] [Google Scholar]
- 2.Perry RL, Rudnick MA. Molecular mechanisms regulating myogenic determination and differentiation. Front Biosci. 2000;5:D750–D767. doi: 10.2741/perry. [DOI] [PubMed] [Google Scholar]
- 3.Glass DJ. Skeletal muscle hypertrophy and atrophy signaling pathways. Int J Biochem Cell Biol. 2005;37:1974–1984. doi: 10.1016/j.biocel.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 4.Jackman RW, Kandarian SC. The molecular basis of skeletal muscle atrophy. Am J Physiol Cell Physiol. 2004;287:C834–C843. doi: 10.1152/ajpcell.00579.2003. [DOI] [PubMed] [Google Scholar]
- 5.Li H, Malhotra S, Kumar A. Nuclear factor-kappa B signaling in skeletal muscle atrophy. J Mol Med. 2008;86:1113–1126. doi: 10.1007/s00109-008-0373-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kandarian SC, Stevenson EJ. Molecular events in skeletal muscle during disuse atrophy. Exerc Sport Sci Rev. 2002;30:111–1116. doi: 10.1097/00003677-200207000-00004. [DOI] [PubMed] [Google Scholar]
- 7.Emery AE. The muscular dystrophies. Lancet. 2002;359:687–695. doi: 10.1016/S0140-6736(02)07815-7. [DOI] [PubMed] [Google Scholar]
- 8.Cao PR, Kim HJ, Lecker SH. Ubiquitin-protein ligases in muscle wasting. Int J Biochem Cell Biol. 2005;37:2088–2097. doi: 10.1016/j.biocel.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 9.Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 10.Giresi PG, Stevenson EJ, Theilhaber J, Koncarevic A, Parkington J, Fielding RA, Kandarian SC. Identification of a molecular signature of sarcopenia. Physiol Genomics. 2005;21:253–263. doi: 10.1152/physiolgenomics.00249.2004. [DOI] [PubMed] [Google Scholar]
- 11.Haslett JN, Sanoudou D, Kho AT, Bennett RR, Greenberg SA, Kohane IS, Beggs AH, Kunkel LM. Gene expression comparison of biopsies from Duchenne muscular dystrophy (DMD) and normal skeletal muscle. Proc Natl Acad Sci U S A. 2002;99:15000–15005. doi: 10.1073/pnas.192571199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Panguluri SK, Bhatnagar S, Kumar A, McCarthy JJ, Srivastava AK, Cooper NG, Lundy RF, Kumar A. Genomic profiling of messenger RNAs and microRNAs reveals potential mechanisms of TWEAK-induced skeletal muscle wasting in mice. PLoS One. 2010;5:e8760. doi: 10.1371/journal.pone.0008760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stevenson EJ, Giresi PG, Koncarevic A, Kandarian SC. Global analysis of gene expression patterns during disuse atrophy in rat skeletal muscle. J Physiol. 2003;551:33–48. doi: 10.1113/jphysiol.2003.044701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delgado I, Huang X, Jones S, Zhang L, Hatcher R, Gao B, Zhang P. Dynamic gene expression during the onset of myoblast differentiation in vitro. Genomics. 2003;82:109–121. doi: 10.1016/s0888-7543(03)00104-6. [DOI] [PubMed] [Google Scholar]
- 15.Rajeevan MS, Ranamukhaarachchi DG, Vernon SD, Unger ER. Use of real-time quantitative PCR to validate the results of cDNA array and differential display PCR technologies. Methods. 2001;25:443–451. doi: 10.1006/meth.2001.1266. [DOI] [PubMed] [Google Scholar]