

Background: Autophagy and cathepsin B-mediated trypsin generation may be deleterious in acute pancreatitis. The role of ARF1 in the process is unknown.
Results: BFA-mediated ARF1 inhibition prevents caerulein-induced processing of procathepsin B and perturbs autophagic maturation.
Conclusion: ARF1-dependent trafficking of procathepsin B and autophagic maturation result in trypsinogen activation.
Significance: ARF1 plays a significant role in acute pancreatitis.
Keywords: ARF, Autophagy, Cell Biology, Enzymes, Golgi
Abstract
Several studies have suggested that autophagy might play a deleterious role in acute pancreatitis via intra-acinar activation of digestive enzymes. The prototype for this phenomenon is cathepsin B-mediated trypsin generation. To determine the organellar basis of this process, we investigated the subcellular distribution of the cathepsin B precursor, procathepsin B. We found that procathepsin B is enriched in Golgi-containing microsomes, suggesting a role for the ADP-ribosylation (ARF)-dependent trafficking of cathepsin B. Indeed, caerulein treatment increased processing of procathepsin B, whereas a known ARF inhibitor brefeldin A (BFA) prevented this. Similar treatment did not affect processing of procathepsin L. BFA-mediated ARF1 inhibition resulted in reduced cathepsin B activity and consequently reduced trypsinogen activation. However, formation of light chain 3 (LC3-II) was not affected, suggesting that BFA did not prevent autophagy induction. Instead, sucrose density gradient centrifugation and electron microscopy showed that BFA arrested caerulein-induced autophagosomal maturation. Therefore, ARF1-dependent trafficking of procathepsin B and the maturation of autophagosomes results in cathepsin B-mediated trypsinogen activation induced by caerulein.
Introduction
Chiari (1) proposed more than a century ago that pancreatitis is an autodigestive disease. Several models of acute pancreatitis have shown intra-acinar activation of the zymogen trypsinogen to trypsin as playing an important role in disease pathogenesis (2–4). This process is thought to be important in pancreatitis because trypsin is a “master activator” of the digestive enzyme cascade. The role of lysosomal proteases in the pathogenesis of pancreatitis is supported by studies showing a marked increase in activity of the lysosomal enzyme cathepsin B (which is known to activate trypsinogen to trypsin (5)) in the zymogen fraction during pancreatitis (6, 7). In addition, the formation of large heterogeneous vacuoles containing lysosomal and digestive enzymes in the post-Golgi compartment (8, 9) has previously been noted in pancreatitis.
Acinar cells contain both procathepsin B (10), which has an N terminus inhibitory propeptide (11), and the glycosylated active forms of cathepsin B (12). Procathepsin B is known to inhibit cathepsin B activity via the propeptide (11). Removal of this procathepsin B propeptide and subsequent activation to cathepsin B is secured in acidic compartments (13) only after procathepsin B glycosylation and sorting out in the trans-Golgi network. Although the co-localization of cathepsin B with trypsinogen in the autophagosome-like vacuoles was hypothesized to play an important role in pancreatitis (10, 14, 15) and studies have shown that autophagy is essential to the generation of intracellular trypsin (16), the regulation of procathepsin B trafficking in this phenomenon and how the organelles involved in trypsinogen activation contribute to autophagosome formation or maturation are unknown.
Golgi membranes seem to be involved in autophagosome maturation (17) and, by extension, proteins involved in recruiting adaptor proteins and lipid-modifying enzymes to the Golgi, such as ARFs2 may be involved in this. BFA is a noncompetitive specific inhibitor of ARF GTPases (18, 19) and prevents GTP exchange mediated by the guanine nucleotide exchange factors of ARFs. The function of ARF1 in recruiting adaptor proteins and lipid-modifying enzymes on the Golgi is well established (20), whereas ARF6 is typically active on the cell surface (21), regulating phenomena such as endocytosis. Because previous studies showed that BFA disrupts Golgi transport and dismantles the Golgi stacks in pancreatic acinar cells (22), we chose to mechanistically study the organellar basis of procathepsin B processing in trypsinogen activation along with post-Golgi trafficking in the progression of autophagy during acute pancreatitis.
EXPERIMENTAL PROCEDURES
Animals and Animal Procedures
8-week-old CD-1 mice or 80–100-gm Sprague-Dawley rats were purchased from Charles River Laboratories (Wilmington, MA) and housed with a 12-h light/dark cycle, at temperatures from 21 to 25 °C, fed standard laboratory chow, and allowed to drink ad libitum. Animals were acclimatized for at least 2 days before use. Caerulein and the cathepsin B substrate (Z-Arg-Arg-MCA) were purchased from Bachem (King of Prussia, PA). Specific antibodies and reagent sources are described below. All other reagents and chemicals were purchased from Sigma. Animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh (Pittsburgh, PA).
Preparation and Use of Pancreatic Acini
Acini were harvested as described previously (23) and stored at 4 °C until further use. Viability before use was more than 95%, as indicated by trypan blue exclusion. When indicated, acini were treated with 50 μm BFA at 37 °C for 2 h, whereas controls were kept in the same conditions without BFA. Results of in vitro studies are reported as averages from at least three independent experiments.
Western Blotting, RT-PCR, and Real-time PCR
Proteins were extracted from pancreas or acini after homogenization with a Potter-Elvehjem homogenizer in homogenization buffer containing 50 mm Tris at pH 7.2, 150 mm NaCl, 0.5 mm EDTA, 1 mm EGTA, 2 mm dithiothreitol, 1 mm Na3VO4, 25 mm NaF, 1% Nonidet P-40, and Complete protease inhibitor mixture (Roche Diagnostics). Lysates were boiled in 1× Laemmli sample buffer before Western blot analysis according to standard procedures as described previously (23) using the following antibodies: GM130, p115, and syntaxin 6 (BD Transduction Laboratories), ARF1 and ARF6 (Thermo Fisher Scientific Inc.), LC3 (Cell Signaling Technology, Boston, MA), cathepsin B (Sigma-Aldrich), cathepsin L and cathepsin B antibody raised against its propeptide domain (Abcam, Cambridge, MA), chymotrypsin (Millipore, Billerica, MA), and actin (Santa Cruz Biotechnology Inc., Santa Cruz, CA). Polyclonal antibody against pancreatic lipases was a kind gift from Dr. Mark Lowe (University of Pittsburgh). RNA from cells or tissue was isolated using TRIzol reagent (Invitrogen), and real-time PCR was conducted as described previously (24) using proprietary primers for CXCL1, CXCL2, TNF-α, and 18S RNA from Applied Biosystems (Invitrogen).
GST Pulldown Assays
Binding assays with the glutathione resin-bound GST-GGA3-PBD fusion protein were performed either on protein lysates from murine or rat pancreas or on isolated acini using active ARF1 (or ARF6) pulldown and detection kit from Thermo Fisher Scientific Inc. Binding assays with GTPγS or GDP served as an internal control according to the manufacturer's protocol.
Activity Assays
Cathepsin B activity was assayed spectrofluorometrically at 38 °C in 0.1 m sodium phosphate buffer (pH 6.0) containing 5 mm dithiothreitol and EDTA using the substrate Z-Arg-Arg-MCA (25). Trypsin was measured in acinar cell homogenates as described previously (4, 7). Cathepsin L activity was measured according to Wartmann et al. (40). Both cathepsin B and cathepsin L assays were normalized to DNA and measured overall cathepsin B or cathepsin L activities irrespective of the isoforms. Amylase release into the medium was measured as described previously (4, 26). Effects of procathepsin B propeptide (amino acids 25–60) (Bachem, Torrance, CA) on cathepsin B activity were studied by preincubating various concentrations of the peptide with acinar lysates for 15 min as described previously (27).
Models of Pancreatitis
Pancreatitis was induced in rats and mice using caerulein. Mice were given hourly injections (×10) of caerulein (50 mcg/kg) intraperitoneally and sacrificed 1 h after the last injection as described previously (4, 28). Pancreatitis was induced in rats with a single dose of 20 mcg/kg of caerulein given intraperitoneally as described previously (23). Rats were sacrificed 6 h after induction of pancreatitis. Blood and pancreas were harvested as described previously (4, 28). BFA (25 mg/kg) or its vehicle (0.1 ml, 50% dimethyl sulfoxide (DMSO)) was administered by intraperitoneal injection 1 h before the induction of pancreatitis as described previously (29). Animal studies included 6–8 rodents per experimental group.
Sucrose Gradient Centrifugation
Fractions were prepared from mouse pancreata homogenized in 0.3 m sucrose and spun in a ST 40R centrifuge (Thermo Fisher Scientific Inc.) as described previously (30). The zymogen fraction was separated from the postnuclear supernatant after centrifugation at 3,000 × g for 10 min. Mitochondrial and microsomal fractions were precipitated from zymogen supernatant at 7,800 × g and 40,000 rpm, respectively. The cytosolic fraction represented the supernatant of the microsomal fraction. For Golgi fractions analysis, purified microsomes were loaded onto 2.5 m sucrose gradient cushion according to previously described protocol in Ref. 30. The individual submicrosomal sucrose gradient fractions were collected from the top of the tube and boiled in 1× Laemmli buffer before Western blot analysis.
Immunofluorescence Microscopy
Immunofluorescence microscopy was done on pancreatic tissue cryosections after embedding in Tissue-Tek® OCT (optimal cutting temperature compound) (Sakura Finetek USA, Inc., Torrance, CA) or on acini plated on plain glass coverslips. These were fixed with 2% paraformaldehyde and processed as described previously (23, 31). After blocking with 5% normal goat serum, tissue cryosections were incubated with primary antibodies (GM130 1:200, p115 1:200, or ARF1 1:50) for 1 h at room temperature, washed, and incubated with secondary antibodies (Alexa Fluor 488- or Alexa Fluor 594-conjugated, diluted 1:500) rhodamine-conjugated phalloidin (100 nm) (Invitrogen) with or without DRAQ5 (1:5000) for 30 min. After washing and mounting in ProLong Gold (Invitrogen), confocal imaging (1 μm thick) was done using a Zeiss LSM510 confocal microscope (Thornwood, NJ), and processed using Adobe Photoshop 6.0 (Adobe Systems, Mountain View, CA).
Electron Microscopy
Pancreatic tissue was cut into <2 μm pieces immediately after harvesting, fixed in Karnovsky's fixative, and processed as described previously (32). Sections (70 nm) were imaged on a Jeol JEM-1011 transmission electron microscope. For quantification of autophagosome contents, all areas that could be imaged from each experimental group at 4,000× were taken, and autophagosomes were classified into empty, those containing both dense and non-dense content, and those containing only dense granules.
Statistical Analysis
All values are presented as means ± S.E. There were at least three independent experiments done for each all data presented from in vitro studies. Differences between two groups were analyzed by unpaired Student's t test. A two-tailed p value of less than 0.05 was considered to indicate statistical significance. For electron microscopy analysis, differences between two groups were compared using a Fisher's exact test.
RESULTS
ARF1 Inhibition Prevents Procathepsin B Processing and Trafficking from the Golgi, Reducing Caerulein-induced Trypsinogen Activation
The Golgi stacks under control or basal conditions were compact, with a crescent-like arrangement and no visible vesiculation (Fig. 1A1). Caerulein (100 nm) caused the Golgi to extend more apically in an antegrade fashion (i.e. toward the apical actin) (Fig. 1A2). Treatment of control acini with BFA resulted in dismantling and vesiculation of the Golgi (Fig. 1A3) in resting acini, as described previously by Farquhar and co-workers (33). BFA reduced caerulein-induced antegrade Golgi extension (Fig. 1, A2 and A4). Although we observed constitutive ARF1 activity under basal conditions (Fig. 1B), which is consistent with the changes noted in Fig. 1A3 and by Farquhar and co-workers (33), a further increase in ARF1 activity was noted in acinar cells within 2 min of adding caerulein (Fig. 1B), although this effect was blocked by BFA. Caerulein did not activate ARF6 (Fig. 1C). BFA also prevented caerulein-induced trypsin generation in mice (Fig. 2A; 2.3 ± 0.1 versus 1.0 ± 0.1-fold control, p = 0.01) and rat acini (Fig. 2B; 18.6 ± 4.0 versus 10.3 ± 1.3-fold control, p < 0.04). Thus, through its inhibition of ARF1, BFA dismantled the Golgi stacks and prevented the caerulein-induced antegrade Golgi extension, resulting in reduced trypsinogen activation. These data suggest that BFA prevents trypsinogen activation via an ARF1-dependent, Golgi-based phenomena and not an ARF6-dependent phenomena, such as endocytosis (21, 34), which is also involved in trypsinogen activation (35, 36). Because cathepsin B can activate trypsinogen to trypsin (5), we examined the effect of BFA on trafficking of cathepsin B and its precursor procathepsin B.
FIGURE 1.

BFA dismantles the Golgi by inhibiting ARF1 activation. A1–A4, immunostaining of pancreatic acini with antibodies against the Golgi marker, P115 (shown in green) and actin (shown in red) under basal conditions (A1) or treated with 100 nm caerulein (CER) alone for 15 min (A2); 50 μm BFA for 2 h (A3); or BFA followed by CER treatment (A4). These changes demonstrate that BFA dismantles the Golgi without causing major changes in actin distribution. Arrows indicate basal relocation of the F-actin. Note that BFA did not prevent caerulein-induced blebbing (A2 and A4). A and B, WBs of ARF1 (A) and ARF6 (B) pulldowns from pancreatic acini after treatment with 100 nm CER at the indicated times show basal activity (Bas) and an increase in ARF1 but not ARF6 activation. BFA (50 μm) blocks CER-induced ARF1 activation (+BFA, 1B). C, no activation of ARF6 was noted. The positive control GTPγS, but not GDP, induces both ARF1 and ARF6 activation.
FIGURE 2.

BFA prevents trypsinogen activation and retains procathepsin B during treatment of pancreatic acini with 100 nm CER in vitro. A and B, trypsin activity assay in acinar cells after 30 min under CER stimulation demonstrates that BFA prevents CER-induced trypsinogen activation in both mouse (A) and rat acini (B). Data are presented as -fold change when compared with untreated acini (CON). C, cathepsin B activity assayed under conditions described in A and B shows that BFA reduces cathepsin B activation quantified as the ratio of the cathepsin B activity in the zymogen (1,300 × g) versus the lysosomal (12,000 × g) fraction. BAS, basal activity. D, WB of cathepsin B isoforms present in pancreatic acini shows that the 34-kDa procathepsin B (CathB) isoform is present in total cell lysate under basal condition and in acini pretreated with BFA followed by stimulation with CER but not in cells treated with CER alone. E1–E3, densitometry (E1) of the protein bands from WB shown in D demonstrates the reduction in the amount of active 25- and 29-kDa cathepsin B (CB) isoforms present in acini under CER-treated conditions, whereas the activity of cathepsin B in total cell lysates remains unchanged (E2). Histograms (E3) represent the quantification of the cathepsin B activity per the average protein content of the active, 25-kDa, and 29-kDa cathepsin B isoform under the same treatment condition as described in D from at least three independent experiments. The histograms show a dramatic increase in the cathepsin B activity/protein upon stimulation with CER, which is significantly inhibited by 50 μm BFA. F, quantification of the inhibitory effect of procathepsin B peptide on cathepsin B activity in acinar cell lysates. * indicates a significant (p < 0.05) decrease in activity at the corresponding peptide concentration compared to the activity in its absence.
Previous studies have shown an increase in cathepsin B activity in the zymogen fraction to be associated with trypsinogen activation (4, 6, 10, 37, 38). We found that 100 nm caerulein significantly increased cathepsin B activity in the zymogen fraction (p < 0.05 versus basal; Fig. 2C), which was prevented by BFA (Fig. 2C). On Western blot analysis of acinar homogenates, however, supraphysiologic caerulein caused a reduction in the amount of active cathepsin B, i.e. its 25- and 29-kDa bands, in both mice (Fig. 2, D and E1) and rat acini (supplemental Fig. 1A) (39), although there was no change in overall cathepsin B activity (Fig. 2E2). On the contrary, calculation of cathepsin B activity per amount of protein showed a significant increase with 100 nm caerulein, and this increase was significantly blunted by BFA (280 ± 37.4% versus 150 ± 29% control, p = 0.035) (Fig. 2E3). We also observed that caerulein caused complete processing of the 34-kDa procathepsin B that contains an inhibitory propeptide (Fig. 2D) and predominantly localizes to the Golgi-containing fraction under control conditions (Fig. 3B1). This caerulein-mediated procathepsin B processing to mature cathepsin B was prevented by BFA (Fig. 2D). Because this BFA-mediated retention of procathepsin B was associated with a decrease in cathepsin B activity (Fig. 2, C and E3), we tested to see whether the propeptide, which may be formed during the maturation of cathepsin B, may have an inhibitory role. The dose-dependent inhibition of cathepsin B activity by the propeptide fragment confirmed its inhibitory role and that of procathepsin B in our system (Fig. 2F).
FIGURE 3.

BFA blocks ARF1 activation, retains procathepsin B in the microsomal fraction, and prevents trypsinogen activation during caerulein-induced pancreatitis in vivo. A, WB of ARF1 pulldowns from mice with 6-h CER-induced acute pancreatitis using GST-GGA3-PBD fusion protein show an increase in ARF1 activation. The activation is completely blocked by BFA. CON, control. B1–B3, WBs of subcellular fractions from pancreas of control mice (B1), mice with 6-h CER-induced pancreatitis (B2, Ptts.), and mice pretreated with BFA during 6-h CER pancreatitis (B3, Ptts. + BFA) show procathepsin B (ProcathB) being retained in a microsomal fraction (Micro) in control and BFA-treated mice but not in mice with 6-h pancreatitis. To assess the distribution of zymogens (Zymo) and microsomes in subcellular fractions, the antibodies against chymotrypsin (Chymo), a protein marker of zymogen granules, and GM130, a Golgi marker, were used. Cathepsin B antibody was used to assess its localization in subcellular fractions under the different treatment conditions. PNS, postnuclear supernatant; Mito, mitochondrial fraction. C and D, trypsin activity assay done on pancreatic tissue from mouse (C) and rat (D) treated under conditions described in A and B1–B3. Histograms represent trypsin activity per DNA content in mouse and rat pancreatic homogenates. E, WB analysis of mouse pancreata shows an induction of autophagy in murine pancreas during 10-h CER-induced pancreatitis that is not prevented by BFA treatment. F, histograms represent the means of at least three densitometries of WB shown in E.
Recently, pharmacological inhibition of cathepsin L has been shown to increase the amount of active trypsin in rodent models of acute pancreatitis (10) and prevent trypsinogen activation in humans (40). This suggested that an imbalance between cathepsin L and cathepsin B could play a role in intra-acinar trypsinogen activation in pancreatitis. We, therefore, compared the processing and maturation of procathepsin L with procathepsin B in acinar cells treated with 100 nm caerulein with or without BFA. Our experiments consistently demonstrated that although the amount of 34-kDa form of procathepsin B was greatly reduced by supraphysiologic caerulein (Figs. 2D and Fig. 6B), equal amounts of the unprocessed 42-kDa form of procathepsin L were present in all acinar cell lysates regardless of the treatment (see Fig. 6C). There were no changes in the protein level of the mature 25-kDa form of cathepsin L (see Fig. 6C). Likewise, calculation of cathepsin L activity per amount of DNA showed no changes with 100 nm caerulein (see Fig. 6A) with or without BFA. Therefore, it is likely that BFA affects caerulein-induced trypsinogen activation via altering the processing of procathepsin B and not procathepsin L.
FIGURE 6.
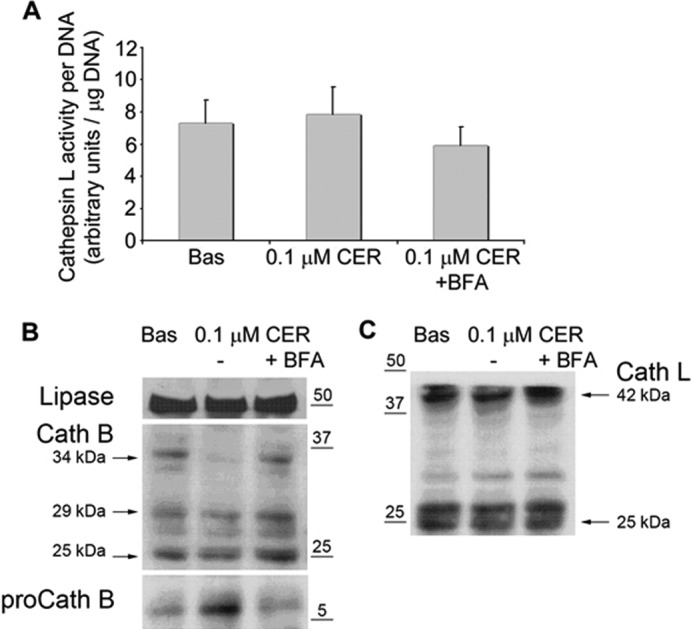
Supraphysiologic caerulein affects neither cathepsin L activity nor protein amount or its precursor procathepsin L, whereas depleting procathepsin B. A, cathepsin L activity under the mentioned conditions shows no significant changes. Bas, basal activity. B, WB of lysates from murine pancreatic acini treated for 30 min with 0.1 μm caerulein (0.1 μm CER) shows the depletion of 34-kDa procathepsin B, which is prevented by 50 μm BFA. Concomitant with the depletion of procathepsin B (proCath B) by 0.1 μm caerulein, its propeptide appears in the acinar cell homogenates in the form of a 6-kDa fragment. Lipase is shown as a loading control. Cath B, cathepsin B. C, WB of lysates from murine pancreatic acini treated for 30 min with 0.1 μm caerulein shows that this does not induce depletion of 42-kDa procathepsin L or change the amounts of mature (25 kDa) cathepsin L (Cath L).
To better characterize the rapid maturation of procathepsin B caused by supraphysiologic caerulein, we assessed the cell homogenates from acini treated with 100 nm caerulein with or without BFA by Western blotting using an antibody raised against cathepsin B propeptide region. Our experimental data have shown that the loss of the 34-kDa form of procathepsin B was accompanied by an increase in the amount of the 6-kDa propeptide in the lysates stimulated with 100 nm caerulein when compared with untreated controls or lysates pretreated with BFA (see Fig. 6B). As cathepsin B activity per protein amount was significantly increased with supraphysiologic caerulein (Fig. 2E3), it is likely that the stimulation with 100 nm caerulein released cathepsin B inhibition by removing its propeptide from the N terminus of procathepsin B. Interestingly, the early caerulein-induced trypsinogen activation was not accompanied by an increase in LC3-II (supplemental Fig. 1B), which is a marker of autophagic activity.
BFA Inhibits ARF1 in Vivo and Reduces Trypsinogen Activation and Severity of Caerulein-induced Pancreatitis without Preventing Induction of Autophagy
We next sought to determine whether caerulein also activated ARF1, which regulates transport from the endoplasmic reticulum to the Golgi and between Golgi cisternae (41) during pancreatitis. Caerulein-induced pancreatitis at 6 h resulted in activation of ARF1 that was prevented by BFA (Fig. 3A). In control animals, when assessed by immunocytochemistry, ARF1 had a diffuse cytoplasmic appearance (supplemental Fig. 2, B and C, ovals) that shifted to supranuclear areas under pancreatitis conditions (supplemental Fig. 2E, dashed ovals). We then studied the subcellular localization of procathepsin B, which normally concentrates in the Golgi-enriched microsomal fraction (Fig. 3B1). Caerulein-induced ARF1 activation was associated with loss of procathepsin B from the microsomal fraction. This was prevented by BFA (Fig. 3B3), which also reduced trypsinogen activation in both rat and mouse caerulein-induced pancreatitis (Fig. 3, C and D). BFA also reduced the parameters of local severity (i.e. necrosis (Fig. 4, A and B), edema (Fig. 4C), and serum amylase (Fig. 4D)) without affecting mRNA up-regulation of the inflammatory mediators TNF-α, CXCL1, and CXCL2 in pancreatic tissue (Fig. 4, E1–E3) in mouse caerulein pancreatitis; BFA also reduced the severity of rat caerulein pancreatitis (data not shown). Interestingly, although BFA prevented the translocation of the zymogen marker chymotrypsin to the microsomal fraction (Fig. 3B2, discussed below), it did not prevent the induction of autophagy, as evidenced by the formation of LC3-II (Fig. 3, E and F).
FIGURE 4.

BFA reduces severity of CER-induced pancreatitis without affecting inflammatory cytokine up-regulation. A1–A4, BFA reduces pancreatic necrosis. Pancreatic tissue from control mice (A1), mice with 10-h CER-induced pancreatitis (A2), and mice pretreated either with vehicle (A3) or with BFA (A4) was dissected for histology and quantified for the amount of necrotic cell death per total area of pancreatic tissue. Representative images are shown. B, histograms from quantified histology data described in A1–A4 show the decrease (p < 0.01) in the amount of necrotic area in mice treated with BFA relative to vehicle control (CON). VEH, vehicle. C and D, BFA reduces pancreatic edema (C) and serum amylase (D) at the end of 10 h of pancreatitis. E1–E3, BFA does not decrease the severity of pancreatitis by interfering with the transcriptional regulation of TNF-α (E1), CXCL1 (E2), or CXCL2 (E3) in the pancreas. Histograms represent the -fold increase in the amount of TNF-α, CXCL1, and CXCL2 transcripts relative to control as assessed by real-time PCR.
ARF1 Inhibition Prevents Autophagic Maturation during Pancreatitis
Recent studies have shown that autophagy is needed for trypsinogen activation during pancreatitis (10, 16). Our observations above on the blunting effect of BFA on trypsinogen activation (Fig. 3, C and D) and severity of pancreatitis (Fig. 4, A–D) but not induction of autophagy (Fig. 3, E and F) prompted us to ask whether inhibiting ARF1 affected maturation of the autophagosomes. Electron microscopy showed that autophagosomal maturation culminated in the formation of compound vacuoles containing both electron-dense and non-dense components during caerulein-induced pancreatitis (Fig. 5, C and E). BFA significantly altered this process, with the predominant population of vacuoles being empty or containing only electron-dense granules in both rat and mouse models (Fig. 5D and supplemental Fig. 3). High power electron microscopy analysis showed these empty vacuoles to have double membranes (supplemental Fig. 4), consistent with their autophagic origin.
FIGURE 5.

BFA prevents autophagosomal maturation. A–D, representative electron microscopy images (10,000×) of tissue sections from control pancreas (A), pancreas of BFA-treated rat (B), rat with 6-h CER pancreatitis (C), and one with 6-h pancreatitis pretreated with BFA (D). C, pancreatitis results in autophagosomes containing both dense (zymogen dense-like) and non-dense granule components. D, BFA results in several of them being empty. E, the table shows quantification of the autophagosomal contents measured on electron microscopy images. There is a significant increase in the number of empty autophagosomes (p < 0.03) and autophagosomes containing only dense granules (gran., p < 0.001) in the CER + BFA group when compared with the CER group. F1–F3, WBs of submicrosomal fractions from control mice (F1), mice with 6-h CER-induced pancreatitis (F2), and pretreated with BFA before CER pancreatitis (F3). F1, WB of pancreatic lysates after their isopycnic centrifugation in 20% (fraction 4) to 50% (fractions 9 and 10) sucrose gradient demonstrate the distribution of zymogen marker chymotrypsin (Chymo), the endolysosomal marker syntaxin 6, the autophagosomal marker LC3-II, and the Golgi marker GM130 under control conditions. F2, 6-h CER treatment induces co-localization of chymotrypsin, syntaxin 6, and LC3-II with the Golgi marker GM130 in heavy fractions (fractions 7–9) demonstrated by isopycnic centrifugation of pancreata from animals with pancreatitis. The WB shows a biochemical equivalent to the EM images in C. F3, BFA pretreatment results in changes in the autophagosomal density leading to a shift of LC3-II to the lighter sucrose density fractions with the loss of chymotrypsin co-localization with the Golgi marker GM130. This is consistent with the increase in empty autophagosomes noted by EM (D).
We independently verified these findings in subfractionation of microsomes. This revealed that although the zymogen marker chymotrypsin is absent, the endolysosomal marker syntaxin 6 (42) enriches in fractions lighter than the Golgi marker GM130 under control conditions (Fig. 5F1). During pancreatitis, syntaxin 6 co-localizes with GM130 and chymotrypsin (consistent with relocation of chymotrypsin noted in Fig. 3B2) in LC3-II-enriched fractions during caerulein-induced pancreatitis (Fig. 5F2). This suggests that co-localization of digestive enzymes and lysosomal hydrolases (6, 9, 38, 43) occurs in mature autophagosomes during pancreatitis. As predicted by the morphological effects we saw with electron microscopy, BFA prevented the entry of the Golgi marker GM130 and chymotrypsin into the LC3-II-rich compartments but did not affect syntaxin 6 (Fig. 5, E and F3). Our studies also demonstrate that supraphysiologic caerulein affects neither cathepsin L activity nor the protein amount or its precursor procathepsin L, whereas it depletes procathepsin B (Fig. 6).
DISCUSSION
Our studies show that trypsinogen activation during acute pancreatitis occurs by two different mechanisms. During the initial 30 min following caerulein administration, procathepsin B, which contains the inhibitory propeptide, undergoes increased processing independent of autophagy. The procathepsin B maturation releases the inhibition of cathepsin B activity, leading to trypsinogen activation. In this phase, we observed that ARF1 inhibition by BFA dismantled the Golgi and prevented its antegrade extension (as would be induced by caerulein), leading to the retention of procathepsin B and its inhibition of trypsinogen activation. The fact that the 34-kDa procathepsin B can inhibit cathepsin B activity has been previously demonstrated in a study, where an addition of mature cathepsin B in catalytic amounts to a non-autoactivatable variant of proenzyme did not result in the significant processing of procathepsin B (44). The concept is also supported by an early finding of intermolecular in trans complementation of protein activity by an unprocessed protein prosequence shown for the subtilisin family of proteases (45).
During the later phase of pathogenic trypsinogen activation, at 6 h after caerulein, ARF1 is activated, promoting the formation of double-membrane compound vacuoles containing both zymogen granules and microsomal components. In this phase, BFA prevented autophagic maturation at the step when the double-membrane autophagic vacuoles were empty or contained only zymogen granules. Fig. 7 presents a schema illustrating these distinct phenomena.
FIGURE 7.

Two distinct pathways regulate trypsinogen activation. During the first 30 min after supraphysiologic caerulein administration, ARF1 inhibition by BFA results in the retention of the inhibitory procathepsin B (shown in red circles) and consequent reduction of cathepsin B (shown in green circles) activity. 6 h later, after autophagy induction, autophagy maturation is arrested, and consequent trypsin generation is prevented by ARF1 inhibition. PAS, Pre-autophagosomal structure.
Although caerulein-induced maturation of procathepsin B at 30 min may contribute to the increase in cathepsin B activity in the zymogen fraction (Fig. 1C) and subsequent trypsinogen activation, how this occurs, i.e. whether by accelerated processing or alternative trafficking of procathepsin B, remains to be determined. The accumulation of the 6-kDa propeptide, along with procathepsin B processing, and the increase in cathepsin B activity per amount of protein in response to supraphysiologic caerulein favors accelerated processing; however, the subcellular organelles to which the propeptide is trafficked remain unknown. Similarly, the reasons for reduced cathepsin B amounts in acinar lysates despite caerulein-induced maturation of procathepsin B remain to be explored. Although this may be due to cathepsin B degradation, secretion into the extracellular environment (46) remains a possibility.
The dismantling of Golgi by BFA may be associated with trapping of procathepsin B in the rough endoplasmic reticulum, thus preventing procathepsin B maturation and subsequent activation; this argues against a preformed mature compartment (e.g. lysosomes) contributing to cathepsin B-induced trypsinogen activation. However, BFA did not fully prevent the caerulein-induced increase in cathepsin B activity (Fig. 2E3), so there may be other mechanisms, such as endocytosis (36), that contribute to early trypsinogen activation. Although ARF6 is known to regulate endocytosis (21), we did not detect any caerulein-induced activation of ARF6 in pancreatic acini at 2 and 30 min (Fig. 1C). Additionally, because BFA induced morphological changes in control acini (Fig. 1A3), there may be ongoing ARF-dependent Golgi trafficking in resting cells, as has been shown previously (33).
Recent studies have shown that intra-acinar trypsinogen activation may not be due to a cathepsin B-mediated process alone and that the balance between cathepsin B and cathepsin L, as activating and inactivating enzymes, may determine the final level of trypsinogen activation (10, 40). In our experimental settings, we observed neither overall changes in the cathepsin L activity nor changes in amounts of the 42-kDa procathepsin L or 25-kDa mature cathepsin L with supraphysiologic caerulein. Thus, cathepsin L did not replicate the increase in cathepsin B activity associated with the maturation of procathepsin B under the same conditions. Our data do not refute the role of cathepsin L in trypsinogen inactivation but clearly demonstrate that its processing is not affected by BFA. Therefore, the effect of BFA on trypsinogen activation results from its effect on the activity of cathepsin B as discussed above.
The novel finding that BFA inhibition of ARF1 prevents autophagic maturation and trypsinogen activation noted at 6 h of pancreatitis reconciles the historically relevant phenomenon of co-localization during pancreatitis (6, 9, 38, 43) to a complex multistep process and demonstrates that this maturation resulting in the formation of macroautophagic vacuoles containing dense and microsomal components is required for this late trypsin generation during pancreatitis. Although recent studies suggest a role for autophagy (16) or its derangement (10) in the generation of intracellular trypsin, the induction of autophagy (e.g. by fasting (47) or cessation of camostat feeding (48)) results in pancreatic atrophy (47) but not in pancreatitis or the generation of trypsin (10). Interestingly, Nevalainen and Janigan (47) did not find zymogen granules in autophagosomes generated during fasting. Similarly, although LAMP-2-deficient mice, which have impaired lysosomal-autophagosomal fusion, experience massive autophagy and atrophy of the pancreas (49), pancreatitis has not been documented during the lifespan of these mice. At 6 h of caerulein-induced acute pancreatitis, however, we see the formation of mature autophagosomes and the generation of trypsin, concurrent with pancreatitis (4, 50). Our data therefore suggest that autophagic maturation is required for late trypsin generation during pancreatitis and that induction of autophagy alone is not sufficient for this.
Although the pathologic role of trypsin generated during pancreatitis has been argued (51), the reduction in morphologic changes of necrosis by BFA (Fig. 4, A and B) supports the role of active intracellular trypsin generated as a consequence of autophagy contributing to pancreatic necrosis in this study. Recent studies showing necrosis in mice with acinar cells carrying an autoactivating trypsinogen (52) support this phenomenon. This is additionally supported by the finding that SPINK3 (the murine homolog of human SPINK1) knock-out mice have massive autophagy (53), generate more trypsin (54), and have elevated serum amylase.
Supraphysiologic caerulein induces numerous deleterious phenomena other than autophagy in acinar cells, including actin reorganization, blebbing (23, 31), mitochondrial depolarization (28, 55, 56), sustained elevations in cytosolic calcium (55), and generation of inflammatory mediators (24, 57). In our studies, BFA did not prevent up-regulation of inflammatory mediator mRNA (i.e. TNF-α) or of the neutrophil attractant chemokines CXCL1 or CXCL2 and did not prevent F-actin localization or blebbing or change the pattern of caerulein-induced amylase secretion (supplemental Fig. 5). These findings indicate that BFA acted selectively on the processing of procathepsin B and the organellar trafficking events described above. Although BFA could indeed have affected similar phenomena in other cells, including inflammatory cells involved in pancreatitis, the acinar cell-specific role of ARF1 in regulating autophagic maturation and the severity of pancreatitis would remain to be determined when such animals are available because genetic deletion of ARF1 (Knockout Mouse Project Repository) or ARF6 (58) is embryonically lethal, and cultured acinar cells that may be adenovirally transfected with ARFs or their mutants do not activate trypsinogen (24), whereas maintaining polarity.
In summary, we demonstrate that ARF1 regulates trypsinogen activation by two different mechanisms in two different phases: by procathepsin B processing during an early phase and by the transfer of microsomal contents into maturing autophagosomes during the later phase. The relevance of these mechanisms of trypsinogen activation to other diverse etiologies of pancreatitis, some of which are triggered by drugs that induce autophagy (e.g. l-asparaginase) (59), whereas others are caused by poorly understood mechanisms (e.g. alcohol), would be interesting to explore in the future.
Supplementary Material
This work was supported by a startup package from the University of Pittsburgh Department of Medicine (to V. P. S.).

This article contains supplemental Figs. 1–5.
- ARF
- ADP-ribosylation factor
- BFA
- brefeldin A
- CXCL
- CXC ligand
- LC3
- microtubule-associated protein light chain 3
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- Z
- benzyloxycarbonyl
- MCA
- 4-methyl-coumarin-7-amide
- CER
- caerulein
- WB
- Western blot.
REFERENCES
- 1. Chiari H. (1906) About the relationship between the pancreas and fat necrosis. Zbl. Pathol. 17, 798–799 [Google Scholar]
- 2. Waldthaler A., Schütte K., Malfertheiner P. (2010) Causes and mechanisms in acute pancreatitis. Dig. Dis. 28, 364–372 [DOI] [PubMed] [Google Scholar]
- 3. Halangk W., Lerch M. M., Brandt-Nedelev B., Roth W., Ruthenbuerger M., Reinheckel T., Domschke W., Lippert H., Peters C., Deussing J. (2000) Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J. Clin. Invest. 106, 773–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Singh V. P., Saluja A. K., Bhagat L., van Acker G. J., Song A. M., Soltoff S. P., Cantley L. C., Steer M. L. (2001) Phosphatidylinositol 3-kinase-dependent activation of trypsinogen modulates the severity of acute pancreatitis. J. Clin. Invest. 108, 1387–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Figarella C., Miszczuk-Jamska B., Barrett A. J. (1988) Possible lysosomal activation of pancreatic zymogens. Activation of both human trypsinogens by cathepsin B and spontaneous acid. Activation of human trypsinogen 1. Biol. Chem. Hoppe Seyler 369, (suppl.) 293–298 [PubMed] [Google Scholar]
- 6. Van Acker G. J., Weiss E., Steer M. L., Perides G. (2007) Cause-effect relationships between zymogen activation and other early events in secretagogue-induced acute pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G1738–G1746 [DOI] [PubMed] [Google Scholar]
- 7. Saluja A. K., Donovan E. A., Yamanaka K., Yamaguchi Y., Hofbauer B., Steer M. L. (1997) Cerulein-induced in vitro activation of trypsinogen in rat pancreatic acini is mediated by cathepsin B. Gastroenterology 113, 304–310 [DOI] [PubMed] [Google Scholar]
- 8. Hiraoka T., Watanabe E., Mochinaga M., Tashiro S., Miyauchi Y., Nakamura I., Yokoyama I. (1984) Intraoperative irradiation combined with radical resection for cancer of the head of the pancreas. World J. Surg. 8, 766–771 [DOI] [PubMed] [Google Scholar]
- 9. Saito I., Hashimoto S., Saluja A., Steer M. L., Meldolesi J. (1987) Intracellular transport of pancreatic zymogens during caerulein supramaximal stimulation. Am. J. Physiol. 253, G517–G526 [DOI] [PubMed] [Google Scholar]
- 10. Mareninova O. A., Hermann K., French S. W., O'Konski M. S., Pandol S. J., Webster P., Erickson A. H., Katunuma N., Gorelick F. S., Gukovsky I., Gukovskaya A. S. (2009) Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. J. Clin. Invest. 119, 3340–3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fox T., de Miguel E., Mort J. S., Storer A. C. (1992) Potent slow-binding inhibition of cathepsin B by its propeptide. Biochemistry 31, 12571–12576 [DOI] [PubMed] [Google Scholar]
- 12. Caglic D., Pungercar J. R., Pejler G., Turk V., Turk B. (2007) Glycosaminoglycans facilitate procathepsin B activation through disruption of propeptide-mature enzyme interactions. J. Biol. Chem. 282, 33076–33085 [DOI] [PubMed] [Google Scholar]
- 13. Mort J. S., Buttle D. J. (1997) Cathepsin B. Int. J. Biochem. Cell Biol. 29, 715–720 [DOI] [PubMed] [Google Scholar]
- 14. Hofbauer B., Saluja A. K., Lerch M. M., Bhagat L., Bhatia M., Lee H. S., Frossard J. L., Adler G., Steer M. L. (1998) Intra-acinar cell activation of trypsinogen during caerulein-induced pancreatitis in rats. Am. J. Physiol. 275, G352–G362 [DOI] [PubMed] [Google Scholar]
- 15. Steer M. L., Meldolesi J., Figarella C. (1984) Pancreatitis: the role of lysosomes. Dig. Dis. Sci. 29, 934–938 [DOI] [PubMed] [Google Scholar]
- 16. Hashimoto D., Ohmuraya M., Hirota M., Yamamoto A., Suyama K., Ida S., Okumura Y., Takahashi E., Kido H., Araki K., Baba H., Mizushima N., Yamamura K. (2008) Involvement of autophagy in trypsinogen activation within the pancreatic acinar cells. J. Cell Biol. 181, 1065–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Geng J., Klionsky D. J. (2010) The Golgi as a potential membrane source for autophagy. Autophagy 6, 950–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Donaldson J. G., Finazzi D., Klausner R. D. (1992) Brefeldin A inhibits Golgi membrane-catalyzed exchange of guanine nucleotide onto ARF protein. Nature 360, 350–352 [DOI] [PubMed] [Google Scholar]
- 19. Peyroche A., Antonny B., Robineau S., Acker J., Cherfils J., Jackson C. L. (1999) Brefeldin A acts to stabilize an abortive ARF-GDP-Sec7 domain protein complex: involvement of specific residues of the Sec7 domain. Mol. Cell 3, 275–285 [DOI] [PubMed] [Google Scholar]
- 20. Lippincott-Schwartz J., Cole N. B., Donaldson J. G. (1998) Building a secretory apparatus: role of ARF1/COPI in Golgi biogenesis and maintenance. Histochem. Cell Biol. 109, 449–462 [DOI] [PubMed] [Google Scholar]
- 21. Donaldson J. G. (2003) Multiple roles for Arf6: sorting, structuring, and signaling at the plasma membrane. J. Biol. Chem. 278, 41573–41576 [DOI] [PubMed] [Google Scholar]
- 22. Hendricks L. C., McClanahan S. L., McCaffery M., Palade G. E., Farquhar M. G. (1992) Golgi proteins persist in the tubulovesicular remnants found in brefeldin A-treated pancreatic acinar cells. Eur. J. Cell Biol. 58, 202–213 [PubMed] [Google Scholar]
- 23. Singh V. P., McNiven M. A. (2008) Src-mediated cortactin phosphorylation regulates actin localization and injurious blebbing in acinar cells. Mol. Biol. Cell 19, 2339–2347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Orlichenko L. S., Behari J., Yeh T. H., Liu S., Stolz D. B., Saluja A. K., Singh V. P. (2010) Transcriptional regulation of CXC-ELR chemokines KC and MIP-2 in mouse pancreatic acini. Am. J. Physiol. Gastrointest. Liver Physiol 299, G867–G876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mach L., Mort J. S., Glössl J. (1994) Maturation of human procathepsin B: proenzyme activation and proteolytic processing of the precursor to the mature proteinase, in vitro, are primarily unimolecular processes. J. Biol. Chem. 269, 13030–13035 [PubMed] [Google Scholar]
- 26. Bhagat L., Singh V. P., Song A. M., van Acker G. J., Agrawal S., Steer M. L., Saluja A. K. (2002) Thermal stress-induced HSP70 mediates protection against intrapancreatic trypsinogen activation and acute pancreatitis in rats. Gastroenterology 122, 156–165 [DOI] [PubMed] [Google Scholar]
- 27. Chagas J. R., Ferrer-Di Martino M., Gauthier F., Lalmanach G. (1996) Inhibition of cathepsin B by its propeptide: use of overlapping peptides to identify a critical segment. FEBS Lett. 392, 233–236 [DOI] [PubMed] [Google Scholar]
- 28. Singh V. P., Bren G. D., Algeciras-Schimnich A., Schnepple D., Navina S., Rizza S. A., Dawra R. K., Saluja A. K., Chari S. T., Vege S. S., Badley A. D. (2009) Nelfinavir/ritonavir reduces acinar injury but not inflammation during mouse caerulein pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol 296, G1040–G1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sausville E. A., Duncan K. L., Senderowicz A., Plowman J., Randazzo P. A., Kahn R., Malspeis L., Grever M. R. (1996) Antiproliferative effect in vitro and antitumor activity in vivo of brefeldin A. Cancer J. Sci. Am. 2, 52–58 [PubMed] [Google Scholar]
- 30. Saraste J., Palade G. E., Farquhar M. G. (1986) Temperature-sensitive steps in the transport of secretory proteins through the Golgi complex in exocrine pancreatic cells. Proc. Natl. Acad. Sci. U.S.A. 83, 6425–6429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Torgerson R. R., McNiven M. A. (1998) The actin-myosin cytoskeleton mediates reversible agonist-induced membrane blebbing. J. Cell Sci. 111, 2911–2922 [DOI] [PubMed] [Google Scholar]
- 32. Stolz D. B., Ross M. A., Salem H. M., Mars W. M., Michalopoulos G. K., Enomoto K. (1999) Cationic colloidal silica membrane perturbation as a means of examining changes at the sinusoidal surface during liver regeneration. Am. J. Pathol. 155, 1487–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hendricks L. C., McClanahan S. L., Palade G. E., Farquhar M. G. (1992) Brefeldin A affects early events but does not affect late events along the exocytic pathway in pancreatic acinar cells. Proc. Natl. Acad. Sci. U.S.A. 89, 7242–7246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Naslavsky N., Weigert R., Donaldson J. G. (2003) Convergence of non-clathrin- and clathrin-derived endosomes involves Arf6 inactivation and changes in phosphoinositides. Mol. Biol. Cell 14, 417–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Raraty M., Ward J., Erdemli G., Vaillant C., Neoptolemos J. P., Sutton R., Petersen O. H. (2000) Calcium-dependent enzyme activation and vacuole formation in the apical granular region of pancreatic acinar cells. Proc. Natl. Acad. Sci. U.S.A. 97, 13126–13131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sherwood M. W., Prior I. A., Voronina S. G., Barrow S. L., Woodsmith J. D., Gerasimenko O. V., Petersen O. H., Tepikin A. V. (2007) Activation of trypsinogen in large endocytic vacuoles of pancreatic acinar cells. Proc. Natl. Acad. Sci. U.S.A. 104, 5674–5679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Willemer S., Bialek R., Adler G. (1990) Localization of lysosomal and digestive enzymes in cytoplasmic vacuoles in caerulein-pancreatitis. Histochemistry 94, 161–170 [DOI] [PubMed] [Google Scholar]
- 38. Saluja A., Saluja M., Villa A., Leli U., Rutledge P., Meldolesi J., Steer M. (1989) Pancreatic duct obstruction in rabbits causes digestive zymogen and lysosomal enzyme colocalization. J. Clin. Invest. 84, 1260–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jane D. T., Morvay L., Dasilva L., Cavallo-Medved D., Sloane B. F., Dufresne M. J. (2006) Cathepsin B localizes to plasma membrane caveolae of differentiating myoblasts and is secreted in an active form at physiological pH. Biol. Chem. 387, 223–234 [DOI] [PubMed] [Google Scholar]
- 40. Wartmann T., Mayerle J., Kähne T., Sahin-Tóth M., Ruthenbürger M., Matthias R., Kruse A., Reinheckel T., Peters C., Weiss F. U., Sendler M., Lippert H., Schulz H. U., Aghdassi A., Dummer A., Teller S., Halangk W., Lerch M. M. (2010) Cathepsin L inactivates human trypsinogen, whereas cathepsin L deletion reduces the severity of pancreatitis in mice. Gastroenterology 138, 726–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. D'Souza-Schorey C., Chavrier P. (2006) ARF proteins: roles in membrane traffic and beyond. Nat. Rev. Mol. Cell Biol. 7, 347–358 [DOI] [PubMed] [Google Scholar]
- 42. Wendler F., Tooze S. (2001) Syntaxin 6: the promiscuous behavior of a SNARE protein. Traffic 2, 606–611 [DOI] [PubMed] [Google Scholar]
- 43. Watanabe O., Baccino F. M., Steer M. L., Meldolesi J. (1984) Supramaximal caerulein stimulation and ultrastructure of rat pancreatic acinar cell: early morphological changes during development of experimental pancreatitis. Am. J. Physiol. 246, G457–G467 [DOI] [PubMed] [Google Scholar]
- 44. Mach L., Schwihla H., Stüwe K., Rowan A. D., Mort J. S., Glössl J. (1993) Activation of procathepsin B in human hepatoma cells: the conversion into the mature enzyme relies on the action of cathepsin B itself. Biochem. J. 293, 437–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhu X. L., Ohta Y., Jordan F., Inouye M. (1989) Pro-sequence of subtilisin can guide the refolding of denatured subtilisin in an intermolecular process. Nature 339, 483–484 [DOI] [PubMed] [Google Scholar]
- 46. Hirano T., Saluja A., Ramarao P., Lerch M. M., Saluja M., Steer M. L. (1991) Apical secretion of lysosomal enzymes in rabbit pancreas occurs via a secretagogue regulated pathway and is increased after pancreatic duct obstruction. J. Clin. Invest. 87, 865–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nevalainen T. J., Janigan D. T. (1974) Degeneration of mouse pancreatic acinar cells during fasting. Virchows Arch. B Cell Pathol. 15, 107–118 [DOI] [PubMed] [Google Scholar]
- 48. Crozier S. J., Sans M. D., Lang C. H., D'Alecy L. G., Ernst S. A., Williams J. A. (2008) CCK-induced pancreatic growth is not limited by mitogenic capacity in mice. Am. J. Physiol. Gastrointest. Liver Physiol 294, G1148–G1157 [DOI] [PubMed] [Google Scholar]
- 49. Tanaka Y., Guhde G., Suter A., Eskelinen E. L., Hartmann D., Lüllmann-Rauch R., Janssen P. M., Blanz J., von Figura K., Saftig P. (2000) Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 406, 902–906 [DOI] [PubMed] [Google Scholar]
- 50. Van Acker G. J., Saluja A. K., Bhagat L., Singh V. P., Song A. M., Steer M. L. (2002) Cathepsin B inhibition prevents trypsinogen activation and reduces pancreatitis severity. Am. J. Physiol. Gastrointest. Liver Physiol 283, G794–800 [DOI] [PubMed] [Google Scholar]
- 51. Klöppel G., Dreyer T., Willemer S., Kern H. F., Adler G. (1986) Human acute pancreatitis: its pathogenesis in the light of immunocytochemical and ultrastructural findings in acinar cells. Virchows Arch. A Pathol. Anat. Histopathol. 409, 791–803 [DOI] [PubMed] [Google Scholar]
- 52. Gaiser S., Daniluk J., Liu Y., Tsou L., Chu J., Lee W., Longnecker D. S., Logsdon C. D., Ji B. (2011) Intracellular activation of trypsinogen in transgenic mice induces acute but not chronic pancreatitis. Gut 60, 1379–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ohmuraya M., Hirota M., Araki M., Mizushima N., Matsui M., Mizumoto T., Haruna K., Kume S., Takeya M., Ogawa M., Araki K., Yamamura K. (2005) Autophagic cell death of pancreatic acinar cells in serine protease inhibitor Kazal type 3-deficient mice. Gastroenterology 129, 696–705 [DOI] [PubMed] [Google Scholar]
- 54. Ohmuraya M., Hirota M., Araki K., Baba H., Yamamura K. (2006) Enhanced trypsin activity in pancreatic acinar cells deficient for serine protease inhibitor Kazal type 3. Pancreas 33, 104–106 [DOI] [PubMed] [Google Scholar]
- 55. Sutton R., Criddle D., Raraty M. G., Tepikin A., Neoptolemos J. P., Petersen O. H. (2003) Signal transduction, calcium, and acute pancreatitis. Pancreatology 3, 497–505 [DOI] [PubMed] [Google Scholar]
- 56. Gukovskaya A. S., Gukovsky I., Jung Y., Mouria M., Pandol S. J. (2002) Cholecystokinin induces caspase activation and mitochondrial dysfunction in pancreatic acinar cells: roles in cell injury processes of pancreatitis. J. Biol. Chem. 277, 22595–22604 [DOI] [PubMed] [Google Scholar]
- 57. Grady T., Liang P., Ernst S. A., Logsdon C. D. (1997) Chemokine gene expression in rat pancreatic acinar cells is an early event associated with acute pancreatitis. Gastroenterology 113, 1966–1975 [DOI] [PubMed] [Google Scholar]
- 58. Suzuki T., Kanai Y., Hara T., Sasaki J., Sasaki T., Kohara M., Maehama T., Taya C., Shitara H., Yonekawa H., Frohman M. A., Yokozeki T., Kanaho Y. (2006) Crucial role of the small GTPase ARF6 in hepatic cord formation during liver development. Mol. Cell. Biol. 26, 6149–6156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bunpo P., Dudley A., Cundiff J. K., Cavener D. R., Wek R. C., Anthony T. G. (2009) GCN2 protein kinase is required to activate amino acid deprivation responses in mice treated with the anticancer agent l-asparaginase. J. Biol. Chem. 284, 32742–32749 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.