

Background: Mitochondrial thymidine kinase 2 phosphorylates thymidine and deoxycytidine and is essential for mitochondrial function.
Results: S-Glutathionylation of TK2 reduced activity in vitro and in vivo and affected its stability in H2O2-treated mitochondria and human cells.
Conclusion: Oxidative stress induces mitochondrial TK2 S-glutathionylation and down-regulation.
Significance: S-Glutathionylation is a new TK2 regulatory mechanism possibly contributing to mitochondrial diseases and aging.
Keywords: Glutathione, Glutathionylation, Mitochondrial Diseases, Mitochondrial Metabolism, Oxidative Stress, Protein Degradation, Mitochondrial DNA Precursor Synthesis, Thymidine Kinase 2
Abstract
Protein glutathionylation in response to oxidative stress can affect both the stability and activity of target proteins. Mitochondrial thymidine kinase 2 (TK2) is a key enzyme in mitochondrial DNA precursor synthesis. Using an antibody specific for glutathione (GSH), S-glutathionylated TK2 was detected after the addition of glutathione disulfide (GSSG) but not GSH. This was reversed by the addition of dithiothreitol, suggesting that S-glutathionylation of TK2 is reversible. Site-directed mutagenesis of the cysteine residues and subsequent analysis of mutant enzymes demonstrated that Cys-189 and Cys-264 were specifically glutathionylated by GSSG. These cysteine residues do not appear to be part of the active site, as demonstrated by kinetic studies of the mutant enzymes. Treatment of isolated rat mitochondria with hydrogen peroxide resulted in S-glutathionylation of added recombinant TK2. Treatment of intact cells with hydrogen peroxide led to reduction of mitochondrial TK2 activity and protein levels, as well as S-glutathionylation of TK2. Furthermore, the addition of S-glutathionylated recombinant TK2 to mitochondria isolated from hydrogen peroxide-treated cells led to degradation of the S-glutathionylated TK2, which was not observed with unmodified TK2. S-Glutathionylation on Cys-189 was responsible for the observed selective degradation of TK2 in mitochondria. These results strongly suggest that oxidative damage-induced S-glutathionylation and degradation of TK2 have significant impact on mitochondrial DNA precursor synthesis.
Introduction
Mitochondrial thymidine kinase 2 (TK2)2 is a salvage enzyme responsible for the initial phosphorylation of thymidine (dT) and deoxycytidine (dC) in mitochondrial DNA (mtDNA) precursor synthesis. Deficiency in TK2 activity is associated with the devastating mitochondrial DNA depletion syndrome, and the disease phenotype extends to the involvement of multiple organs beside skeletal muscle, as described in initial reports (1–5). TK2 is expressed in all tissues at low levels; however, little is known regarding TK2 regulation at the transcriptional, translational, and post-translational levels.
Glutathione (GSH) is a ubiquitous low molecular weight non-protein thiol-containing molecule found at millimolar concentrations in eukaryotic cells and is involved in numerous cellular processes, particularly in redox buffering. The ratio of GSH to glutathione disulfide (GSSG) contributes to the redox potential of the cell and thereby to redox homeostasis (6). Oxidative stress leads to a decreased GSH/GSSG ratio and formation of mixed disulfide bonds between GSH or GSSG and redox-sensitive cysteine residues within proteins (7–9).
Protein thiol groups can interact with GSH or GSSG via two different pathways, thiol-disulfide exchange between a free protein thiol group with GSSG or protein thiol oxidation by reactive oxygen species (ROS), yielding a thiyl radical that reacts with a glutathionylate anion to eventually form a mixed disulfide (7–9). This modification of protein reactive cysteines has emerged as a central mechanism by which changes in the intracellular redox status may be transduced into functional responses. Like protein phosphorylation, glutathionylation can modulate enzyme activities, alter transcription profiles, modify protein-protein interactions, and regulate adaptive cell signaling (9).
Mitochondria are the sites for the generation of ROS and reactive nitrogen species. Hydrogen peroxide (H2O2) and hydroxyl radicals are produced during mitochondrial respiration and lipid oxidation. The levels of ROS and H2O2 increase under oxidative stress or during the normal aging process. Normally, intramitochondrial GSH/GSSG ratios are in favor of GSH; however, under oxidative stress or redox-sensing, ROS oxidizes GSH to GSSG. Increased GSSG concentrations lead to protein glutathionylation, which affects either the activity or stability of target proteins (9, 10). For instance, S-glutathionylated recombinant p53 is selectively degraded in an ATP-dependent manner when incubated with proteasomes prepared from rabbit reticulocytes (11).
Mature mitochondrial TK2 contains 5 cysteine residues, and freshly purified recombinant human TK2 appears as a single monomeric band of ∼28 kDa on non-reducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE); however, under oxidative conditions such as prolonged storage, the enzyme forms a high molecular weight complex as seen in SDS-PAGE analysis (12). Here, we demonstrate that TK2 activity is sensitive to reducing and oxidizing agents and that the addition of GSSG results in site-specific S-glutathionylation of TK2 on two of its five cysteine residues. The consequences of this post-translational modification on TK2 activity and stability were also investigated. Our results suggest an important new mechanism for the regulation of mtDNA precursor synthesis.
EXPERIMENTAL PROCEDURES
Materials
Radioactive thymidine ([methyl-3H]dT, 20 Ci/mmol) was purchased from PerkinElmer Life Sciences, and deoxycytidine ([5-3H]dC, 27 Ci/mmol) was from Moravek Biochemicals Inc. Mouse monoclonal antibodies against GSH and cytochrome c oxidase subunit IV (COX IV) were from Abcam. The anti-His-tag antibody was from GE Healthcare. A goat anti-TK2 antibody was purchased from Santa Cruz Biotechnology.
A polyclonal rabbit anti-human TK2 antibody was produced using a synthetic peptide (NRDRILTPENRKHC) chosen from the C-terminal sequence of human mitochondrial TK2 as the antigen (Genscript Inc). The TK2-specific antibody was purified from rabbit antisera by affinity chromatography on Sepharose resin coupled with the peptide antigen following a standard protocol (CNBr activated Sepharose; GE Healthcare). TK2 activity was determined by a radiochemical assay using the DE-81 filter paper method with tritium-labeled dT or dC as the substrate, as previously described (13).
Site-directed Mutagenesis, Expression, and Purification
An N-terminal truncated TK2 protein starting from amino acid 51 of the full-length human TK2 sequence (GenBankTM accession number NM_004614) has been previously characterized and showed kinetic properties identical to those of the native mitochondrial form of TK2 (2, 13); therefore, it was used as wild-type (wt) TK2. The site-directed mutagenesis procedure was essentially as described (2) and the mutant cDNA was subcloned into the pEXP5NT vector (Invitrogen). The mutations were verified by sequencing. The recombinant protein contained an N-terminal fusion 6× histidine tag and a tobacco etch virus cleavage site.
The plasmids containing the desired mutations were transformed into the BL21 (DE3) pLysS strain of Escherichia coli. Induction was performed in the presence of 0.05 mm isopropyl β-d-thiogalactopyranoside for 2 h at 37 °C or for 6 h at 25 °C. Wild-type TK2 was expressed in parallel as a control. Recombinant proteins were purified essentially as described (2).
S-Glutathionylation of Recombinant TK2
Recombinant TK2 (4–6 μg) was mixed with various concentrations of DTT (dithiothreitol), GSH, or GSSG and incubated either at room temperature or at 37 °C for 30–60 min. The samples were then analyzed by 12% non-reducing SDS-PAGE and Western blot using anti-GSH antibody.
The Effects of DTT, IAM, and Diamide on S-Glutathionylation of TK2
Recombinant TK2 (6 μg) was incubated at 37 °C for 15 min in the presence of DTT (0.5, 1, and 2 mm), IAM (10, 50, 100, and 1000 μm) or diamide (10, 50, 100, and 1000 μm), and then GSSG (10 mm) was added and incubated for additional 45 min. The samples were then analyzed by non-reducing SDS-PAGE and Western blot using the anti GSH antibody.
Determination of S-Glutathionylated TK2 Activity
Recombinant TK2 (4 μg) was incubated with 5 mm GSSG at 37 °C for 30 min, and then the mixture was diluted with enzyme dilution buffer (25 mm Tris/HCl, pH 7.6, 0.5 mg ml−1 BSA, and 1 mm MgCl2). Two ng of the S-glutathionylated TK2 was used in activity determination using 10 μm [3H]dT as substrate in the presence of various concentrations of DTT, IAM, and diamide.
Western Blot Analysis
Samples of recombinant TK2 treated with reducing or oxidizing agents or mitochondrial proteins were separated on 12% SDS-polyacrylamide gels and transferred to polyvinylidene difluoride membranes. The membranes were then probed for 2 h at room temperature or at 4 °C overnight with the appropriate primary antibodies: rabbit anti-human TK2 (1:2000), goat anti-TK2 (1:200), mouse anti-GSH (1:150), mouse anti-cytochrome c oxidase subunit 4 (1:500 dilution), or mouse anti-His (1:2000 dilution) antibodies. After washing, the membranes were incubated with appropriate secondary antibodies and detected by the enhanced chemiluminescence immunodetection system (GE Healthcare). Band intensity was quantified using the Quantity One software (Bio-Rad).
H2O2 Treatment of Isolated Rat Mitochondria
Rat (Sprague-Dawley; ∼250 g) liver or brain mitochondria were isolated using differential centrifugation according to the standard protocol (14). H2O2 was added to the mitochondria preparations (5 mg of protein) followed by incubation at 37 °C for 30 min, and mitochondrial proteins were extracted by sonication in an ice-water bath. Recombinant TK2 (2 μg) was added, and the mitochondrial extracts were incubated at 37 °C for an additional 30 min. After this, Ni2+-Sepharose resin was added to the reaction mixture, which was then incubated at 4 °C for 60 min. Ni2+-Sepharose was pelleted by centrifugation at 16,000 × g for 1 min. After 3 washes with 30 mm imidazole, the bound proteins were eluted with 300 mm imidazole and analyzed directly by SDS-PAGE and Western blot using anti-GSH and anti-TK2 antibodies.
Cell Culture and Mitochondria Isolation
U2OS (human osteosarcoma cell line; ATCC HTB-96TM) were cultured in McCoys 5A medium supplemented with 10% fetal calf serum at 37 °C with 5% CO2. Cells were incubated with various concentrations of hydrogen peroxide and harvested after 30 min. About 30 × 106 cells were resuspended in 1 ml of mitochondrial isolation buffer (320 mm sucrose, 5 mm Tris, and 2 mm EGTA, pH 7.4), and mitochondria were isolated essentially as described (15). The mitochondrial pellets were resuspended in 50 μl of mitochondrial isolation buffer containing 0.5% Nonidet P-40. Mitochondrial proteins were extracted by repeated freezing and thawing and by sonication. The protein concentration was determined by the Bio-Rad protein assay using bovine serum albumin as the standard.
Immunoprecipitation
Approximately 2.9 mg of mitochondrial proteins prepared from 4 × 108 U2OS cells treated with 4 mm H2O2 and 1.5 mg of mitochondrial proteins prepared from 2 × 108 untreated U2OS cells were first incubated with protein A-Sepharose to remove nonspecific binding to protein A-Sepharose. Rabbit anti-human TK2 antibody (10 μg) was then added, and the mixture was incubated at 4 °C overnight. Phenylmethylsulfonyl fluoride (PMSF) was added to the above mixture at a final concentration of 0.1 mm. The antigen-antibody complexes were pulled down by incubation with protein A-Sepharose at 4 °C for 2 h. After 3 washes with phosphate-buffered saline, the bound proteins were released by the addition of 50 μl of non-reducing SDS sample buffer and heating at 96 °C for 3 min. After a brief centrifugation, 25 μl of each supernatant was analyzed by 12% SDS-PAGE and Western blot using the anti-GSH and goat anti-TK2 antibodies.
Degradation of S-Glutathionylated Recombinant TK2
Mitochondrial extracts prepared from U2OS cells treated with 4 mm H2O2 and from untreated cells were used. Recombinant TK2 (140 ng) (wt, C189A, and C264A mutants) were incubated with 5 mm GSSG at 37 °C for 60 min to ensure complete glutathionylation. The treated and untreated TK2 proteins were then mixed with 35 μg of mitochondrial extract in the presence or absence of 0.1 mm PMSF in a total of 35 μl and incubated at 37 °C. At 0, 15, 30, 45, and 60 min, 5-μl aliquots were withdrawn, mixed with reducing SDS sample buffer, and resolved on 12% SDS-PAGE. Finally, Western blot analysis was performed using the anti-His antibody to detect TK2 protein and the anti-COX IV antibody for a loading control and to estimate protein stability in general.
RESULTS
S-Glutathionylation of Recombinant TK2 and the Effects of Thiol-modifying Agents
To study the effect of reducing and oxidizing agents on TK2 activity, we compared TK2 activities determined in the presence of DTT, GSH, and GSSG to the activity in the absence of any agents. As shown in Fig. 1A, the addition of DTT or GSH resulted in an increase in TK2 activity. At low concentrations, GSSG had only minor effects, but ∼25% inhibition was observed at high concentrations (Fig. 1A). TK2 activities were significantly higher in the presence of DTT or GSH than in the presence of GSSG (Fig. 1A). Pre-incubation with 5 mm GSSG on ice for 30 min resulted in a 55% decrease in TK2 activity.
FIGURE 1.

A, shown are the effects of reducing/oxidizing agent on TK2 activity. TK2 activity was determined with [3H]dT as the substrate in the presence of various concentrations of DTT, GSH, and GSSG in the reaction mixtures. Activities in the presence of DTT, GSH, and GSSG are relative to the activity in the absence any agent (1038 nmol min−1 mg−1). B, concentration-dependent S-glutathionylation of recombinant TK2 is shown. Recombinant TK2 (4 μg) was mixed with GSSG or GSH at the indicated concentrations and incubated at room temperature for 30 min. To assess the effects of thiol-modifying agents, 6 μg of TK2 was incubated at 37 °C for 15 min in the presence of DTT/IAM/diamide at the indicated concentrations, and then GSSG (10 mm) was added and incubated for additional 45 min. SDS-PAGE analysis was carried out under non-reducing conditions. C, shown is the effect of thiol-modifying agents on the activity of S-glutathionylated TK2. Recombinant TK2 was treated with 5 mm GSSG at 37 °C for 30 min, and then the S-glutathionylated TK2 activity was measured using 10 μm [3H]dT as substrate in the presence of various concentrations of DTT, IAM, and diamide. Activities in the presence of thiol-modifying agents are relative to the activity of S-glutathionylated TK2 (445 nmol·min−1 mg−1). D, identification of cysteine residues involved in S-glutathionylation by site-directed mutagenesis and Western blot analysis using the anti-GSH antibody is shown.
GSH and GSSG are known to be able to modify protein thiol groups by introducing disulfide bonds between the protein thiols and GSSG/GSH. Human mitochondrial TK2 contains 5 cysteines (Fig. 2), which are potential targets for S-glutathionylation. To examine whether direct S-glutathionylation occurs in the presence of GSSG or GSH, the recombinant TK2 protein was incubated with various concentrations of GSSG and GSH at room temperature for 30 min. The samples were then subjected to non-reducing SDS-PAGE analysis. Two bands, one corresponding to the TK2 monomer and one with a higher molecular mass, were observed in the presence of GSSG. However, only one band corresponding to the TK2 monomer was observed in the presence of GSH (data not shown). Western blot analysis using an anti-GSH antibody revealed concentration-dependent S-glutathionylation of the TK2 protein in the presence of GSSG, whereas no S-glutathionylation of TK2 was seen in the presence of GSH (Fig. 1B), suggesting an exchange of the protein thiol group with GSSG. Complete S-glutathionylation of TK2 can be achieved by incubation with 5 mm GSSG at 37 °C for 30 min (data not shown).
FIGURE 2.

A, shown is a TK2 structure model with all five cysteines indicated. The model was built based on the D. melanogaster deoxynucleoside kinase structure (PDB entry 1OT3). B, shown is the amino acid sequence of human TK2. The arrow indicates the first amino acid of mitochondrial TK2. The positions of the cysteines in mature mitochondrial TK2 are marked.
Treatment with reducing agents such as DTT decreased the level of TK2 S-glutathionylation, demonstrating that S-glutathionylation is a reversible process (Fig. 1B). Blocking of cysteine residues by the alkylating agent IAM also reduced TK2 S-glutathionylation (Fig. 1B). However, treatment with diamide, a thiol oxidizing agent, enhanced the glutathionylation of TK2 (Fig. 1B).
The effects of DTT, IAM, and diamide on S-glutathionylated TK2 activity were also studied. The addition of DTT to S-glutathionylated TK2 resulted in an almost 50% increase in TK2 activity, whereas the addition of the alkylating agent IAM strongly inhibited TK2 activity. The addition of diamide resulted in a >90% loss of TK2 activity at 20 mm (Fig. 1C). These results indicate that the removal of the -SSG group restores TK2 activity, whereas cysteine modifications by IAM or diamide result in loss of activity.
Site-specific S-Glutathionylation of TK2
To identify the cysteine residues involved in the S-glutathionylation of TK2, each of the five cysteines was individually substituted with an alanine by site-directed mutagenesis, and the mutant enzymes were expressed in E. coli and purified. Equal amounts of purified mutant proteins were incubated with GSSG, and the level of S-glutathionylation was assessed by Western blot analysis. All five cysteine to alanine mutants showed S-glutathionylation; three of them had similar intensity, whereas the other two e.g. C189A and C264A mutants had significantly lower signal intensity (Fig. 1D), indicating that more than one cysteine is involved. Therefore, both Cys-189 and Cys-264 were mutated to alanine, and the double mutant (C189A/C264A) enzyme showed no S-glutathionylation when incubated with either GSSG or GSH (Fig. 1D), demonstrating that Cys-189 and Cys-264 are the specific cysteine residues involved in S-glutathionylation.
S-Glutathionylation of Recombinant TK2 in Isolated Rat Liver Mitochondria
Freshly isolated rat liver and brain mitochondria were treated with different concentrations of H2O2, and the mitochondrial proteins extracted. We were unable to detect any endogenous TK2 protein, native or S-glutathionylated, upon direct Western blot analysis of mitochondrial proteins, possibly due to the low abundance of the TK2 protein and/or low sensitivity of the anti-GSH antibody. Therefore, recombinant TK2 was added to these mitochondrial extracts that had been treated with different concentrations of H2O2. Thereafter, the Ni2+-Sepharose resin was used to pull down recombinant TK2 from the reaction mixtures. The bound proteins were eluted with imidazole and analyzed by SDS-PAGE and Western blot using the anti-GSH and anti-TK2 antibodies. S-Glutathionylated TK2 was detected with liver mitochondria but not with brain mitochondria (Fig. 3).
FIGURE 3.
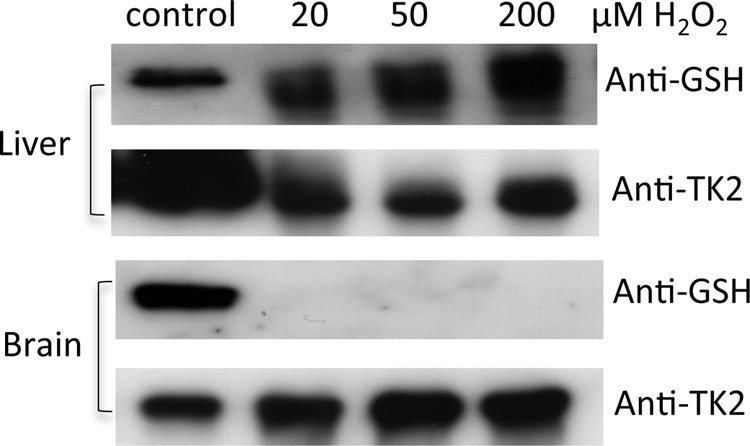
S-Glutathionylation of recombinant TK2 in mitochondria treated with H2O2. Freshly isolated rat liver and brain mitochondria (5 mg protein) were treated with different concentrations of H2O2. Mitochondria were then disrupted by repeated freezing and thawing and mixed with recombinant TK2 (2 μg) and incubated. Ni2+-Sepharose was used to pull down recombinant TK2, which was then analyzed by SDS-PAGE and Western blot using anti-GSH and anti-TK2 antibodies. S-Glutathionylated TK2 was used as a control in the Western blot analysis.
Oxidative Stress-induced TK2 S-Glutathionylation and Decreased Protein Abundance in U2OS Cells
H2O2 is known to induce oxidative stress and protein S-glutathionylation in cultured cells (18). We treated U2OS (human osteosarcoma cell line) cells with H2O2 at 37 °C for 30 min, and then the cells were harvested, and mitochondria were isolated. TK2 activity was determined using [3H]dT as substrate, and TK2 protein levels were determined by Western blot analysis using the rabbit anti-human TK2 antibody. TK2 activity decreased in a concentration-dependent manner (Fig. 4A), and the level of TK2 protein also decreased with increasing H2O2 concentrations (Fig. 4B). The levels of COX IV, however, were unchanged (Fig. 4B). We observed a linear correlation between TK2 activity and protein levels in mitochondria isolated from cells treated with H2O2, with an R value of 0.96 using Pearson correlation analysis. This strongly suggests that the reduced TK2 activity is due to a decreased TK2 protein level, which is most likely the result of selective degradation of TK2 as COX IV levels were unchanged.
FIGURE 4.
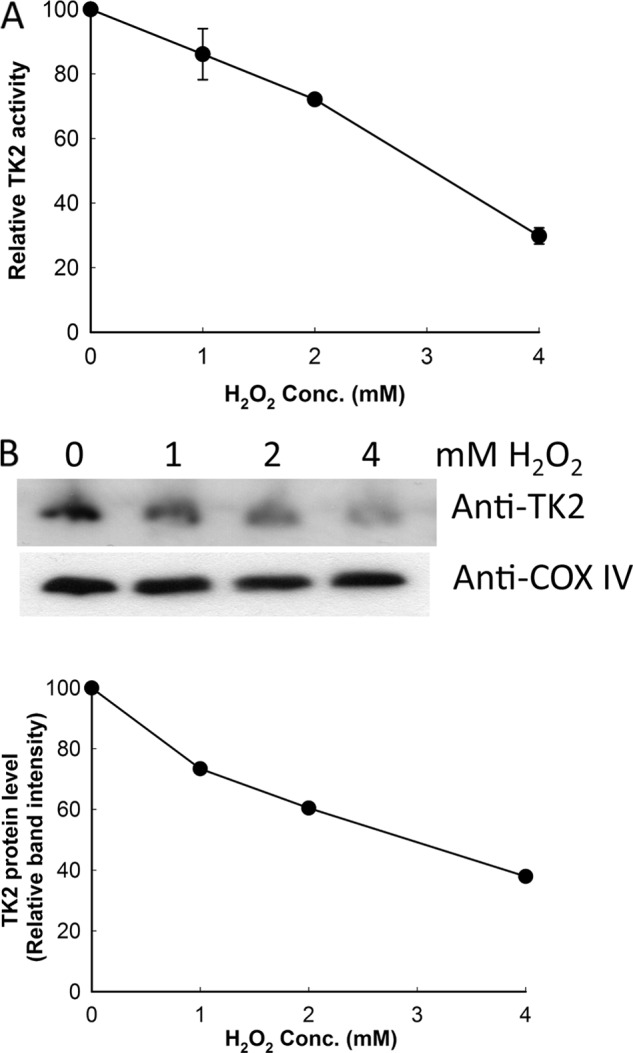
The levels of TK2 activity (A) and protein (B) in mitochondria isolated from U2OS cells treated with H2O2. TK2 activity levels are relative to the values obtained from mitochondria isolated from untreated cells (145.7 pmol min−1 mg−1) set to 100. An equal amount of mitochondrial proteins (30 μg) was resolved on 12% SDS-PAGE, and the TK2 protein level was determined by Western blot using the rabbit anti-TK2 antibody. The same membrane was reprobed with the anti-COX IV antibody and used as a loading control. Band intensity was quantified, and the level of TK2 protein was normalized to the level of COX IV in each sample and shown as levels relative to untreated cells (set to 100). The experiments were repeated at least six times and one representative blot is shown.
Under these conditions we did not detect any S-glutathionylated TK2 in mitochondria prepared from U2OS cells, possibly due to the low abundance of TK2 protein and the limited sensitivity of the anti-GSH antibody. Therefore, we repeated the 4 mm H2O2 treatment again and compared with untreated cells but this time with a large number of cells and an immunoprecipitation method using the rabbit anti-human TK2 antibody to capture the TK2 protein. PMSF was added to the isolated mitochondria to inhibit protease activity. In a subsequent Western blot analysis using the anti-GSH antibody and a goat anti-TK2 antibody, we were able to detect total TK2 protein content and the amount of S-glutathionylated TK2 in these cells. As shown in Fig. 5A, S-glutathionylated TK2 was detected in H2O2-treated cells at levels at least 3–5 times higher than in untreated cells. The level of total TK2 protein was significantly higher in untreated cells, although the number of cells used was only half that of the treated cells (Fig. 5A). The TK2 activity and protein levels as well as the COX IV level were as expected (Fig. 5B). Thus, mitochondrial TK2 is S-glutathionylated under oxidative stress.
FIGURE 5.
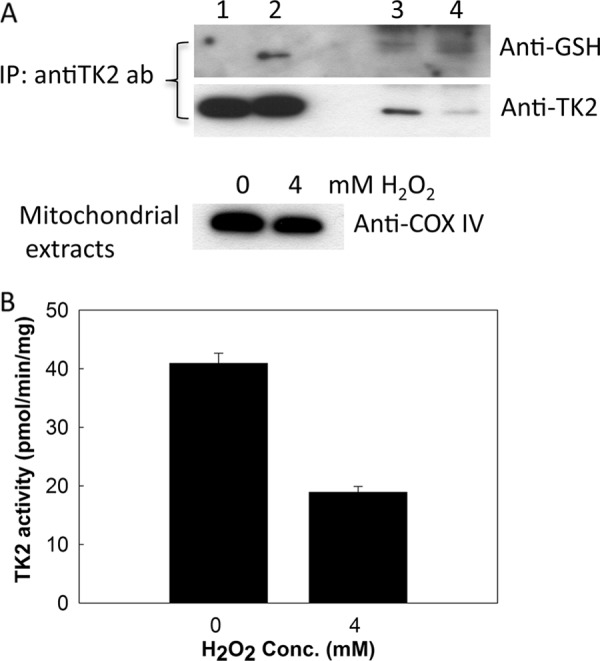
H2O2-induced S-glutathionylation of mitochondrial TK2 in U2OS cells. A, mitochondrial proteins prepared from ∼4 × 108 U2OS cells treated with 4 mm H2O2 (2.9 mg protein) and from 2 × 108 untreated U2OS cells (1.5 mg protein) were used in immunoprecipitation (IP) using the rabbit anti-human TK2 antibody (ab). See “Experimental Procedures” for details. Lane 1, untreated recombinant TK2 (100 ng); lane 2, S-glutathionylated recombinant TK2 (100 ng); lane 3, mitochondrial proteins (corresponded to 0.75 mg of mitochondrial protein) from untreated U2OS cells after IP; lane 4, mitochondrial proteins (corresponded to 1.45 mg of mitochondrial protein) from H2O2-treated U2OS cells after IP. The level of COX IV was determined for mitochondria prepared from the H2O2-treated and untreated U2OS cells. Mitochondrial protein (15 μg) was analyzed by SDS-PAGE and Western blot using the anti-COX IV antibody. B, TK2 activities in mitochondria prepared from H2O2-treated and untreated U2OS cells were determined using [3H]dT as substrate.
S-Glutathionylation-dependent Degradation of Recombinant TK2
Recombinant TK2 untreated and treated with GSSG was mixed with mitochondrial extracts prepared from U2OS cells treated with H2O2 and incubated at 37 °C. At each time interval, aliquots were removed and analyzed by SDS-PAGE and Western blot analysis using the anti-His-tag antibody to detect recombinant TK2 and the anti-COX IV antibody to detect COX IV protein. With unmodified TK2 there was no change in the band intensity; however, a time-dependent decrease of the S-glutathionylated recombinant TK2 was observed (Fig. 6A). In the presence of a serine protease inhibitor, e.g. PMSF, the degradation of S-glutathionylated TK2 was strongly inhibited (Fig. 6A). The control protein, COX IV, was at similar levels at all time points (Fig. 6A). When the GSSG-treated and untreated TK2 were mixed with mitochondria isolated from untreated U2OS cells, we did not observed any significant degradation (Fig. 6B). Thus, S-glutathionylation apparently led to selective degradation of TK2 in mitochondria under conditions of oxidative stress.
FIGURE 6.
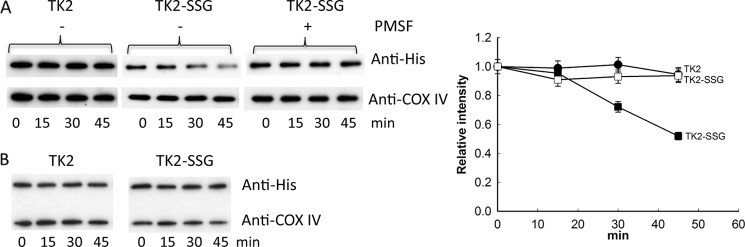
Selective degradation of recombinant TK2 upon S-glutathionylation. A, recombinant TK1, untreated (TK2), and TK2 treated with GSSG (TK2-SSG) were mixed with mitochondrial extracts prepared from U2OS cells treated with 4 mm H2O2 in the absence or presence of the serine protease inhibitor PMSF. At each time point, an aliquot was drawn and analyzed using reducing 12% SDS-PAGE and Western blot using the anti-His and anti-COX IV antibodies to determine the levels of TK2 and COX IV; see “Experimental Procedures” for details. TK2 band intensity was quantified and expressed as intensity relative to the zero time point. Shown are untreated TK2 in the absence of PMSF (●) and TK2-SSG in the absence (■) and presence (□) of PMSF. B, recombinant TK2, untreated (TK2), and TK2 treated with GSSG (TK2-SSG) were mixed with mitochondrial extracts prepared from untreated U2OS cells and analyzed. This experiment was repeated at least four times, and one representative blot is shown.
S-Glutathionylation on Cys-189 Is Responsible for the Observed Degradation in Mitochondria Isolated from H2O2-treated Cells
To investigate which of the cysteines are important for S-glutathionylation-dependent degradation, we mixed S-glutathionylated C189A and C264A mutants with mitochondria isolated from cells treated with 4 mm H2O2 and incubated at 37 °C. At each time point, aliquots were removed and analyzed by Western blot using the anti-His antibody to detect recombinant TK2. As a control, the untreated C189A and C264A mutants were also mixed with these mitochondria and analyzed. As shown in Fig. 7, there was a slight decrease in band intensity at the last time point for C189A, C189A-SSG, and C264A (Fig. 7). However, there was a significant time-dependent decrease in the intensity of the C264A-SSG band (Fig. 7). These results indicated that S-glutathionylation on Cys-189 is responsible for the observed degradation.
FIGURE 7.
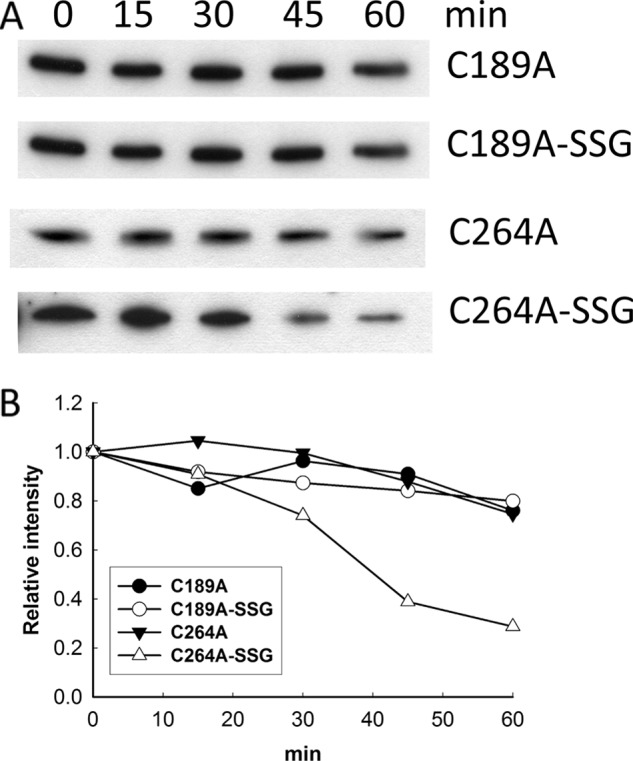
Proteolytic degradation of S-glutathionylated C189A and C264A mutants. A, untreated C189A and C264A mutants and those treated with GSSG (C189A-SSG and C264A-SSG) were mixed with mitochondrial extracts prepared from 4 mm H2O2-treated U2OS cells and incubated at 37 °C. At each time point, aliquots were drawn and analyzed. See “Experimental Procedures.” B, band intensity was quantified and is represented as intensity relative to the zero time point.
The Role of the Cysteines in Catalysis
The results described above demonstrate that modification of cysteine residues affects TK2 stability and activity. Therefore, we characterized all the mutant enzymes (the Cys to Ala and Cys to Ser mutants) to evaluate the role of the cysteines in catalysis.
Mutations of Cys to Ala or Ser had no significant effect on the Km values for dT with the mutant enzymes. However, the kcat values for C66S and C189S were significantly lower, whereas C197S had a higher kcat value than that of the wt enzyme. These mutations resulted in lower catalytic efficiencies (kcat/Km values) for C54A, C189A, C197A, C264A, C54S, C66S, C189S, and C264S, whereas C197S and C66A had higher catalytic efficiency than the wt enzyme (Table 1).
TABLE 1.
Kinetic parameters of TK2 cysteine mutants with dT and ATP
Assays were performed with three different batches of enzyme preparation, and the data are presented as the mean ± S.E. With variable dT concentrations (0.5–160 μm), ATP concentration was maintained at 2 mm. When ATP was the variable substrate (1–2000 μm), dT concentration was maintained at 50 μm.
| Variable dT |
Variable ATP/fixed dT |
|||||
|---|---|---|---|---|---|---|
| Km | kcat | kcat/Km | Km | kcat | kcat/Km | |
| μm | s−1 | m−1s−1×10−4 | μm | s−1 | m−1s−1×10−4 | |
| Wild type | 2.8 ± 0.3 | 0.38 ± 0.05 | 14 | 1.8 ± 0.2 | 0.39 ± 0.09 | 21 |
| C54A | 2.7 ± 0.2 | 0.25 ± 0.01 | 9.2 | 3.1 ± 1.5 | 0.25 ± 0.01 | 8.1 |
| C66A | 1.8 ± 0.1 | 0.27 ± 0.06 | 15 | 1.7 ± 0.1 | 0.22 ± 0.03 | 13 |
| C189A | 3.5 ± 0.1 | 0.31 ± 0.05 | 8.8 | 2.7 ± 0.5 | 0.29 ± 0.03 | 11 |
| C197A | 4.6 ± 0.8 | 0.30 ± 0.02 | 6.6 | 3.5 ± 0.3 | 0.26 ± 0.01 | 7.4 |
| C264A | 2.7 ± 0.6 | 0.31 ± 0.05 | 12 | 1.8 ± 0.4 | 0.30 ± 0.05 | 17 |
| C54S | 3.3 ± 0.7 | 0.26 ± 0.05 | 7.9 | 4.7 ± 0.7 | 0.23 ± 0.03 | 4.8 |
| C66S | 4.2 ± 0.8 | 0.08 ± 0.01 | 1.0 | 7.1 ± 0.8 | 0.08 ± 0.01 | 1.0 |
| C189S | 2.3 ± 0.4 | 0.08 ± 0.01 | 3.2 | 2.4 ± 0.6 | 0.09 ± 0.01 | 3.7 |
| C197S | 2.5 ± 0.7 | 0.57 ± 0.26 | 22 | 4.0 ± 1.3 | 0.52 ± 0.21 | 13 |
| C264S | 3.0 ± 0.6 | 0.26 ± 0.04 | 8.7 | 2.1 ± 0.3 | 0.24 ± 0.08 | 11 |
The Km values for ATP in the presence of excess dT showed similar Km values as the wild-type enzyme for all mutants, except for C54S and C66S, which had higher Km values. The kcat values for ATP were lower for all mutants except C197S, which had a higher kcat value than that of wt enzyme. All the mutants had lower kcat/Km values than the wt enzyme (Table 1).
With dC as the variable substrate, the C189A, C197A, C54S, C189S, and C264S mutants had lower Km values, whereas the remaining mutants had similar Km values as the wt enzyme. The kcat values were low for C54A, C54S, C66S, and C189S, but C197S had a higher kcat value than that of the wt enzyme. Accordingly, the C54A, C66A, C264A, C54S, C66S, and C189S mutants had lower efficiencies compared with the wt enzyme, whereas the C189A, C197A, and C264S mutants had efficiencies similar to that of the wt enzyme, and C197S had the highest catalytic efficiency (Table 2).
TABLE 2.
Kinetic parameters of TK2 cysteine mutants with dC and ATP
Assays were performed with three different batches of enzyme preparation, and the data are presented as the mean ± S.E. With variable dC concentrations (0.5–160 μm), ATP concentration was maintained at 2 mm. When ATP was the variable substrate (1–2000 μm), dC concentration was maintained at 50 μm.
| Variable dC |
variable ATP/fixed dC |
|||||
|---|---|---|---|---|---|---|
| Km | kcat | kcat/Km | Km | kcat | kcat/Km | |
| μm | s−1 | m−1s−1×10−4 | μm | s−1 | m−1s−1×10−4 | |
| Wild type | 11.7 ± 1.0 | 0.39 ± 0.05 | 3.3 | 2.7 ± 0.0 | 0.27 ± 0.05 | 10 |
| C54A | 10.3 ± 0.5 | 0.15 ± 0.01 | 1.5 | 3.9 ± 1.9 | 0.12 ± 0.01 | 3.1 |
| C66A | 13.7 ± 0.2 | 0.27 ± 0.06 | 1.9 | 3.1 ± 0.2 | 0.20 ± 0.04 | 6.3 |
| C189A | 6.6 ± 1.0 | 0.24 ± 0.04 | 3.6 | 6.2 ± 1.6 | 0.21 ± 0.04 | 3.3 |
| C197A | 8.9 ± 0.2 | 0.30 ± 0.01 | 3.4 | 9.5 ± 1.6 | 0.26 ± 0.01 | 2.7 |
| C264A | 12.6 ± 1.3 | 0.31 ± 0.01 | 2.5 | 3.0 ± 0.6 | 0.23 ± 0.01 | 7.8 |
| C54S | 8.6 ± 0.9 | 0.14 ± 0.01 | 1.7 | 9.7 ± 0.7 | 0.11 ± 0.01 | 1.1 |
| C66S | 14.4 ± 0.4 | 0.07 ± 0.01 | 0.5 | 10.1 ± 0.8 | 0.05 ± 0.01 | 0.5 |
| C189S | 8.5 ± 0.4 | 0.10 ± 0.03 | 1.2 | 4.4 ± 0.6 | 0.06 ± 0.01 | 1.5 |
| C197S | 11.9 ± 0.9 | 0.74 ± 0.28 | 6.2 | 6.8 ± 3.6 | 0.56 ± 0.19 | 8.2 |
| C264S | 7.4 ± 1.3 | 0.26 ± 0.04 | 3.5 | 1.9 ± 0.6 | 0.26 ± 0.03 | 14 |
The Km value for ATP in the presence of excess dC was unchanged in the case of C54A, C66A, C264A, C189S, C197S, and C264S, but Cys189A, C197A, C54S, and C66S had higher Km values for ATP than did the wt enzyme. The kcat values were lower for all mutants except for C197S, which had a higher kcat value (Table 2).
The structure of TK2 is not available; therefore, a model was generated based on the structure of the homologous Drosophila melanogaster deoxynucleoside kinase (16) in an attempt to explain the role of the cysteine residues in substrate binding and catalysis (Fig. 2). All 5 cysteine residues most likely are not in direct contact with dT or dC; therefore, the mutations to either Ala or Ser had only minor effects on the Km values. Cys-54, Cys-66, Cys-189, and Cys-197 are in the proximity of the two ATP binding structure elements, e.g. the p-loop and the lid region. Thus, substitution of these cysteines with Ala or Ser would most likely affect ATP binding and catalysis (Tables 1 and 2).
In summary, Cys to Ser mutations mainly affected catalysis (low kcat values) especially in the case of Cys-66 and Cys-189. This suggested that replacing the -SH group with an -OH group in these positions may introduce hydrogen bonds that may alter the bonding pattern and the interactions between the enzyme and the substrate that affect catalysis. This also explained why S-glutathionylation on Cys-189 affected TK2 activity. However, the C197S mutation resulted in significant increases in Vmax (kcat) values with both substrates; this could be due to a hydrogen bond introduced by the -OH group that may stabilize the lid region structure and facilitate catalysis. Kinetic studies with these mutants indicated that the amino acid residues involved in binding and catalysis for dT and dC are different, and this may contribute to the different kinetic behavior of TK2 with dT and dC previously observed (2, 13, 17).
DISCUSSION
Mitochondrial TK2 has been studied extensively in recent years, and studies of enzyme properties have helped to elucidate the role of TK2 in mitochondrial diseases associated with mutations in the TK2 gene and the mitochondrial toxicity of nucleoside analogs used in antiviral therapy (2, 5, 13, 19–21). With recombinant enzyme preparations, we have demonstrated here that TK2 activity can be modulated by reducing or oxidizing agents and that such modifications lead to large variations of the activity in vitro. Incubation of the TK2 protein with GSSG resulted in S-glutathionylation of TK2, which can be reversed by the addition of reducing agents. Furthermore, S-glutathionylated TK2 had reduced activity. Using site-directed mutagenesis, Cys-189 and Cys-264 were identified as the residues that are specifically glutathionylated in the presence of GSSG. These results suggest that TK2 activity in vivo is influenced by cellular redox status. Therefore, we studied TK2 activity and protein expression levels in mitochondria isolated from U2OS cells treated with H2O2 and found a concentration-dependent decrease in TK2 activity. This was correlated with the level of TK2 protein detected in these mitochondrial extracts, suggesting that increased oxidative pressure can affect both TK2 activity and protein levels.
Mitochondrial GSH/GSSG plays an important role in redox buffering and redox signaling. In humans, glutathione depletion has been associated with renal, hepatic, and brain tissue damage along with degradation of mitochondria (22, 23). Severe impairment of glutathione homeostasis and changes in glutathione-dependent antioxidant defense have been found to be associated with Friedreich's ataxia (24). In yeast, glutathione depletion is known to cause irreversible respiratory incompetence and a complete loss of mtDNA (25).
The levels of mitochondrial GSH and GSSG differ from tissue to tissue, and mitochondrial GSH/GSSG ratios decrease during oxidation and can be restored by respiratory substrate and active respiration (10). Defective respiration can cause the accumulation of oxidative products such as hydrogen peroxide, superoxide, etc. It has been estimated that as much as 1% of all oxygen consumed may result in the formation of ROS, which cause oxidation of proteins and increased GSSG levels in mitochondria (26). This process leads to S-glutathionylation of a large number of proteins and affects their function and/or stability (7–9). A recent study showed that treatment of isolated rat mitochondria with H2O2 led to a rapid drop in GSH levels, S-glutathionylation of mitochondrial ATP synthase and succinyl-CoA transferase, and decreased catalytic activities (10). We detected S-glutathionylation of recombinant TK2 in rat liver mitochondria that were treated with H2O2 but not in rat brain mitochondria under similar conditions. These data suggested that increased oxidative pressure can lead to S-glutathionylation of TK2 in mitochondria and that mitochondria from different tissues may have different redox buffering or ROS scavenging capacities.
Under oxidative conditions, protein thiols can be oxidized to thiyl radicals, which strongly react with oxygen leading to higher levels of cysteine oxidation (9). Oxidatively damaged proteins must be degraded or removed to avoid the accumulation of protein aggregates that compromise cellular function. Mitochondrial ATP-dependent proteases such as the matrix-located Lon protease and Clp-like proteases are believed to act as a quality control system responsible for the degradation of oxidatively damaged proteins (27–31).
H2O2 treatment of U2OS cells resulted in a decrease in TK2 activity and protein levels. We also detected S-glutathionylated endogenous TK2 protein in H2O2-treated cells. The decrease in TK2 protein levels determined by Western blot analysis indicated mitochondrial-specific proteolytic degradation of TK2 under oxidative conditions, as the level of COX IV protein was not affected by H2O2 treatment. Using recombinant TK2 protein and mitochondria prepared from H2O2-treated cells, we demonstrated that selective mitochondrial degradation of TK2 appears to be S-glutathionylation-dependent and occurs only in mitochondria isolated from H2O2-treated cells and that S-glutathionylation on Cys-189 is responsible for the selective degradation of TK2. These results strongly suggest that oxidative damage of the TK2 protein and modification of its cysteine residues by glutathionylation result in proteolytic degradation and, therefore, reduced activity. To our knowledge this is the first report showing that S-glutathionylation leads to selective degradation in the mitochondria of cells under oxidative stress. We do not know which protease is involved in the degradation of TK2 and what implications this may have on mitochondrial function. It is likely that the relevant proteases are either induced or activated in response to oxidative stress and are involved in the redox regulation of mtDNA replication and maintenance (32, 33).
Enzyme kinetic studies of cysteine mutants and structure modeling have confirmed that these cysteine residues are not likely to be directly involved in the binding of substrates. Mutations of the two cysteine residues involved in S-glutathionylation (Cys-189 and Cys-264) to either Ala or Ser had no effect on the Km values for dT, whereas the C189A, C189S, and C264S mutants had lower Km values for dC than did the wt enzyme. These results suggest that these residues are not in direct contact with dT but may interact with dC. Cys-189 is located at one of the α-helices that form the lid region. Therefore, S-glutathionylation of these residues would mainly have a structural effect on the enzyme, leading to lower catalytic activity.
According to sequence alignment, Cys-189 is conserved in TK2, deoxyguanosine kinase, and deoxycytidine kinase (supplemental Fig. S1), indicating that this cysteine residue in deoxyguanosine kinase and deoxycytidine kinase is also a potential target for redox regulation through S-glutathionylation. Our preliminary data indicate that recombinant deoxyguanosine kinase can be glutathionylated in the presence of GSSG, suggesting that glutathionylation may be a common mechanism in the redox regulation of these deoxynucleoside kinases.
Increased mitochondrial production of ROS and mitochondrial protein oxidation has been implicated in the normal aging process and in the development cancer (28, 34). Long term exposure to therapeutic nucleoside analogs such as AZT can increase cellular oxidative stress in certain cell/tissue types, which is reflected in decreased GSH levels and perturbation of the mitochondrial membrane potential (35). Such oxidative stress caused by nucleoside analogs may affect/modify many mitochondrial proteins including TK2 and thus lead to mitochondrial dysfunction. Our results suggest that TK2 is sensitive to the cellular/mitochondrial redox status. This redox regulation of the TK2 protein may play an important role in mitochondrial DNA replication and maintenance, contributing to mitochondrial dysfunction in chemotherapy with nucleoside analogs and during the normal aging process.
Supplementary Material
Acknowledgment
We thank Dr. Elena Sjuvarsson for help with cell cultures.
This work was supported by a grant from the Swedish Research Council and in part by a grant from the Swedish Research Council for Environment, Agricultural Sciences, and Spatial Planning.

This article contains supplemental Fig. S1.
- TK2
- mitochondrial thymidine kinase 2
- ROS
- reactive oxygen species
- IAM
- iodoacetamide
- diamide
- diazenedicarboxylic acid bis(N,N-dimethylamide)
- COX IV
- cytochrome oxidase subunit IV.
REFERENCES
- 1. Saada A., Shaag A., Mandel H., Nevo Y., Eriksson S., Elpeleg O. (2001) Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nat. Genet. 29, 342–344 [DOI] [PubMed] [Google Scholar]
- 2. Wang L., Saada A., Eriksson S. (2003) Kinetic properties of mutant human thymidine kinase 2 suggest a mechanism for mitochondrial DNA depletion myopathy. J. Biol. Chem. 278, 6963–6968 [DOI] [PubMed] [Google Scholar]
- 3. Oskoui M., Davidzon G., Pascual J., Erazo R., Gurgel-Giannetti J., Krishna S., Bonilla E., De Vivo D. C., Shanske S., DiMauro S. (2006) Clinical spectrum of mitochondrial DNA depletion due to mutations in the thymidine kinase 2 gene. Arch. Neurol. 63, 1122–1126 [DOI] [PubMed] [Google Scholar]
- 4. Götz A., Isohanni P., Pihko H., Paetau A., Herva R., Saarenpää-Heikkilä O., Valanne L., Marjavaara S., Suomalainen A. (2008) Thymidine kinase 2 defects can cause multi-tissue mtDNA depletion syndrome. Brain 131, 2841–2850 [DOI] [PubMed] [Google Scholar]
- 5. Tyynismaa H., Sun R., Ahola-Erkkilä S., Almusa H., Pöyhönen R., Korpela M., Honkaniemi J., Isohanni P., Paetau A., Wang L., Suomalainen A. (2012) Thymidine kinase 2 mutations in autosomal recessive progressive external ophthalmoplegia with multiple mitochondrial DNA deletions. Hum. Mol. Genet. 21, 66–75 [DOI] [PubMed] [Google Scholar]
- 6. Meister A., Anderson M. (1983) Glutathione. Annu. Rev. Biochem. 52, 711–760 [DOI] [PubMed] [Google Scholar]
- 7. Hurd T. R., Costa N. J., Dahm C. C., Beer S. M., Brown S. E., Filipovska A., Murphy M. P. (2005) Glutathionylation of mitochondrial proteins. Antioxid. Redox Signal 7, 999–1010 [DOI] [PubMed] [Google Scholar]
- 8. Hurd T. R., Filipovska A., Costa N. J., Dahm C. C., Murphy M. P. (2005) Disulfide formation on mitochondrial protein thiols. Biochem. Soc. Trans. 33, 1390–1393 [DOI] [PubMed] [Google Scholar]
- 9. Cooper A. J., Pinto J. T., Callery P. S. (2011) Reversible and irreversible protein glutathionylation. Biological and clinical aspects. Expert Opin. Drug Metab. Toxicol. 7, 891–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Garcia J., Han D., Sancheti H., Yap L. P., Kaplowitz N., Cadenas E. (2010) Regulation of mitochondrial glutathione redox status and protein glutathionylation by respiratory substrates. J. Biol. Chem. 285, 39646–39654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yusuf M. A., Chuang T., Bhat G. J., Srivenugopal K. S. (2010) Cys-141 glutathionylation of human p53. Studies using specific polyclonal antibodies in cancer samples and cell lines. Free Radic. Biol. Med. 49, 908–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sun R., Eriksson S., Wang L. (2010) Identification and characterization of mitochondrial factors modulating thymidine kinase 2 activity. Nucleosides Nucleotides Nucleic Acids 29, 382–385 [DOI] [PubMed] [Google Scholar]
- 13. Wang L., Munch-Petersen B., Herrström Sjöberg A., Hellman U., Bergman T., Jörnvall H., Eriksson S. (1999) Human thymidine kinase 2. Molecular cloning and characterization of the enzyme activity with antiviral and cytostatic nucleoside substrates. FEBS Lett. 443, 170–174 [DOI] [PubMed] [Google Scholar]
- 14. Graham J. C. (1999) Current Protocols in Cell Biology, pp. 3.3.1–3.3.15, John Wiley & Sons, Inc., New York [Google Scholar]
- 15. Palacino J. J., Sagi D., Goldberg M. S., Krauss S., Motz C., Wacker M., Klose J., Shen J. (2004) Mitochondrial dysfunction and oxidative damage in parkin-deficient Mice. J. Biol. Chem. 279, 18614–18622 [DOI] [PubMed] [Google Scholar]
- 16. Johansson K., Ramaswamy S., Ljungcrantz C., Knecht W., Piskur J., Munch-Petersen B., Eriksson S., Eklund H. (2001) Structural basis for substrate specificities of cellular deoxyribonucleoside kinases. Nat. Struct. Biol. 8, 616–620 [DOI] [PubMed] [Google Scholar]
- 17. Munch-Petersen B., Cloos L., Tyrsted G., Eriksson S. (1991) Diverging substrate specificity of pure human thymidine kinases 1 and 2 against antiviral dideoxynucleosides. J. Biol. Chem. 266, 9032–9038 [PubMed] [Google Scholar]
- 18. Kil I. S., Park J. W. (2005) Regulation of mitochondrial NADP+-dependent isocitrate dehydrogenase activity by glutathionylation. J. Biol. Chem. 280, 10846–10854 [DOI] [PubMed] [Google Scholar]
- 19. Wang L., Limongelli A., Vila M. R., Carrara F., Zeviani M., Eriksson S. (2005) Molecular insight into mitochondrial DNA depletion syndrome in two patients with novel mutations in the deoxyguanosine kinase and thymidine kinase 2 genes. Mol. Genet. Metab. 84, 75–82 [DOI] [PubMed] [Google Scholar]
- 20. Lesko N., Naess K., Wibom R., Solaroli N., Nennesmo I., von Döbeln U., Karlsson A., Larsson N. G. (2010) Two novel mutations in thymidine kinase 2 cause early onset fatal encephalomyopathy and severe mtDNA depletion. Neuromuscul. Disord. 20, 198–203 [DOI] [PubMed] [Google Scholar]
- 21. Wang L., Sun R., Eriksson S. (2011) The kinetic effects on thymidine kinase 2 by enzyme bound dTTP may explain the mitochondrial side effects of antiviral nucleoside analogs. Antimicrob. Agents Chemother. 55, 2552–2558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mårtensson J., Lai J. C., Meister A. (1990) High affinity transport of glutathione is part of a multicomponent system essential for mitochondrial function. Proc. Natl. Acad. Sci. U.S.A. 87, 7185–7189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jain A., Mårtensson J., Stole E., Auld P. A., Meister A. (1991) Glutathione deficiency leads to mitochondrial damage in brain. Proc. Natl. Acad. Sci. U.S.A. 88, 1913–1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bulteau A. L., Planamente S., Jornea L., Dur A., Lesuisse E., Camadro J. M., Auchère F. (2012) Changes in mitochondrial glutathione levels and protein thiol oxidation in deltayfh1 yeast cells and the lymphoblasts of patients with Friedreich's ataxia. Biochim. Biophys. Acta 1822, 212–225 [DOI] [PubMed] [Google Scholar]
- 25. Ayer A., Tan S. X., Grant C. M., Meyer A. J., Dawes I. W., Perrone G. G. (2010) The critical role of glutathione in maintenance of the mitochondrial genome. Free Radic. Biol. Med. 49, 1956–1968 [DOI] [PubMed] [Google Scholar]
- 26. Fariss M. W., Chan C. B., Patel M., Van Houten B., Orrenius S. (2005) Role of mitochondria in toxic oxidative stress. Mol. Interv. 5, 94–111 [DOI] [PubMed] [Google Scholar]
- 27. Bota D. A., Davies K. (2002) Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nat. Cell Biol. 4, 674–680 [DOI] [PubMed] [Google Scholar]
- 28. Bulteau A. L., Szweda L. I., Friguet B. (2006) Mitochondrial protein oxidation and degradation in response to oxidative stress and aging. Exp. Gerontol. 41, 653–657 [DOI] [PubMed] [Google Scholar]
- 29. Tatsuta T., Langer T. (2008) Quality control of mitochondria. Protection against neurodegeneration and aging. EMBO J. 27, 306–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ugarte N., Petropoulos I., Friguet B. (2010) Oxidized mitochondrial protein degradation and repair in aging and oxidative stress. Antioxid. Redox Signal. 13, 539–549 [DOI] [PubMed] [Google Scholar]
- 31. Baker B. M., Haynes C. M. (2011) Mitochondrial protein quality control during biogenesis and aging. TIBS 36, 254–261 [DOI] [PubMed] [Google Scholar]
- 32. Sabens Liedhegner E. A., Gao X. H., Mieyal J. J. (2012) Mechanism of altered redox regulation in neurodegenerative diseases. Focus on S-glutathionylation. Antioxid. Redox Signal 16, 543–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Anathy V., Roberson E. C., Guala A. S., Godburn K. E., Budd R. C., Janssen-Heininger Y. M. (2012) Redox-based regualtion of apoptosis. S-Glutathionylation as a regulatory mechanism to control cell death. Antioxid. Redox Signal. 16, 496–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Verschoor M. L., Wilson L. A., Singh G. (2010) Mechanisms associated with mitochondrial-generated reactive oxygen species in cancer. Can J. Physiol. Pharmacol. 88, 204–219 [DOI] [PubMed] [Google Scholar]
- 35. Kline E. R., Bassit L., Hernandez-Santiago B. I., Detorio M. A., Liang B., Kleinhenz D. J., Walp E. R., Dikalov S., Jones D. P., Schinazi R. F., Sutliff R. L. (2009) Long term exposure to AZT, but not D4T, increases endothelial cell oxidative stress and mitochondrial dysfunction. Cardiovasc. Toxicol. 9, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.