

Background: The roles of conformational changes of actin in myosin motility are unclear.
Results: A G146V mutation in actin, which perturbed its conformation, impaired force generation by myosin II, but not by myosin V.
Conclusion: Conformational changes of actin involving Gly-146 have critical roles in motility of myosin II, but not of myosin V.
Significance: The mechanism of motility may be different between myosin types.
Keywords: Actin, Molecular motors, Muscle, Mutant, Myosin
Abstract
The G146V mutation in actin is dominant lethal in yeast. G146V actin filaments bind cofilin only minimally, presumably because cofilin binding requires the large and small actin domains to twist with respect to one another around the hinge region containing Gly-146, and the mutation inhibits that twisting motion. A number of studies have suggested that force generation by myosin also requires actin filaments to undergo conformational changes. This prompted us to examine the effects of the G146V mutation on myosin motility. When compared with wild-type actin filaments, G146V filaments showed a 78% slower gliding velocity and a 70% smaller stall force on surfaces coated with skeletal heavy meromyosin. In contrast, the G146V mutation had no effect on either gliding velocity or stall force on myosin V surfaces. Kinetic analyses of actin-myosin binding and ATPase activity indicated that the weaker affinity of actin filaments for myosin heads carrying ADP, as well as reduced actin-activated ATPase activity, are the cause of the diminished motility seen with skeletal myosin. Interestingly, the G146V mutation disrupted cooperative binding of myosin II heads to actin filaments. These data suggest that myosin-induced conformational changes in the actin filaments, presumably around the hinge region, are involved in mediating the motility of skeletal myosin but not myosin V and that the specific structural requirements for the actin subunits, and thus the mechanism of motility, differ among myosin classes.
Introduction
It is well known that actin filaments exhibit structural polymorphism. Electron microscopic and biochemical analyses have shown that the structures of actin subunits within filaments differ depending on the bound nucleotides, interactions with binding proteins, and the age of the filaments. An actin molecule consists of large and small domains, with a deep nucleotide-binding cleft in between (1). The cleft is narrower when ADP and a Pi analog are bound than when only ADP is bound (2). The subunit conformation also changes as time elapses after polymerization. In “young” filaments (i.e. immediately after polymerization), there is a larger population of actin subunits assuming a twisted conformation, in which the large and small domains are rotated with respect to one another, than in “old” filaments (3). Consequently, young filaments are less stable and depolymerize more rapidly than old filaments (4). Interestingly, cofilin binding also causes twisting between the large and small domains (5, 6) as in young filaments, and these conformational changes in the actin subunits have been implicated in the severing activity of cofilin (7). Thus, the conformations of the actin subunits are inseparably connected to the properties of the filaments and to the functions of actin-binding proteins.
Myosin also affects the conformation and flexibility of actin filaments (8–10). Conversely, chemically cross-linking actin filaments (11, 12), which presumably impairs their flexibility and their ability to assume a proper conformation, inhibits their ability to move on surfaces of skeletal heavy meromyosin (sk HMM),4 although they bind to HMM and stimulate its ATPase activity. This suggests that the flexibility of the actin filaments and their capacity for conformational changes are important determinants of myosin motility. In addition, transient phosphorescence anisotropy measurements indicated that the conformational dynamics of actin filaments depend on the myosin class (13). This raises the possibility that the effects of changes in the conformation of actin filaments on myosin motility differ among different classes of myosin.
We previously reported that the G146V mutation in actin inhibits the binding of cofilin, presumably by perturbing the twisting motion between the large and small domains, as Gly-146 is situated within the hinge region between the two domains, and cofilin prefers to bind to the twisted conformation (14). In the present study, we investigated the interaction between actin G146V filaments and sk myosin II (nonprocessive motor) and myosin V (processive motor) in light of the suggested importance of actin filament conformation for myosin motility. We found that the gliding velocity and stall force of sk HMM were significantly reduced by the G146V mutation, presumably reflecting the weaker affinity of the mutant filaments for the myosin heads carrying ADP. In contrast, neither the gliding velocity nor the stall force for myosin V was detectably affected by the mutation. These results suggest that the importance of the actin subunit conformation for motility is myosin class-specific.
EXPERIMENTAL PROCEDURES
Protein Preparations
Recombinant Dictyostelium actin, GFP-fused Dictyostelium HMM (GFP-Dd HMM), endogenous Dictyostelium myosin II, and recombinant human myosin V HMM were purified as described previously (15–18). Skeletal HMM and S1 were prepared by limited proteolysis (19).
In Vitro Motility Assay
Gliding assays on sk HMM or S1 using nitrocellulose-coated coverslips were performed as described previously (20). To remove molecules that bind irreversibly to actin filaments, sk HMM was ultracentrifuged with phalloidin (Ph)-stabilized actin filaments (either wild-type (WT) or mutant) in assay buffer (AB: 10 mm HEPES (pH 7.4), 4 mm MgCl2, 1 mm EGTA, 25 mm KCl, 0.5 mm DTT) containing 1 mm ATP. The supernatants containing sk HMM at a concentration of 0.13 mg/ml or 60 μg/ml S1 were introduced into a flow chamber with a nitrocellulose-coated surface. Motility was initiated by adding ATP solution (AB/BSA containing 1 mm ATP, 10 mm DTT, 200 μg/ml glucose oxidase, 30 μg/ml catalase, and 3 mg/ml glucose). Actin gliding over myosin V HMM immobilized on a nitrocellulose-coated glass surface was observed as described previously (21), using assay buffer 2 (AB2: 25 mm HEPES (pH 7.4), 30 mm KCl, 5 mm MgCl2, 1 mm EGTA) containing an oxygen scavenger system (216 μg/ml glucose oxidase, 36 μg/ml catalase, 4.5 mg/ml glucose), 0.1% (v/v) 2-mercaptoethanol, and 2 mm ATP. The gliding velocity of each actin filament was determined by manually tracking fluorescent spots using the ImageJ software (National Institutes of Health).
Single Molecule Myosin V force Measurement Using Optical Trapping Microscopy
A flow chamber was made using chemically etched glass with 1-μm pedestals (Institute of Microchemical Technology, Kawasaki, Japan) and an 18 × 18-mm coverslip, as described previously (21). Myosin V HMM was fixed to a glass surface coated with α-casein. Actin filaments were labeled with rhodamine phalloidin (RhPh) and biotin-Ph and then introduced into the flow chamber containing AB2, 10 μm ATP, 0.1 mg/ml calmodulin, an oxygen scavenger system, and 1% 2-mercaptoethanol. When an actin filament became tethered between two optically trapped streptavidin-coated microspheres (1 μm φ), it was brought into contact with myosin V HMM fixed on a pedestal. Microsphere displacements were then recorded at a sampling rate of 20 kHz using a quadrant photodiode detector (S994-13, Hamamatsu Photonics, Hamamatsu, Japan). The stall force was calculated by multiplying the microsphere displacement by the trap stiffness (0.01–0.02 pN/nm), which according to the equipartition theorem can be determined from the variance of the Brownian motion of the trapped bead.
Multiple Molecule sk HMM Force Measurement Using Optical Trapping Microscopy
sk HMM was adsorbed onto the etched glass by introducing 0.05 mg/ml sk HMM solution into a flow chamber with the 1-μm pedestals described above. After coating the glass surface with α-casein, labeled actin filaments in AB2, with 2 mm ATP and an oxygen scavenger system, were introduced into the chamber. An actin filament tethered between two optically trapped, streptavidin-coated microspheres was brought into contact with multiple sk HMM molecules fixed on the pedestal. The microspheres were pulled and stalled by sk HMM, and the load exerted on the microspheres under the stalled condition was calculated as described above. All force measurements were performed at 23 °C.
Measurement of Actin-activated S1 MgATPase
The actin-activated MgATPase activities of sk S1 and myosin V HMM were measured using an EnzChek phosphate assay kit (Invitrogen). Various concentrations (0–30 μm) of Ph-stabilized actin filaments and 20 nm sk S1 in myoII ATPase buffer (10 mm HEPES (pH 7.4), 3 mm MgCl2, 0.5 mm EGTA, 2.5 mm KCl, 0.5 mg/ml BSA, 0.5 mm DTT) or 15 nm myosin V HMM in myoV ATPase buffer (10 mm HEPES (pH 7.4), 2 mm MgCl2, 1 mm EGTA, 25 mm KCl, 0.5 mg/ml BSA, 0.2 mm DTT) were incubated with 0.2 mm 2-amino-6-mercapto-7-methylpurine riboside and 1 unit/ml purine nucleoside phosphorylase for 5 min at 23 °C, after which the reaction was started by the addition of 1 mm ATP. The amount of released Pi was monitored continually for 5 min based on the absorbance at 360 nm.
Stopped Flow Analyses
The rates of ATP-induced dissociation of the rigor sk S1-actin filament or sk S1·ADP-actin filament complexes were measured at 26 °C by monitoring light scattering at 340 nm using a stopped flow system (SX18MV, Applied Photophysics, Leatherhead, UK). Rigor sk S1-actin filament complexes were formed by mixing 0.7 μm sk S1 and 0.5 μm Ph-stabilized actin filaments in AB and were dissociated by rapidly adding an equal volume of 20–100 μm ATP solution in AB. sk S1·ADP-actin filament complexes were formed by mixing 1.0 μm sk S1 and 1.4 μm Ph-stabilized actin filaments in AB containing 0.6 mm ADP and then dissociated by rapidly adding an equal volume of AB containing 10 mm ATP.
Co-sedimentation Assays
Ph-stabilized 2.5 μm actin filaments in AB containing 0.4 unit/ml apyrase (grade VII, Sigma) or 2 mm ADP, 0.4 unit/ml hexokinase (Wako, Osaka, Japan) and 10 mm glucose were incubated for 1 h at room temperature to completely remove ATP. Various concentrations of sk S1 (1–6 μm) were then added and ultracentrifuged (100,000 × g, 10 min, 25 °C) after incubation for 10 min at 25 °C. The resultant pellets and supernatants were subjected to SDS-PAGE along with known amounts of sk S1 serving as standards and were stained with Coomassie Blue. The amounts of sk S1 in the bands were determined using the ImageJ software (National Institutes of Health). The dissociation constants (Kd) were determined by fitting the data to the previously described equation (22). To measure the strength of the weak binding between sk S1 and actin filaments, 20 μm Ph-stabilized actin filaments and 0.27 μm sk S1 were mixed in low salt buffer (10 mm Tris-Cl (pH 7.4), 3 mm MgCl2, 0.4 mm EGTA, 1 mg/ml BSA, and 3 mm ATP) and ultracentrifuged immediately. The concentrations of sk S1 in the supernatants were determined by measuring the NH4-CDTA ATPase activity (22) using the malachite green method (23) to measure phosphate.
Observations of Cooperative Binding of Dictyostelium HMM to actin filaments
WT or G146V actin filaments (10 μm) were polymerized in the presence of 50 mm KCl, 25 mm imidazole chlorine (pH 7.4), 4 mm MgCl2, 1 mm DTT, and 0.2 mm ATP at 25 °C for 1 h. The resultant actin filaments were dialyzed against a 5,000-fold excess of magnesium buffer (50 mm KCl, 25 mm imidazole chlorine (pH 7.4), 4 mm MgCl2, 1 mm DTT) and were labeled by RhPh at a molar ratio of 1:80 (RhPh:actin) on ice overnight. Labeled actin filaments (40 nm) were mixed with various concentrations of GFP-fused Dd HMM in magnesium buffer in the presence of 0.05–0.1 μm ATP. After incubation for 10–20 min at 25 °C, an aliquot was placed on a glass slide, overlaid with a coverslip, and observed with a fluorescence microscope (BX60, Olympus) equipped with an EB-CCD camera (C7190, Hamamatsu Photonics) and an image processor (ARGUS-10, Hamamatsu).
RESULTS
Interaction between Gly-146 Mutant Filaments and Skeletal Myosin
We initially assessed the ability of three Gly-146 mutant (G146A, G146T, and G146V) actin filaments to interact with sk HMM. The mutant filaments all labeled with RhPh normally, and their length distributions were similar to that of WT filaments. When subjected to in vitro motility assays, all three mutants formed rigor bonds normally on sk HMM-coated surfaces and moved smoothly like WT filaments over the surfaces after the addition of ATP (Fig. 1A). The gliding velocity of the G146A filaments at 24 °C (1.8 ± 0.29 μm/s) on sk HMM surfaces was slightly slower than that of the WT filaments (2.9 ± 0.40 μm/s), whereas the velocities of the G146T (1.0 ± 0.11 μm/s) and G146V filaments (0.65 ± 0.15 μm/s) were significantly slower (average ± S.D., n > 147 for each). The severity of the inhibitory effects, such that G146A < G146T < G146V, suggests that the bulkiness, rather than hydrophobicity, of the side chain at actin residue position 146 impairs the motility of sk myosin II. The inhibitory effect was also seen over the sk S1 surfaces (WT: 1.29 ± 0.13 μm/s versus G146V: 0.13 ± 0.03 μm/s, n > 60 for each). We also measured the gliding velocities of WT and G146V filaments on surfaces coated with Dictyostelium myosin II. Upon the addition of ATP, G146V filaments detached from the surface without noticeable unidirectional movement, whereas WT filaments moved smoothly under the same conditions (1.2 ± 0.2 μm/s at 30 °C). G146V filaments failed to move continuously on Dictyostelium myosin II surfaces even in the absence of KCl or in the presence of 0.5% methylcellulose.
FIGURE 1.
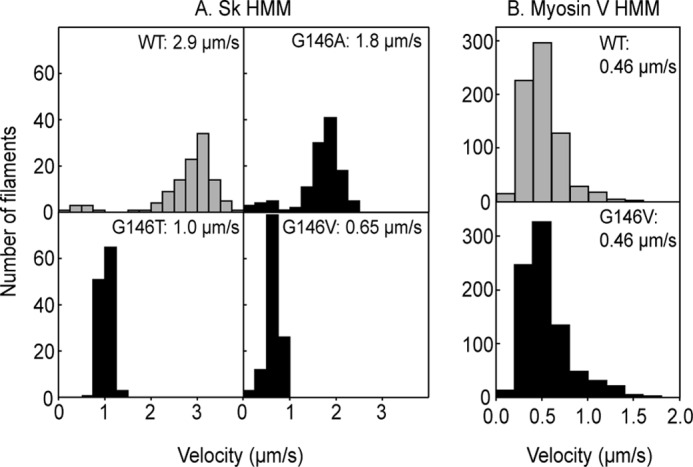
Distributions of gliding velocities of WT and G146 mutant actin filaments on sk HMM surfaces at 24 °C (A) and on myosin V HMM surfaces at 23 °C (B). Histograms indicate the velocity distributions for WT and Gly-146 mutant actin filaments, respectively. The numbers are the averages.
To understand the mechanism underlying the diminished motility of Gly-146 mutant actin filaments on myosin II, we first measured the stall force for single G146V filaments on an sk HMM surface using a dual optical trap nanometry system (Fig. 2A and supplemental Fig. 1; note that this was a multiple motor system). We found that stall force for G146V filaments was significantly smaller than for WT filaments (WT: 0.54 ± 0.30 pN versus G146V: 0.17 ± 0.12 pN; Fig. 2B). In addition, because the duration of the strong binding state is a key determinant of gliding velocity, the rate constants of ATP-induced dissociation of nucleotide-free sk S1 and sk S1·ADP from G146V filaments were measured using a stopped flow system (Fig. 3). Dissociation of the nucleotide-free (rigor) S1-actin filament complex by ATP can be described as follows.
 |
The first step, binding of ATP to the S1-actin filament complex, is in rapid equilibrium, and the association constant is given as K1. The second step is the dissociation of the actin filament from S1 upon the binding of ATP, and the dissociation rate constant is given as k2. Thus, the rate for the ATP-induced dissociation reaction (kobs) can be expressed as K1k2[ATP]/(1 + K1[ATP]) (24). Resolution of K1 and k2 was difficult at high ATP concentrations as the dissociation was very fast. However, we were able to measure the apparent rate constant at low ATP concentrations (Fig. 3A). Under these conditions, the rate (kobs) was approximated by K1k2 [ATP], and the slope of the relation between the rate constant and the ATP concentration gave K1k2, which revealed the K1k2 values for WT filaments (2.2 μm−1s−1) and G146V filaments (2.4 μm−1s−1) to be similar.
FIGURE 2.

Stall force produced by multiple sk HMM molecules or single myosin V HMM molecules. A and C, stall forces were measured on pedestal surfaces coated with sk HMM (A) or with a single myosin V HMM molecule (C) using dual optical traps. B and D, mean stall forces and standard deviations for multiple sk HMM (B) and single myosin V HMM (D) molecules were derived from Gaussian fitting of the distribution of stall forces.
FIGURE 3.
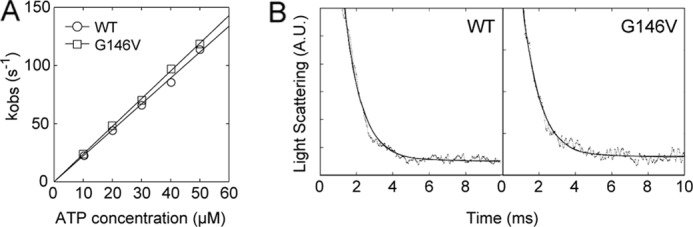
ATP-induced dissociation of sk S1-actin filament complexes and ADP from sk S1·ADP-actin filament complexes. A, dissociation rate for rigor complexes of S1 and actin filaments increased linearly as the ATP concentration was increased from 10 to 50 μm. B, typical data of ATP-induced dissociation of ADP from sk S1·ADP-actin filament complexes. A. U., absorbance units.
The ADP dissociation rate constant from the sk S1·ADP-actin filament complex was estimated by adding a saturating concentration (5 mm) of ATP to sk S1·ADP-actin filament complexes in the presence of 0.3 mm ADP (Fig. 3B). Because the equilibrium constant between sk S1-actin filament complexes and ADP is 120 μm (25), most of the sk S1-actin filament complexes under our experimental conditions initially carried bound ADP. The dissociation rate for G146V filaments (1,010 ± 90 s−1, average ± S.D., n = 5) did not significantly differ from that of WT filaments (1,110 ± 130 s−1). The dissociation rate between sk S1 and actin filaments in the presence of saturating ATP concentration is very fast: 5,000 s−1 (24). Thus, this result suggests that the dissociation rates of ADP from sk S1-actin filaments are similar between WT and G146V filaments.
To estimate the binding affinity of G146V filaments for sk S1, we carried out a set of co-sedimentation assays in the presence or absence of nucleotides (Fig. 4). The dissociation constants (Kd) in AB, which contained 25 mm KCl and no nucleotides, were estimated by fitting the data to the equation described in Ref. 22 (WT: 7.4 ± 5.9 nm versus G146V: 1.3 ± 6.0 nm; Fig. 4A). We believe that the observed difference is not significant as the standard errors are very large when compared with the mean values. In the presence of ADP, the Kd for sk S1 binding to G146V filaments (870 ± 110 nm) was significantly (p < 0.01, Student's t test) higher than for sk S1 binding to WT filaments (340 ± 7.0 nm), as estimated by fitting the data in Fig. 4B. The Kd for sk S1 binding to G146V filaments (27.2 μm, n = 3) was also higher than that of WT filaments (11.0 μm, n = 2) in the presence of ADP and 150 mm NaCl. In the presence of ATP, the affinities of sk S1 for G146V or WT filaments did not greatly differ (Fig. 4C).
FIGURE 4.
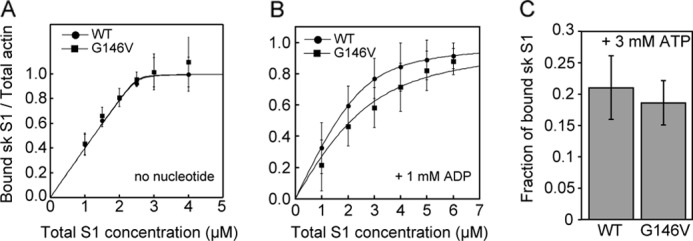
Co-sedimentation in the presence and absence of nucleotides. A and B, binding of sk S1 to actin filaments in the absence of nucleotide (A) and in the presence of 1 mm ADP (B). C, fraction of bound sk S1 in the presence of ATP. Error bars indicate S.D. (n = 3–5).
In the actin-activated sk S1 MgATPase activity measurements (Fig. 5A), the Kapp for G146V filaments was only slightly higher than for WT filaments (WT: 10.4 μm versus G146V: 14.8 μm). This is consistent with the above affinity data in the presence of ATP. By contrast, the Vmax for G146V filaments was reduced to about one-third of that for WT filaments (WT: 9.2 s−1 head−1 versus G146V: 3.4 s−1 head−1).
FIGURE 5.
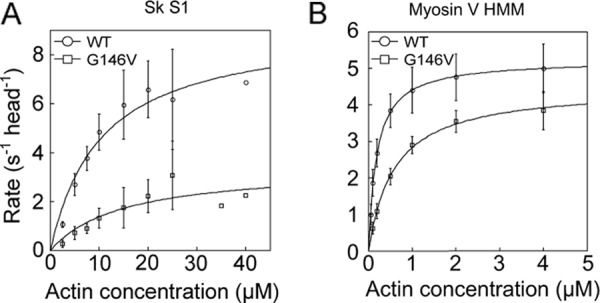
Actin-activated MgATPase activities of sk S1 (A) and myosin V HMM (B). Reactions were performed at 23 °C. Data were fitted to the Michaelis-Menten equation. Error bars indicate S.D. Three to seven measurements were made at each actin concentration, except for 35 and 40 μm actin in the assay using sk S1. At those concentrations, measurements were made only once due to the limited availability of the recombinant actin.
We previously reported that GFP-Dd HMM bound cooperatively to skeletal actin filaments in the presence of 0.1 μm ATP, meaning that some actin filaments were sparsely bound with GFP-Dd HMM, whereas other filaments remained bare (26). We thus examined whether the G146V mutation impairs this cooperative binding between myosin II and actin filaments. In the presence of 0.1 μm ATP and 4 or 8 nm GFP-Dd HMM, none of the WT filaments were initially bound with detectable amounts of GFP-Dd HMM (not shown), but after incubation for 10–20 min, some WT actin filaments were sparsely bound with GFP-Dd HMM, whereas others remained bare (Fig. 6, top). The requirement of an incubation period suggested that the low concentration of ATP must be depleted by the HMM ATPase activity to establish the cooperative binding, presumably in the ADP-bound state. In contrast, cooperative binding of GFP-Dd HMM to G146V filaments was not observed under any of the conditions we tested (Fig. 6, middle and bottom). We suggested that the cooperative binding involves myosin-induced cooperative conformational changes of actin subunits (26). Thus, the G146V mutation in actin appears to disturb conformational changes of actin filaments that are necessary for the cooperative binding of myosin II.
FIGURE 6.
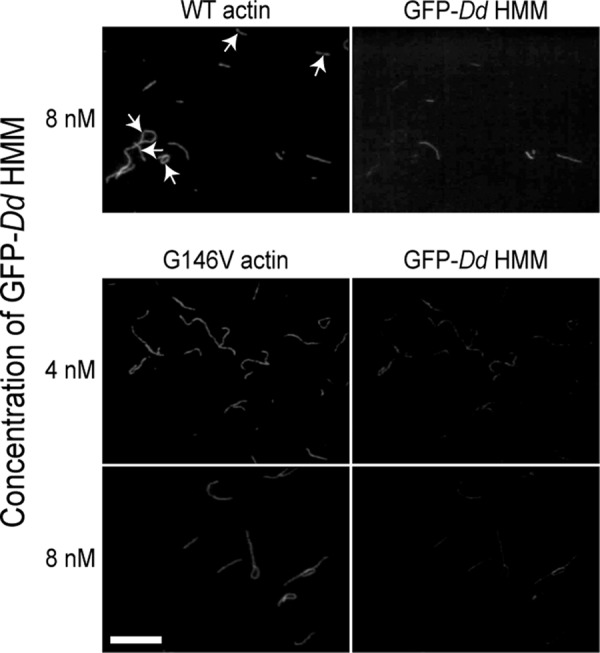
Cooperativity of binding between WT or G146V actin filaments and myosin heads. Each pair of micrographs shows the fluorescence of RhPh-labeled actin filaments (left) and GFP-Dd HMM bound to actin filaments (right). Actin filaments without bound GFP-Dd HMM are marked by arrows. Bar indicates 10 μm.
Interaction between Actin G146V Filaments and Myosin V
For comparison, we also assessed the impact of actin G146V mutation on the motility of a different class of myosin by measuring the gliding velocity, stall force, and actin-activated MgATPase activity of processive, double-headed, truncated myosin V, often referred to as myosin V HMM. Interestingly, there was no detectable difference between the gliding velocities of WT and G146V actin filaments on myosin V HMM surfaces or between the stall forces measured with single myosin V HMM molecules (gliding velocities, WT: 0.46 ± 0.13 μm/s versus G146V: 0.46 ± 0.13 μm/s; Fig. 1B; stall forces, WT: 1.2 ± 0.21 pN versus G146V: 1.1 ± 0.14 pN; Fig. 2D). In ATPase measurements, the Kapp for G146V filaments was reduced when compared with WT filaments, but the Vmax for G146V and WT filaments did not significantly differ (WT: Kapp, 0.19 μm; Vmax, 5.2 s−1 versus G146V: Kapp, 0.59 μm; Vmax, 4.5 s−1; Fig. 5B).
DISCUSSION
Mechanism by which Actin Gly-146 Mutation Impairs Myosin II Motility
We found that a side chain at actin position 146 impairs the motility of myosin II (Fig. 1A). Thus, we performed a series of biochemical and biophysical analyses to understand the molecular mechanism of this inhibition. In a simple model, the gliding velocity of the actin-myosin system is given by the step size divided by the duration of the strongly bound state between actin and myosin heads (27). Thus, the slower motility of the Gly-146 mutant filaments could be due to a longer duration of the strong binding state. However, ATP-induced dissociation rates of the rigor sk S1-actin filaments and ADP dissociation rates from sk S1·ADP-actin filaments were similar between WT and G146V filaments (Fig. 3), suggesting that this is not the case.
Because the in vitro gliding velocity of actin filaments is also affected by the balance between active force and resistive load (28), the slower velocity may be a result of decreased active force. Indeed, the 70% reduction of the force generated by G146V filaments through interaction with sk HMM (Fig. 2B) parallels the 78% reduction in gliding velocity.
To investigate why the actin G146V mutation reduced force generation, we first measured the affinity of sk S1 for G146V filaments in the presence and absence of nucleotides. We found that G146V filaments exhibited significantly less affinity for sk S1 than did WT filaments in the presence of ADP (Fig. 4B). The reduced affinity for sk S1·ADP could contribute to the reduction in force generation by G146V filaments as the myosin·ADP-actin filament is the tension-generating state (27), and strong binding in this state is needed to bear tension or move actin filaments against a resistive load. This is also consistent with our finding that upon the addition of ATP, G146V filaments detached from surfaces coated with Dictyostelium myosin II.
It is also noteworthy that the G146V mutation reduced the Vmax of the actin-activated MgATPase activity of sk S1 to 37% of the WT value (Fig. 5A). ATP hydrolysis and Pi release are the rate-limiting steps in the actin-activated MgATPase cycle of sk S1 (29). Of the two rate-limiting steps, ATP hydrolysis is not known to be affected by actin filaments. We therefore suggest that the G146V mutation slows the Pi release and the accompanying transition from the weak to the strong binding state. This would reduce the fraction of strongly bound, force-generating heads among a given population, which would account for the reduced force generation and gliding velocity in our multiple motor system.
We speculate that both the weaker affinity of the strong binding force-generating S1·ADP-actin filament complexes and the slower transition from the weak to the strong binding state contribute to the observed motility defect, but we presently have no data to suggest the relative contributions of the two mechanisms. On the one hand, the relatively large difference in Vmax between S1 ATPase activities stimulated by WT and G146V actin filaments suggests that the latter mechanism plays a more important role. However, on the other hand, we can say that the reduced affinity of actin filaments for S1·ADP likely shifts the equilibrium between the weak and strong binding states toward the weak binding state, thereby reducing the rate of the weak-to-strong transition, and consequently, the Vmax. In this scenario, the primary defect in the interaction between sk S1 and G146V filaments is the reduced affinity between the actin filaments and S1·ADP.
Structural Basis for the Diminished Force Generation
A simple structural explanation for the reduced affinity between myosin II and G146V actin would be that the bulky side chain at actin position 146 disrupts the strong binding site for myosin as this residue is within the region implicated in hydrophobic interaction with sk S1 (30). Related to this possibility, it is worth mentioning a previous mutational study in which Ile-341, near Gly-146 in the hydrophobic myosin-binding surface of yeast actin, was mutated to Ala, with the aim of evaluating the impact of simple reduction of the hydrophobic affinity in the strong actin-myosin interaction (31). The I341A mutation increased the Kd for sk S1·ADP by 15-fold (22), but decreased the gliding velocity on HMM surfaces by only ∼40% (31). These numbers are considerably disproportionate to the corresponding values of G146V actin (which had a 2-fold increase in Kd for S1·ADP and a 70% decrease in the gliding velocity), suggesting that an additional or alternative mechanism, other than simple disruption of the binding site, should be considered to explain the severely disrupted motility of the G146V actin filaments accompanying a mild decrease in affinity for S1·ADP.
Monomeric actin molecules assume a more flattened configuration after incorporation into filaments (32–34). However, recent detailed electron microscopic observations of a population of filaments revealed that there is a tilted state of the subunits in which the large and small domains are twisted relative to one another (6, 35). Evidence also suggests that the motion of the small domain is involved in myosin function. For example, skeletal and smooth muscle myosins in a strong binding state (rigor or ADP-bound state) affect the structure of subdomain 2 within the small domain (36). Cross-linking Glu-41 in subdomain 2 to Cys-374 within the small domain of the adjacent subunit diminishes flexibility of the filaments (37) and is accompanied by a motility defect with skeletal myosin (12). We previously suggested that the G146V mutation inhibits cofilin binding by hindering the twisting motion of the small domain relative to the large domain (14). Similarly, we suggest that the diminished force generated by myosin II with G146V filaments is due to perturbation of the motion of the small domain.
Myosin has been shown to induce cooperative conformational changes within actin filaments (10, 38), including cooperative restriction of the mobility of the actin subunits by strongly bound myosin heads (22). In addition, myosin II heads bind to actin filaments in a cooperative manner (27, 39), indicating that cooperative conformational changes induced by the binding of the first myosin head to one actin subunit accelerate binding of other myosin heads to neighboring actin subunits. In contrast, G146V filaments, with defective motility with myosin II, failed to bind Dd HMM cooperatively (Fig. 6). We therefore speculate that the myosin-induced, Gly-146-dependent cooperative conformational changes in the actin filaments facilitate motility with myosin II by increasing the overall affinity for S1·ADP and/or accelerating the transition from the weak to the strong binding state. Detailed structural analyses of G146V filaments are required to test this hypothesis.
G146V Mutation Differentially Affects Myosin II and V
Actin G146V mutation had no detectable effect on force generation, gliding velocity, or Vmax of actin-induced MgATPase activity with myosin V, which is in sharp contrast to its effects on myosin II (Figs. 1, 2, and 5). There are two possible explanations for this difference in the inhibitory effects of actin G146V mutation. One is that because the rate-limiting step in the myosin V ATPase cycle is ADP release (40), the slower transition from weak to strong binding may not be deleterious for this myosin. Consistent with this possibility, Kubota et al. (41) reported that the M47A/E360H mutation of Dictyostelium actin slows down the transition of the leading head from the weak to the strong binding state, but it does not reduce gliding velocity. The second possibility is that the conformational states of the actin subunits required for myosin V motility differ from those required for myosin II motility. Supporting this possibility, transient phosphorescence anisotropy measurements indicated that the conformational dynamics of actin filaments interacting with myosin II differ from those of filaments interacting with myosin V (13). This raises the possibility that the structural requirements of the actin subunits for motility of different classes of myosin may be qualitatively different, particularly between fast, nonprocessive classes and slow, processive classes. In this scenario, the contributions of actin filaments to the motilities of myosin II and V are not identical, and the conformational changes involving the large and small domains of the actin subunits required for specific aspects of myosin motility likely differ.
Supplementary Material
Acknowledgment
We thank Dr. K. Sutoh of the University of Tokyo (present address: Waseda University) for the use of a stopped flow apparatus.
This work was supported in part by Grants-in-aid for scientific research from the Ministry of Science, Culture, Sports and Technology (to T. Q. P. U. and T. Q. P. N.).

This article contains supplemental Fig. 1.
- sk HMM
- skeletal heavy meromyosin
- sk S1
- skeletal S1 protein
- GFP-Dd HMM
- GFP-fused Dictyostelium HMM
- Ph
- phalloidin
- pN
- piconewtons
- RhPh
- rhodamine phalloidin
- AB
- assay buffer
- CDTA
- 1,2-cyclohexylenedinitrilotetraacetic acid.
REFERENCES
- 1. Kabsch W., Mannherz H. G., Suck D., Pai E. F., Holmes K. C. (1990) Atomic structure of the actin:DNase I complex. Nature 347, 37–44 [DOI] [PubMed] [Google Scholar]
- 2. Belmont L. D., Orlova A., Drubin D. G., Egelman E. H. (1999) A change in actin conformation associated with filament instability after Pi release. Proc. Natl. Acad. Sci. U.S.A. 96, 29–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Orlova A., Shvetsov A., Galkin V. E., Kudryashov D. S., Rubenstein P. A., Egelman E. H., Reisler E. (2004) Actin-destabilizing factors disrupt filaments by means of a time reversal of polymerization. Proc. Natl. Acad. Sci. U.S.A. 101, 17664–17668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kueh H. Y., Brieher W. M., Mitchison T. J. (2008) Dynamic stabilization of actin filaments. Proc. Natl. Acad. Sci. U.S.A. 105, 16531–16536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Galkin V. E., VanLoock M. S., Orlova A., Egelman E. H. (2002) A new internal mode in F-actin helps explain the remarkable evolutionary conservation of actin's sequence and structure. Curr. Biol. 12, 570–575 [DOI] [PubMed] [Google Scholar]
- 6. Galkin V. E., Orlova A., Kudryashov D. S., Solodukhin A., Reisler E., Schröder G. F., Egelman E. H. (2011) Remodeling of actin filaments by ADF/cofilin proteins. Proc. Natl. Acad. Sci. U.S.A. 108, 20568–20572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Galkin V. E., Orlova A., Lukoyanova N., Wriggers W., Egelman E. H. (2001) Actin depolymerizing factor stabilizes an existing state of F-actin and can change the tilt of F-actin subunits. J. Cell Biol. 153, 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yanagida T., Nakase M., Nishiyama K., Oosawa F. (1984) Direct observation of motion of single F-actin filaments in the presence of myosin. Nature 307, 58–60 [DOI] [PubMed] [Google Scholar]
- 9. Kozuka J., Yokota H., Arai Y., Ishii Y., Yanagida T. (2006) Dynamic polymorphism of single actin molecules in the actin filament. Nat. Chem. Biol. 2, 83–86 [DOI] [PubMed] [Google Scholar]
- 10. Oosawa F., Fujime S., Ishiwata S., Mihashi K. (1972) Dynamic property of F-actin and thin filament. Cold Spring Harb. Symp. Quant. Biol. 37, 277–285 [Google Scholar]
- 11. Prochniewicz E., Yanagida T. (1990) Inhibition of sliding movement of F-actin by cross-linking emphasizes the role of actin structure in the mechanism of motility. J. Mol. Biol. 216, 761–772 [DOI] [PubMed] [Google Scholar]
- 12. Kim E., Bobkova E., Hegyi G., Muhlrad A., Reisler E. (2002) Actin cross-linking and inhibition of the actomyosin motor. Biochemistry 41, 86–93 [DOI] [PubMed] [Google Scholar]
- 13. Prochniewicz E., Chin H. F., Henn A., Hannemann D. E., Olivares A. O., Thomas D. D., De La Cruz E. M. (2010) Myosin isoform determines the conformational dynamics and cooperativity of actin filaments in the strongly bound actomyosin complex. J. Mol. Biol. 396, 501–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Noguchi T. Q., Toya R., Ueno H., Tokuraku K., Uyeda T. Q. (2010) Screening of novel dominant negative mutant actins using glycine-targeted scanning identifies G146V actin that cooperatively inhibits cofilin binding. Biochem. Biophys. Res. Commun. 396, 1006–1011 [DOI] [PubMed] [Google Scholar]
- 15. Noguchi T. Q., Kanzaki N., Ueno H., Hirose K., Uyeda T. Q. (2007) A novel system for expressing toxic actin mutants in Dictyostelium and purification and characterization of a dominant lethal yeast actin mutant. J. Biol. Chem. 282, 27721–27727 [DOI] [PubMed] [Google Scholar]
- 16. Hachikubo Y., Ito K., Yamamoto K. (2003) Roles of the hydrophobic triplet in the motor domain of myosin in the interaction between myosin and actin. J. Biochem. 134, 165–171 [DOI] [PubMed] [Google Scholar]
- 17. Okada T., Tanaka H., Iwane A. H., Kitamura K., Ikebe M., Yanagida T. (2007) The diffusive search mechanism of processive myosin class-V motor involves directional steps along actin subunits. Biochem. Biophys. Res. Commun. 354, 379–384 [DOI] [PubMed] [Google Scholar]
- 18. Uyeda T. Q., Tokuraku K., Kaseda K., Webb M. R., Patterson B. (2002) Evidence for a novel, strongly bound acto-S1 complex carrying ADP and phosphate stabilized in the G680V mutant of Dictyostelium myosin II. Biochemistry 41, 9525–9534 [DOI] [PubMed] [Google Scholar]
- 19. Margossian S. S., Lowey S. (1982) Preparation of myosin and its subfragments from rabbit skeletal muscle. Methods Enzymol. 85, 55–71 [DOI] [PubMed] [Google Scholar]
- 20. Uyeda T. Q., Kron S. J., Spudich J. A. (1990) Myosin step size: estimation from slow sliding movement of actin over low densities of heavy meromyosin. J. Mol. Biol. 214, 699–710 [DOI] [PubMed] [Google Scholar]
- 21. Komori T., Nishikawa S., Ariga T., Iwane A. H., Yanagida T. (2009) Simultaneous measurement of nucleotide occupancy and mechanical displacement in myosin V, a processive molecular motor. Biophys. J. 96, L04–L06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Prochniewicz E., Thomas D. D. (2001) Site-specific mutations in the myosin-binding sites of actin affect structural transitions that control myosin binding. Biochemistry 40, 13933–13940 [DOI] [PubMed] [Google Scholar]
- 23. Kodama T., Fukui K., Kometani K. (1986) The initial phosphate burst in ATP hydrolysis by myosin and subfragment-1 as studied by a modified malachite green method for determination of inorganic phosphate. J. Biochem. 99, 1465–1472 [DOI] [PubMed] [Google Scholar]
- 24. Millar N. C., Geeves M. A. (1983) The limiting rate of the ATP-mediated dissociation of actin from rabbit skeletal muscle myosin subfragment 1. FEBS Lett. 160, 141–148 [DOI] [PubMed] [Google Scholar]
- 25. Ritchie M. D., Geeves M. A., Woodward S. K., Manstein D. J. (1993) Kinetic characterization of a cytoplasmic myosin motor domain expressed in Dictyostelium discoideum. Proc. Natl. Acad. Sci. U.S.A. 90, 8619–8623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tokuraku K., Kurogi R., Toya R., Uyeda T. Q. (2009) Novel mode of cooperative binding between myosin and Mg2+-actin filaments in the presence of low concentrations of ATP. J. Mol. Biol. 386, 149–162 [DOI] [PubMed] [Google Scholar]
- 27. Spudich J. A. (1994) How molecular motors work. Nature 372, 515–518 [DOI] [PubMed] [Google Scholar]
- 28. Warshaw D. M., Desrosiers J. M., Work S. S., Trybus K. M. (1990) Smooth muscle myosin cross-bridge interactions modulate actin filament sliding velocity in vitro. J. Cell Biol. 111, 453–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. White H. D., Belknap B., Webb M. R. (1997) Kinetics of nucleoside triphosphate cleavage and phosphate release steps by associated rabbit skeletal actomyosin, measured using a novel fluorescent probe for phosphate. Biochemistry 36, 11828–11836 [DOI] [PubMed] [Google Scholar]
- 30. Rayment I., Holden H. M., Whittaker M., Yohn C. B., Lorenz M., Holmes K. C., Milligan R. A. (1993) Structure of the actin-myosin complex and its implications for muscle contraction. Science 261, 58–65 [DOI] [PubMed] [Google Scholar]
- 31. Miller C. J., Doyle T. C., Bobkova E., Botstein D., Reisler E. (1996) Mutational analysis of the role of hydrophobic residues in the 338–348 helix on actin in actomyosin interactions. Biochemistry 35, 3670–3676 [DOI] [PubMed] [Google Scholar]
- 32. Oda T., Iwasa M., Aihara T., Maéda Y., Narita A. (2009) The nature of the globular- to fibrous-actin transition. Nature 457, 441–445 [DOI] [PubMed] [Google Scholar]
- 33. Fujii T., Iwane A. H., Yanagida T., Namba K. (2010) Direct visualization of secondary structures of F-actin by electron cryomicroscopy. Nature 467, 724–728 [DOI] [PubMed] [Google Scholar]
- 34. Murakami K., Yasunaga T., Noguchi T. Q., Gomibuchi Y., Ngo K. X., Uyeda T. Q., Wakabayashi T. (2010) Structural basis for actin assembly, activation of ATP hydrolysis, and delayed phosphate release. Cell 143, 275–287 [DOI] [PubMed] [Google Scholar]
- 35. Galkin V. E., Orlova A., Schröder G. F., Egelman E. H. (2010) Structural polymorphism in F-actin. Nat. Struct. Mol. Biol. 17, 1318–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Volkmann N., Ouyang G., Trybus K. M., DeRosier D. J., Lowey S., Hanein D. (2003) Myosin isoforms show unique conformations in the actin-bound state. Proc. Natl. Acad. Sci. U.S.A. 100, 3227–3232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Orlova A., Galkin V. E., VanLoock M. S., Kim E., Shvetsov A., Reisler E., Egelman E. H. (2001) Probing the structure of F-actin: cross-links constrain atomic models and modify actin dynamics. J. Mol. Biol. 312, 95–106 [DOI] [PubMed] [Google Scholar]
- 38. Miki M., Wahl P., Auchet J. C. (1982) Fluorescence anisotropy of labeled F-actin: influence of divalent cations on the interaction between F-actin and myosin heads. Biochemistry 21, 3661–3665 [DOI] [PubMed] [Google Scholar]
- 39. Orlova A., Egelman E. H. (1997) Cooperative rigor binding of myosin to actin is a function of F-actin structure. J. Mol. Biol. 265, 469–474 [DOI] [PubMed] [Google Scholar]
- 40. De La Cruz E. M., Wells A. L., Rosenfeld S. S., Ostap E. M., Sweeney H. L. (1999) The kinetic mechanism of myosin V. Proc. Natl. Acad. Sci. U.S.A. 96, 13726–13731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kubota H., Mikhailenko S. V., Okabe H., Taguchi H., Ishiwata S. (2009) D-loop of actin differently regulates the motor function of myosins II and V. J. Biol. Chem. 284, 35251–35258 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.