

Background: Few effective treatments exist for human respiratory syncytial virus infection. The absence of small hydrophobic (SH) protein in RSV leads to viral attenuation.
Results: SH protein forms pentamers and shows pH-dependent ion channel activity.
Conclusion: SH protein forms pentameric ion channels.
Significance: The SH protein and its channel activity constitute a potential drug target.
Keywords: Analytical Ultracentrifugation, Biophysics, Ion Channels, Nuclear Magnetic Resonance, Viral Protein, ATR-FTIR, Small Hydrophobic (SH) Protein, Human Respiratory Syncytial Virus (hRSV), Viroporin
Abstract
The small hydrophobic (SH) protein is encoded by the human respiratory syncytial virus. Its absence leads to viral attenuation in the context of whole organisms, and it prevents apoptosis in infected cells. Herein, we have examined the structure of SH protein in detergent micelles and in lipid bilayers, by solution NMR and attenuated total reflection-Fourier transform infrared spectroscopy, respectively. We found that SH protein has a single α-helical transmembrane domain and forms homopentamers in several detergents. In detergent micelles, the transmembrane domain is flanked N-terminally by an α-helix that forms a ring around the lumen of the pore and C-terminally by an extended β-turn. SH protein was found in the plasma membrane of transiently expressing HEK 293 cells, which showed pH-dependent (acid-activated) channel activity. Channel activity was abolished in mutants lacking both native His residues, His22 and His51, but not when either His was present. Herein, we propose that the pentameric model of SH protein presented is a physiologically relevant conformation, albeit probably not the only one, in which SH contributes to RSV infection and replication. Viroporins are short (∼100 amino acids) viral membrane proteins that form oligomers of a defined size, act as proton or ion channels, and in general enhance membrane permeability in the host. However, with some exceptions, their precise biological role of their channel activity is not understood. In general, viroporins resemble poorly specialized proteins but are nevertheless critical for viral fitness. In vivo, viruses lacking viroporins usually exhibit an attenuated or weakened phenotype, altered tropism, and diminished pathological effects. We have chosen to study the SH protein, 64 amino acids long, found in the human respiratory syncytial virus because of the effect of RSV on human health and the lack of adequate antivirals. We show that SH protein forms oligomers that behave as ion channels when activated at low pH. This study adds SH protein to a growing group of viroporins that have been structurally characterized. Although the precise biological role of this pentameric channel is still unknown, this report is nevertheless essential to fill some of the many gaps that exist in the understanding of SH protein function.
Introduction
The SH4 protein is found in human respiratory syncytial virus (hRSV), an enveloped pneumovirus in the paramyxoviridae family that causes lower respiratory tract disease in infants, the elderly, and immunocompromised populations worldwide (1). Up to 64 million reported cases of hRSV infection and 160,000 deaths occur each year. Although the virus was identified almost half a century ago (2), there are still no vaccines or effective antiviral drugs available. hRSV can cause repeated reinfections throughout life, and little is known about its molecular mechanism of pathogenesis.
The hRSV genome composes a nonsegmented negative-stranded RNA of ∼15 kb that transcribes 11 proteins, including the three membrane proteins F, G, and SH. Proteins F and G are key factors during virus entry, attachment, and fusion (3, 4). In contrast, the role of SH protein is less clear. RSV that lacks SH (RSVΔSH) is still viable and still forms syncytia (5–7). However, RSVΔSH was attenuated in in vivo mouse and chimpanzee models (6, 9), which indicates that SH protein is important for RSV pathogenesis.
The SH protein is a 64- (RSV subgroup A) or 65 (RSV subgroup B)-amino acid-long type II integral membrane protein, with the C terminus oriented extracellularly/lumenally in the host and with a single predicted α-helical transmembrane (TM) domain (10). The sequence of SH protein is highly conserved, especially at the TM domain (11, 12). In infected cells, most SH protein accumulates at the membranes of the Golgi complex, but it is also found in the endoplasmic reticulum and plasma membranes (13), which enables patch clamp studies. During infection, SH protein has been shown to exist in several forms, e.g. full-length truncated protein (4.5 kDa), and post-translationally modified by glycosylation and phosphorylation (14, 15), although the full-length unmodified form is the major species (10).
SH protein permeabilizes membranes (16, 17). Its synthetic TM domain (residues 18–43) shows channel activity in synthetic bilayers (18) and forms homopentamers in perfluorooctanoic acid (PFO) gel electrophoresis. In that work, using the technique of linear infrared dichroism (19, 20) and conformational searching methods (21, 22), we provided a low resolution model of the TM homopentamer responsible for channel activity. More recently, an electron microscopy study of an 86-amino acid-long polypeptide containing the 64 amino acids of SH protein visualized in a membrane-mimetic environment (17) identified ring-like features interpreted as having either 5- or 6-fold symmetry.
The use of full-length, tag-free SH protein is essential to resolve possible ambiguities in structure and function. In our hands, synthetic SH protein could not be purified satisfactorily, and native ligation of synthetic peptides produced very low yield. Similarly, SH protein alone or with a His tag could not be expressed in Escherichia coli, presumably due to toxicity. Thus, in this work, full-length SH protein was obtained using maltose-binding protein (MBP) as an N-terminal fusion protein, which in turn has an N-terminal His tag for easier purification. The construct His6-MBP-SH improved expression significantly. Between MBP and SH protein, a tobacco etch virus (TEV) cleavage site was introduced. Once SH protein was cleaved from the construct, it contained only three extra residues (SNA) at the N terminus, which do not interfere with the biophysical properties of the protein. SH protein was characterized by ATR-FTIR when reconstituted into model lipid bilayers and in parallel by solution NMR after isotopic labeling, using detergent micelles. The latter technique allowed reconstruction of the pentameric α-helical bundle that includes the extramembrane regions. We have also investigated the channel activity of the full-length SH protein and some of its mutants, especially at the protonatable His residues, using the whole-cell patch clamp technique.
MATERIALS AND METHODS
Cloning of SH Gene
The nucleotide sequence of SH protein was obtained from NCBI (NC_001803.1), strain S2 ts1C. The SH gene, accession number NP_044594.1 (23), was synthesized, and the purity was determined by agarose gel electrophoresis. The SH gene was cloned into pTBMalE vector (24) with MBP as fusion partner carrying a His tag at the N terminus to produce the construct His-MBP-SH. Mutants were constructed in the same way.
Expression of the His-MBP-SH Construct
The DNA plasmid containing the SH gene was transformed into an E. coli competent cell strain BL-21 (DE3) CodonPlus-RIL strain (Stratagene) for protein overexpression. Cells from a single colony were picked to inoculate 10 ml of LB media with 100 μg/ml ampicillin and 34 μg/ml chloramphenicol and grown overnight at 37 °C with shaking. 8 ml of the overnight culture was then transferred to 800 ml of LB media and grown at 37 °C with shaking to an A600 of 0.6–0.7. For unlabeled samples, cells were induced with 0.4 mm isopropyl β-thiogalactoside and grown at 23 °C overnight with shaking.
For isotopically labeled samples, cells were harvested when A600 0.6–0.7 was reached by centrifugation and washed with M9 minimal media once. The cells were transferred to M9 minimal media containing [15N]ammonium chloride or [13C]glucose. Expression levels in minimal media were enhanced by increasing cell densities through a 4:1 concentrating method (25). The concentrated culture in M9 media was induced with 0.4 mm isopropyl β-thiogalactoside and grown overnight at 23 °C with shaking.
After induction, cells were harvested and resuspended in Ni2+-NTA binding buffer containing 20 mm Tris-HCl, 500 mm NaCl, and 5 mm imidazole, pH 8.0, and then kept frozen at −20 °C overnight. Thawed cells were incubated with 0.2 mg/ml lysozyme and 0.02 mg/ml benzonase for 10 min. Then, octyl-β-d-glucopyranoside was added to the sample to a final concentration of 50 mm. The cells were lysed with a microfluidizer at 15,000 p.s.i. pressure, and the supernatant was collected after centrifugation at 20,000 × g for 30 min and loaded to Econo-column (Bio-Rad) packed with Ni2+-NTA-agarose resin (Qiagen) pre-equilibrated with binding buffer. The proteins were allowed to bind to the nickel resin with gentle shaking at 4 °C overnight.
Purification of His-MBP-SH Construct by Ni2+-NTA Chromatography
The nickel resin with bound protein was washed with 20 column volumes of buffer containing 20 mm Tris-HCl, 500 mm NaCl, and 20 mm imidazole, pH 8.0. Bound proteins were eluted with elution buffer containing 20 mm Tris-HCl, 500 mm NaCl, 500 mm imidazole, pH 8.0, and 30 mm octyl-β-d-glucopyranoside. All fractions collected were stored at 4 °C, whereas fractions containing the His-MBP-SH fusion construct were analyzed by SDS-PAGE.
Expression and Purification of TEV Protease
Expression and purification of TEV protease were done in a similar way for SH protein except that no octyl-β-d-glucopyranoside was added to the sample, as it is a soluble protein. TEV was stored at −20 °C in buffer containing 50% glycerol, 10 mm Tris-HCl, 250 mm NaCl, 500 mm imidazole, 1 mm EDTA, and 5 mm DTT, pH 8.0.
Cleaving of His-MBP-SH Construct by TEV Protease
TEV protease was added to the His-MBP-SH construct at a mass ratio of 1:5 (TEV/fusion protein, mg/ml). The digestion was performed at room temperature with gentle shaking, and the progress of the reaction was monitored by SDS-PAGE. Usually, the reaction was completed in 4–6 h. Digestion was stopped by addition of trichloroacetic acid (TCA) to a final concentration of 6% (v/v)/ and the precipitate was collected by centrifugation at 18,000 × g for 30 min. The pellet was washed twice with water followed by lyophilization.
Purification of SH Protein by Organic Solvent Extraction and RP-HPLC
The cleaved SH protein was separated from the His-MBP fragment using a two-step purification strategy. First, SH protein was extracted by methanol by adding 10 ml of methanol per 1 liter of culture and mixing gently for 2 h at room temperature. The supernatant was collected by centrifugation at 18,000 × g for 30 min and checked by MALDI spectroscopy and SDS-PAGE. SH protein was further purified by injecting the supernatant onto a Zorbax C3–300 Å column connected to an HPLC system. Purified SH protein was eluted with a linear gradient of solvent A (water/TFA, 99.9:0.1, v/v) and solvent D (isopropyl alcohol/acetonitrile/TFA, 80:19.9:0.1, v/v). Pooled fractions were lyophilized, and the purity of the samples was checked by MALDI.
Gel Electrophoresis
Reduced SDS-PAGE or PFO-PAGE using precast Tricine gradient gel (Bio-Rad), with or without heat treatment, was used to test the purity of the preparations. The gel used in Fig. 1C was an in-house 12.5% gel that was run using Tris-glycine buffer, ∼pH 9, a typical Laemmli system. The gel used in Fig. 2A was a commercially available pre-cast gel from Invitrogen, and the NuPAGE® SDS-PAGE was prepared with BisTris buffer at near neutral pH and run using MES running buffer, ∼pH 7.0, to minimize protein modification at extreme pH. SDS or PFO sample buffer, for SDS or PFO-PAGE, respectively, was added to the lyophilized protein to a final protein concentration of 2 μg/μl. After mixing for 1 min followed by heating at 95 °C for 5 min, the sample was loaded onto the gel. Electrophoreses were run at constant voltage of 80 V for 3 h at room temperature, and gels were stained with Coomassie Blue dye.
FIGURE 1.

Overexpression of the His-MBP-SH construct in E. coli and purification of SH protein. A, MW, molecular weight markers; BI, before induction; AI, after induction. The arrow shows the band corresponding to the His-MBP-SH fusion protein migrating at ∼50 kDa. B, distribution of His-MBP-SH fusion protein in cell fractions: SUP, lysate supernatant; CYT, cytoplasmic fraction; MEM, membrane fraction; INC, inclusion body fraction. C, electrophoretic mobility of the His-MBP-SH preparation at room temperature before (0 h) and 4 h after addition of TEV. The arrowhead at 0 h shows the band corresponding to the fusion construct His-MBP-SH, whereas the arrowhead at 4 h shows the band corresponding to cleaved SH protein. The position of the cleaved His-MBP fragment (labeled MBP) and the TEV protease are also shown in the 4-h experiment. D, RP-HPLC showing the peak corresponding to SH protein (arrow). E, MALDI mass spectrum of the HPLC fraction corresponding to SH protein (D, arrow). F, sequence of the final purified SH protein; TEV cleavage resulted in additional N-terminal three residues, SNA.
FIGURE 2.
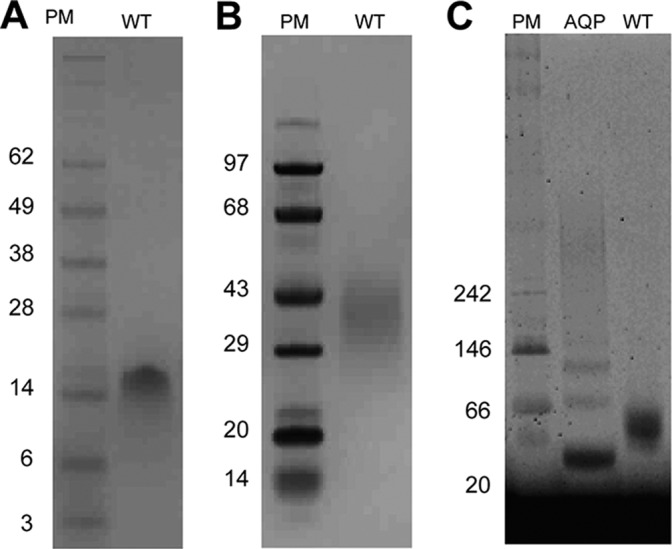
Gel electrophoresis analysis of SH protein. Wild type (WT) protein (Fig. 1F) was analyzed by SDS-PAGE (A), PFO-PAGE (B), and BN-PAGE (in C14 betaine) (C). A and B, PM, protein markers; WT, wild type (expected molecular weight of 7808); C, BN-PAGE analysis; AQP, aquaporin Z from E. coli.
Analytical Ultracentrifugation
AUC ES experiments were performed in a Beckman XL-1 analytical ultracentrifuge (Beckman Coulter) using a six-channel carbon-epoxy composite centerpiece equipped with quartz windows for the absorbance experiments or sapphire windows for interference experiments. All experiments were performed at 25 °C on samples at three different initial SH protein concentrations (0.3, 0.5, and 0.8 absorbance at 280 nm) solubilized in 5 mm C14 betaine (Sigma) and three different rotor speeds (16,000, 19,500, and 24,000 rpm). The software program Winmatch was used to monitor whether the equilibrium had been reached at each speed, every 3–4 h. Usually, the samples achieved equilibrium in about 20 h. For DPC or C8E5, the detergent concentration was 15 and 33 mm, respectively.
SH protein in methanol was dried under a stream of nitrogen gas and lyophilized for at least 3 h to remove any residual organic solvent. 5 mm C14 betaine in buffer containing 50 mm Tris-HCl, 100 mm NaCl, 29.4% D2O, pH 7.3, was added to the dried protein samples and mixed by 5 min of vortexing. The sample was centrifuged at 15,000 rpm for 5 min before loading into the centerpiece. 110 μl of sample and 120 μl of reference buffer were loaded into the six-channel centerpiece. Data obtained by UV absorption at 280 nm were analyzed with Sedphat by nonlinear least squares curve-fitting of radial concentration profiles using the Levenberg-Marquardt algorithm. Different reversible association models were tried to fit the data to obtain the most suitable model. The monomeric mass, extinction coefficient at 280 nm, and partial specific volumes for each sample were calculated using the program SEDNTERP.
Peptide Synthesis
Peptides corresponding to the TM of SH protein, SH(18–43) and SH(45–64), were investigated by FTIR. Peptide SH(18–43) and two fragments to be used in a native ligation procedure (1–44, as a C-terminal thioester, and 45–64, as an N-terminal cysteine) were obtained by solid-phase synthesis and purified as described previously (18). However, native ligation was not successful due to aggregation of the N-terminal peptide SH(1–44) (data not shown).
Infrared Spectroscopy
FTIR spectra were recorded on a Nicolet Nexus 560 spectrometer (Madison, WI) purged with N2 and equipped with an MCT/A detector cooled with liquid nitrogen. Attenuated total reflection (ATR) spectra were measured with a 25-reflection ATR accessory from Graseby Specac (Kent, UK) and a wire grid polarizer (0.25 micrometers, Graseby Specac). Approximately 100 μl of sample in water with 50:1 lipid/peptide molar ratio were applied onto a trapezoidal (50 × 2 × 20 mm) germanium internal reflection element. The lipids used here were DMPC and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (Avanti Polar Lipids). A dry or D2O-saturated N2 stream flowing through the ATR compartment was used to remove bulk water or to achieve D2O exchange, respectively. After insertion of the plate in the ATR cell, spectra were collected. A total of 200 interferograms collected at a resolution of 4 cm−1 were averaged for every sample and processed with one-point zero filling and Happ-Genzel apodization. The area of amide I (C=O stretching) was obtained by peak integration from 1600 to 1700 cm−1, and the area of amide II (N–H bending, centered at ∼1550 cm−1) was obtained by peak integration from 1510 to 1580 cm−1. No difference in band area was observed employing other means of peak size estimation such as peak fitting and Fourier self-deconvolution. For the hydrogen-deuterium exchange experiment, the kinetics of exchange was calculated by measuring the relative area of amide II relative to the amide I band (26).
Electrophysiological Recordings and Data Analysis
HEK 293 cells were transfected with a plasmid containing the SH protein gene. Expression of wild type and mutants of the full-length SH gene was confirmed using green fluorescent protein (GFP) (27). The full-length SH gene was cloned into the pIRES-AcGFP1 (Aequorea coerulescens green fluorescent protein) vector (Clontech), containing a green fluorescent protein gene, by using restriction enzymes BgIII and PstI. The internal ribosome entry site of the encephalomyocarditis virus permits the translation of two open reading frames from one messenger RNA. Thus, two proteins, in this case SH protein and AcGFP1, are expressed simultaneously from the same bicistronic mRNA transcript.
The cDNA was transiently transfected into HEK 293 cells using the standard calcium phosphate method (28). The vector alone, pIRES-AcGFP1, was also transiently transfected in separate experiments as a control. Whole-cell current was recorded at room temperature using the standard patch clamp technique 48–72 h after transfection. The bath contained the following (mm): 124.0 NaCl, 3.5 KCl, 0.01 NaH2PO4, 26.2 NaHCO3, 1.3 MgSO4, 2.5 CaCl2, and 10.0 d(+)-glucose; gassed with a mixture of 95% O2 and 5% CO2, pH 7.4, and an osmolarity of 300 mosm/kg. The internal solution (pipette solution) contained the following (mm): 135.0 potassium gluconate, 10.0 KCl, 10.0 HEPES buffer, 0.5 EGTA, 2.0 Mg-ATP (pH adjusted to 7.3 with KOH, and an osmolarity of 275–285 mosm /kg). The voltages were uncorrected for a −9 mV junction potential, and actual voltage was obtained by subtracting 9 mV from the reported values. Whole-cell currents, obtained under voltage clamp with an Axonpatch Multiclamp 700B amplifier (Axon Instruments), were filtered at 1–5 kHz and sampled at 5–50 kHz. The access resistance Ra (usually less than 20 MegaOhms) and the capacitive transients were not compensated.
Localization of SH Protein in the Plasma Membrane of HEK 293 Cells
The SH protein wild type (WT) and H22F/H51F (FF mutant) genes carrying a C-terminal FLAG epitope were PCR-amplified using primers 5′-CTCAGATCTATGGAAAATACATCGATAACAATAG-3′ and 5′-CGTCTGCAGCTATTTATCGTCATCATCTTTGTAGTCTGTATTGACTCGAGCTCTTGG-3′. The plasmid containing SH-FLAG was generated by cloning a BglII/PstI-digested PCR product into the BglII/PstI-digested pIRES-AcGFP1 vector.
Human embryonic kidney 293 (HEK 293) cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Invitrogen), supplemented with 10% FBS and 1% penicillin and streptomycin, and maintained in 5% CO2 incubator at 37 °C. For transfection, cells were seeded on the 10-mm Petri dishes containing poly-d-lysine-coated coverslips and grown overnight. A total of 4 μg of pIRES-AcGFP plasmids, with either SHwt or SHFF FLAG-tagged, were transfected into HEK 293 cells using Lipofectamine 2000 kit (Invitrogen) following the protocol of the manufacturer. Transfected HEK 293 cells were grown for 48 h, and then they were fixed in PBS containing 4% paraformaldehyde for 45 min at 4 °C. The fixed cells were washed twice with PBS before blocking by 0.3% BSA/PBS with or without 0.1% Triton X-100 for 30 min. The HEK 293 cells were incubated with Alexa Fluor 647-conjugated anti-FLAG antibody (1:100 dilutions, Cell Signaling Technology) overnight at 4 °C. After washing three times with PBS + 0.3% BSA and one time with PBS, the immunolabeled cells were visualized using a laser scanning confocal microscope (Fluoview BX61; Olympus).
NMR Sample Preparation
15N- and 15N/13C-labeled SH protein was tested in various detergents (SDS, DPC, and DHPC) for best dispersion in an HSQC spectrum. The best results were obtained in d38 DPC. For paramagnetic probe measurements, 0.8 mg of dry 5-doxyl stearic acid (5-DSA) or 16-doxyl stearic acid (16-DSA) was first dissolved separately in 300 μl of methanol. 30 μl of methanol solubilized DSA was then aliquoted into an Eppendorf tube, corresponding to 1 mm in the final NMR samples. The DSA probes were then dried under a stream of dry N2 gas and placed in a vacuum lyophilizer for at least 2 h to remove any residual methanol. The NMR sample containing SH/DPC was added to the NMR tube containing dried DSA and vortexed until the film dissolved completely. The sample was left to equilibrate for at least 2 h before NMR measurements was taken.
For Cu2+ experiments, copper(II) chloride was made up as a 1 mm stock, of which 50 μm Cu2+ (final concentration) was added to the sample. Weakly aligned samples were prepared by soaking a solution of 0.5 mm 15N-labeled SH protein in 100 mm DPC into polyacrylamide gels. Gels were prepared from stock containing 36% w/v acrylamide (Bio-Rad) and 0.94 w/v N,N-methylenebisacrylamide (Bio-Rad), which yields an acrylamide/bisacrylamide molar ratio of 83:1. An 8% polyacrylamide gel was cast using a gel chamber of 5.4 mm inner diameter (New Era Enterprise). After polymerization was complete, gels were washed in large excess of H2O overnight to ensure removal of unreacted components. The gels were then dried to completeness at 37 °C. The protein solution containing SH/DPC was soaked into the dried gels over 2 days to ensure complete re-hydration. The hydrated 8% gel was then radially compressed into a 4.2-mm inner diameter open-ended tube using the gel press assembly of Chou et al. (73) (New Era Enterprise, Inc.) and secured using the supplied support rod and end gel plug.
Approximately 1 mg of lyophilized SH protein was solubilized in 100 μl of methanol and dried under a dry stream of N2 gas, resulting in a thin protein film deposit. The tube was placed in a vacuum lyophilizer for at least 2 h to remove any residual methanol. The thin protein film was then solubilized with sample buffer containing 5 mg of d38 DPC (Avanti Polar Lipids) and 100 mm acetate buffer to a give final detergent concentration of 150 mm and sample, pH 4.4, with the protein/detergent ratio of about 1:200. The sample pH was brought up to 4.4 by titrating with aqueous sodium hydroxide. The sample was subjected to repeated cycles of heating to 318 K, vortexing, cooling, and sonication until the solution was cleared indicating protein reconstitution into detergent micelles. The sample was spun at 13,000 rpm to clear nonreconstituted protein. For copper addition in NMR experiments, the SH protein was reconstituted in 100 mm acetate buffer at pH 4.5 and 160 mm DPC, to a final protein/detergent molar ratio of 1:200.
NMR Spectroscopy
NMR experiments were performed at 37 °C (310 K) using Bruker Avance-II 700 and 600 NMR spectrometers equipped with cryogenic TXI probes (Bruker BioSpin). Complete sequence-specific assignment of backbone was achieved using standard triple resonance experiments (HNCA, HN(CO)CA, CBCACONH, HNCACB, and HNCO), whereas side-chain resonances were assigned using three-dimensional 15N-resolved NOESY-HSQC (200 ms mixing time), three-dimensional 15N-resolved TOCSY-HSQC (45 and 75 ms mixing time), and three-dimensional 13C-resolved NOESY-HSQC (200 ms mixing time).
To identify transmembrane residues, the NMR sample was lyophilized overnight and reconstituted in 99% D2O. Sodium 2,2-dimethyl-2-silapentane-5-sulfonate was used as the internal reference for 1H nuclei. The chemical shifts of 13C and 15N nuclei were calculated from the 1H chemical shifts (29). Analysis of the RDC data and rigid body modeling was done using the program MODULE (30). To determine protein backbone flexibility, 1H-15N heteronuclear steady-state NOEs were obtained by recording spectra with and without 3 s 1H presaturation at 600 MHz.
Structure Reconstruction
The structure of the SH monomer in DPC micelles was calculated using the assignment of 1H, 13C, and 15N resonances and unassigned NOEs as input for the program CYANA (31, 32). Backbone dihedral angles were predicted using chemical shift data and PREDITOR (33). Structure calculations were started from 100 random conformers, using the standard simulated annealing protocol in CYANA. The statistics of meaningful NOE distance constraints in the final CYANA cycle showed a high density of structural constraints per amino acid. Seven cycles of NOE assignment and structure reconstruction resulted in a bundle of 20 conformers. All data sets used for CYANA were reformatted for CNS using PDBstat (34).
The program CNS 1.2 (35) was used to refine the SH structure by incorporating RDCs, dihedral angles, hydrogen bonds, and upper limit distance constraints (from CYANA). Structures were recalculated from an extended strand with random initial velocities using the default simulated annealing protocol of the CNS package. 100 conformers were calculated, and the structure with the lowest energy function after fitting the topology based on rigid body modeling was selected as the representative model for construction of the C5-symmetrical SH pentamers (supplemental Fig. S7).
The pentameric structure of SH was determined using the multibody interface of the HADDOCK webserver (36). The docking was performed from five copies of the representative model (from CNS with the lowest energy function) and inclusion of RDCs for all five monomers using the SANI module for RDC analysis (37). To prevent possible occlusion and stearic hindrance during the docking process, the flexible parts of the protein showing negative HNOE values were removed from the monomer. Center-of-mass restraints, C5 symmetry restraints, and noncrystallographic restraints were imposed on all sequential monomer pairs (AB-BC-CD-DE-EA).
The pentameric model of SH-TM in lipid bilayers obtained previously using site-specific infrared dichroism was used to derive a set of plausible intermonomeric NOEs. These were verified against experimental side-chain NOEs from standalone peaks in 13C HSQC and 13C-resolved NOESY-HSQC spectra resulting in a list of NOEs incompatible with α-helical backbone geometry but exhibiting clearly detectable and unique NOEs. These inter-monomeric NOEs were implemented as the enforced restraints for the HADDOCK procedure to obtain the full atomic description of the pentameric structure.
The lumen dimensions for the pentameric model were calculated using HOLE (38) and visualized using Visual Molecular Dynamics (39). Chemical shift perturbations were monitored using 1H-15N TROSY spectra where the magnitude of the perturbation was calculated using the weighted-average chemical shifts for each amino acid residue as shown in Equation 1 (40).
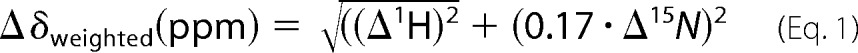 |
RESULTS
Overexpression and Purification of SH Protein from E. coli
We expressed SH protein with its N terminus fused to His6-MBP (41), reaching moderate levels of expression after isopropyl β-thiogalactoside induction, shown by a band migrating at 50 kDa (Fig. 1A, arrow). His6-MBP-SH fusion protein was mainly found in the membrane fraction (Fig. 1B, see arrowhead in lane labeled MEM), which suggests that SH protein in that instance was properly folded and likely in a native conformation. Only a small percentage of the expressed fusion protein appeared in inclusion bodies (Fig. 1B, lane labeled INC). Similar observations regarding the membrane localization of MBP-fused proteins have been reported in cytochrome b6, M2, CorA, and KdpF (41).
The His6-MBP-SH fusion protein construct was purified using Ni2+-NTA resin and subjected to tobacco etch virus (TEV) enzymatic cleavage to remove His6-MBP. Complete cleavage (Fig. 1C) was achieved at room temperature in 4 h, after mixing TEV and His6-MBP-SH at a 1:5 molar ratio under constant shaking. SH protein was extracted from the mixture with methanol, followed by reverse-phase high pressure liquid chromatography (RP-HPLC) purification. The HPLC chromatogram shows an intense peak (Fig. 1D, arrow). The MALDI mass spectrum of that fraction (Fig. 1E) shows a major peak corresponding to pure SH protein, with only three extra N-terminal amino acids (see sequence in Fig. 1F). This purified polypeptide showed channel activity when reconstituted in black lipid membranes (supplemental Fig. S1).
Gel Electrophoresis Analysis of SH Protein
It has been shown that SH protein can be cross-linked in infected cells to produce a ladder of oligomers, from dimers to pentamers, in an SDS gel (10, 15). We have shown previously that the synthetic peptide corresponding to the predicted TM domain of SH protein (residues 18–43) formed monomers in SDS but pentamers in the mild detergent PFO (18). The latter results showed that SDS is not a good detergent to monitor SH protein oligomerization and that milder detergents are required to retain the native interactions between monomers.
This is confirmed when examining the full-length SH protein (∼7.5 kDa) in the presence of SDS (Fig. 2A, WT), which migrates as a single band with an apparent molecular mass of ∼17 kDa, consistent with dimers. However, in the presence of PFO (Fig. 2B, WT), mobility was consistent with a higher molecular weight (∼35–40 kDa), compatible with a pentameric form, and in agreement with the results we reported for SH-TM (18). We therefore conclude that the dimeric form observed for full-length SH protein in SDS is artifactual.
To confirm the pentameric oligomerization of SH protein, we performed a Blue Native (BN)-PAGE analysis with SH protein previously solubilized in zwitterionic 3-(N,N-dimethylmyristylammonio)propanesulfonate (C14 betaine) micelles (Fig. 2C, WT). Because water-soluble molecular weight markers may show abnormal mobility compared with membrane proteins in BN-PAGE (42), we included as an additional molecular weight marker a membrane protein, E. coli aquaporin Z (monomeric size 27 kDa), which forms a ladder with oligomers of increasing size in these conditions (Fig. 2C, lane AQP) (43). WT SH protein migrated as a single band with a mobility between monomeric and dimeric aquaporin Z (i.e. between 30 and 60 kDa), again consistent with a pentamer (∼40 kDa). These results show that WT SH protein appears to form a single oligomer, probably pentameric, in both PFO and in C14 betaine detergents.
Analytical Ultracentrifugation Equilibrium Sedimentation (AUC ES) of WT SH Protein in Detergent Micelles
An independent determination of WT SH protein oligomeric size was performed using AUC ES. Reversible association models corresponding to an equilibrium between monomers and n-mers, where n = 2–7, were used to fit the experimental data.
The monomer/pentamer (1:5) model produced the best fit for the WT SH protein in C14 betaine (Fig. 3, A–C). The residuals are all closely distributed around zero, and χ2 and root mean square deviations values were small, 1.09103 and 0.00704, respectively, suggesting that the 1:5 model is a good description of the experimental data. The calculated standard free energy (ΔGx0) of pentamerization (44) of SH protein in C14 betaine micelles was −16.3, or −3.3 kcal/mol per monomer.
FIGURE 3.

AUC ES data corresponding to SH protein in detergent micelles. A–C, data for WT SH collected at three protein concentrations (80, 50, and 30 μm) and at three different speed as follows: 16,000 (blue), 19,500 (green), and 24,000 (red) rpm, in A–C, respectively. Data for WT was globally best-fitted to a monomer-pentamer equilibrium model. The graph shows both data points (black filled circles) and fitted function (in color). Lower panels represent fit residuals. D, WT oligomeric species distribution in C14 betaine, C8E5, and DPC detergents, where the thick bar on the curve represents the range of protein/detergent molar ratios used in the AUC experiment. The dotted line represents the protein/DPC molar ratio (1:200) used in the NMR experiments (see below) and its corresponding pentamer fraction (∼90%). C-14 betaine, DPC, and C8E5 detergent concentrations were 5, 15, and 33 mm, respectively.
For the range of concentrations used in the AUC experiments (Fig. 3, A–C), the fraction that formed pentamers was 75–87% in C14 betaine, C14SB (Fig. 3D). The same panel shows that SH protein formed even more stable pentamers in DPC and C8E5 micelles, with Kx = 5.15 × 1013 and 1.83 × 108 m−4, which results in ΔGx0 per monomer of −3.74 and −4.9 kcal/mol, respectively.
Thus, results from PFO-PAGE, BN-PAGE, and equilibrium sedimentation indicate that SH protein forms homopentamers in detergent. As the polar headgroup of DPC is more relevant biologically, this detergent was used for further NMR studies (see below).
Secondary Structure of SH Protein Reconstituted in Model Lipid Bilayers
Secondary structure predictions indicate that the α-helical region of SH protein spans residues 16–46, i.e. ∼40% of the molecule (45), whereas the TM domain is predicted to encompass residues ∼20–40 (46).
To test this, we examined the infrared amide I region of full-length SH protein and synthetic peptides SH-TM (18–43), corresponding to the TM domain, and SH(45–64), corresponding to the last 20 C-terminal residues, when incorporated in DMPC bilayers. The amide I band in the infrared spectrum is contributed mainly by peptide backbone C=O stretching vibrations, which are sensitive to hydrogen bonding strength, and therefore to secondary structure.
The amide I of SH protein shows a major peak centered at 1653 cm−1 and a shoulder centered at 1632 cm−1 (Fig. 4A) indicating a mixture of α-helix and β-strand. SH-TM(18–43) (Fig. 4B) shows a narrower band, centered at 1655 cm−1, indicating a mostly α-helical structure. Finally, SH(45–64) shows a main band centered at 1635 cm−1, assigned to β-structure (Fig. 4C). These amide I bands were analyzed using Fourier self-deconvolution, which results in narrower components that can be assigned to a particular secondary structure (Table 1).
FIGURE 4.

Secondary structure and hydrogen/deuterium exchange in lipid bilayers. ATR-FTIR spectra in the amide I region of SH protein and truncations reconstituted in DMPC bilayers. A, full-length SH protein (1–64); B, SH-TM(18–43); C, peptide corresponding to the last 20 C-terminal residues, SH(45–64). The original spectrum (continuous line) and Fourier self-deconvolved (broken line) are shown; D–F, amide II region corresponding to peptides in A–C, before (solid line) and after (dot-dashed) H/D exchange.
TABLE 1.
Analysis of the amide I region in SH and SH-TM
Assignment, wave number (v), and relative areas of the component bands in the amide I region of SH protein and SH-TM were reconstituted in DMPC bilayers (Fig. 4). The standard deviation is a result of three independent experiments. For the SH-C20 peptide, it was estimated that the β-structure constituted at least >60% of the peptide.
| Assignment | SH protein(1–64) |
SH-TM(18–43) |
||
|---|---|---|---|---|
| v | Relative area | v | Relative area | |
| cm−1 | % | cm−1 | % | |
| β-Strand | 1632 | 34.93 ± 1.25 | 1636 | 18.72 ± 0.50 |
| α-Helix | 1653 | 59.51 ± 0.85 | 1654 | 74.01 ± 1.77 |
| Turns | 1672 | 5.56 ± 0.40 | 1670 | 7.28 ± 1.57 |
For full-length SH protein the relative area corresponding to α-helix and β-structure was 60% (∼40 amino acids) and ∼30% (∼20 amino acids), respectively. The remaining of the structure in SH protein is likely to be represented by random coil or turn regions.
For SH-TM(18–43), the α-helical content was calculated to be 74% (∼20 amino acids) and ∼10% (2–3 amino acids) of β-structure, i.e. mostly α-helix as expected from a predicted α-helical TM domain. The full-length SH protein shows a higher content in α-helix relative to the TM, suggesting that in the full-length protein there is an extramembrane α-helical region of ∼20 amino acids. Although it is difficult to extrapolate results obtained for a fragment, one can speculate that this extra α-helical region should be found in the N-terminal extramembrane domain SH(1–17) because the spectrum for SH(45–64) shows >60% β-structure and little or no α-helix (Fig. 4C).
Hydrogen/Deuterium (H/D) Exchange of SH Protein Reconstituted in Model Lipid Bilayers
The amide II band in the samples above was used to monitor protein H/D exchange, as the amide II band is due primarily to the peptide backbone N-H bending vibration. Upon H/D exchange, the peptidic N-H bond becomes N-D and the frequency of amide II downshifts from near 1545 to 1450 cm−1 (∼100 cm−1) (47). In a typical experiment, the percentage of H/D exchange can be measured from the decrease in the intensity of amide II band at 1550 cm−1. The amide II band spectra (Fig. 4, D–F) were recorded either in the presence of H2O or after 1 h exposure to D2O. For full-length SH protein (Fig. 4D), 45 ± 6%, i.e. 30 ± 4 residues of the protein, was protected against H/D exchange, which therefore may include some extramembrane residues. For SH-TM(18–43), 77 ± 8% was resistant to H/D exchange (∼20 ± 2 residues), consistent with the expected number of residues in the α-helical TM domain. Finally, the C-terminal peptide SH(45–64) exchanged almost completely, with >90% (Fig. 4F).
NMR Study of SH Protein in DPC Micelles
NMR spectra were recorded in DPC micelles, as it was found that SH forms stable pentamers in this detergent (Fig. 3D). Reconstitution of SH protein in DPC micelles resulted in well resolved 1H-15N-TROSY-HSQC spectra (Fig. 5A), where most of the resonances could be assigned to individual residues. However, for extramembrane residues 5–14, 57, and 59–64, two or more closely spaced cross-peaks were observed, representing a slow equilibrium of two spectroscopically resolved conformations (see supplemental Fig. S2). This structural heterogeneity was also observed in other solubilization media such as SDS, DHPC micelles, and DMPC/DHPC isotropic bicelles with q = 0.1, in the studied pH range from 4.5 to 6.5 (data not shown). For NMR analysis, only the major component of each cross-peak was used. A comparison of the 1H-15N-TROSY-HSQC spectra in water and in 99% D2O (Fig. 5, A and B, respectively) shows that amide protons in 19 residues are significantly protected from exchange with water (Met23–Leu41). This is consistent with the infrared H/D exchange data for the TM peptide (∼20 residues) obtained in lipid bilayers (Fig. 4) (18). However, this number is lower than the ∼30 residues determined for the full-length SH protein in lipid bilayers by FTIR (Fig. 4B). The origin of this difference is discussed below.
FIGURE 5.

NMR characterization of SH protein in DPC micelles. 1H-15N-TROSY-HSQC of SH protein in H2O (A) and 99.9% D2O (B) in 100 mm acetate buffer at pH 4.5 and 160 mm d38 DPC to a final protein/detergent molar ratio of 1:200. Spectra were acquired at 310 K. Residues at the termini exhibiting doubling of cross-peaks are marked by an asterisk. A, Trp15 indole group is show in inset. C, heteronuclear 1H-15N NOE (HNOE) experiment. P represents proline residues, and NA represents two of the three extra N-terminal residues, SNA. The stretch protected from H/D exchange in B is indicated by a line labeled TM.
The dynamic properties of SH were studied using a 1H,15N steady-state HNOE experiment (Fig. 5C), obtained for the backbone 1H-15N moieties. HNOEs provide information regarding backbone molecular motions in the picosecond-nanosecond time scale and therefore help to identify structured regions (48). HNOE values of the majority of the residues of SH protein correspond to a fully folded protein (residues 5–57), flanked with highly mobile residues at the termini, although the central part of the protein is the most stable (residues ∼12–49).
The structure of the SH protein monomer was calculated using the constraints derived from 1508 NOEs and 126 dihedral angle constraints using CYANA (Table 2) (31, 32). The upper limit, dihedral angle, and hydrogen bond constraints were input into CNS-1.2 (35) and recalculated starting from an extended conformation using the default-simulated annealing protocol.
TABLE 2.
NMR and refinement statistics
| NMR distance and dihedral constraints | |
| Distance constraints | |
| Total NOE | 1508 |
| Intra-residue | 432 |
| Inter-residue | 848 |
| Sequential (| j| = 1) | 377 |
| Medium range (|i − j| <4) | 396 |
| Long range (|i − j| >5) | 75 |
| Intermoleculara | 15 |
| Hydrogen bondsb | 228 |
| Total dihedral angle restraintsb | |
| φ | 63 |
| ψ | 63 |
| Total residual dipolar couplings | 53 |
| Excluding flexible regions | 42 |
| Structure statisticsc | |
| Violations (mean ± S.D.) | |
| Maximum dihedral angle violation | 9.2° (0.057°) |
| Maximum distance constraint violation | 0.50 Å (0.58 Å) |
| Average pairwise root mean square deviation among 20 structures (Å) | |
| Heavy | 0.50 ± 0.06 |
| Backbone | 0.33 ± 0.07 |
| Ramachandran analysisd | |
| Residues in most favored regions | 62.6% |
| Residues in additionally allowed regions | 26.3% |
| Residues in generously allowed regions | 9.5% |
| Residues in disallowed regions | 1.6% |
a Intermolecular NOEs were not included in the total distance constraints for SH monomer calculation.
b Backbone hydrogen bond and dihedral restraints of α-helices and β-turns are applied for regions confirmed to be α-helices or β-turns according to local NOE pattern and chemical shifts (assessed with the PREDITOR program for predicting polypeptide secondary structure).
c Statistics are calculated and averaged over an ensemble of the 20 structures with lowest target function from CYANA.
d Ramachandran analysis indicates that 1.6% of residues are disallowed. This residue (Thr4) is found within the flexible region of the protein and can be safely ignored.
From the NMR model, the total α-helical content of SH protein can be estimated at ∼53%, which is comparable with ∼60% obtained using FTIR data (Table 1). Analysis of intra-monomer NOEs shows that SH protein composes two distinct α-helical segments (Fig. 6, A and B). The first is close to the N terminus (residues 7–16) and is not protected from H/D exchange in DPC micelles (Fig. 5B), although FTIR data suggested otherwise. The second corresponds to the TM domain (residues 21–44), and is almost completely protected from H/D exchange in DPC micelles (Fig. 5B) and in lipid bilayers (Fig. 4).
FIGURE 6.

Secondary structure of SH protein in DPC micelles. A, for α-helices, the peptide backbone is shown in cyan, and the side chains in gold, whereas the arrows depict extended structure. B, sequential and medium range NOE connectivities. C, representative two-dimensional strips from a three-dimensional 15N-resolved NOESY-HSQC showing amide NOEs for residues Tyr47 to Thr54. Red lines show medium range HN/HN cross-peaks defining the β-turn structure, with Phe50 and His51 positioned at the tip of the β-turn.
For both N-terminal and TM-helices, backbone and side-chain heavy atom root mean square deviations of a set of 20 monomeric conformers were 0.50 and 0.80 Å, respectively, suggesting a well defined structure. These two α-helical domains are connected by a stretch of extended conformation (residues ∼17–22).
At the C-terminal extramembrane domain, the structure consists of an extended strand (residues 46–57), susceptible to H/D exchange in DPC micelles (Fig. 5B) and also in lipid bilayers (Fig. 4). The data suggest an overall appearance of an extended β-turn formed by 11 residues, stabilized by hydrogen bonds between amide and carbonyl groups at the tip of the turn structure, which is confirmed by NOEs (Fig. 6C). We note that from FTIR data, the number of residues forming β-structure was slightly higher (∼20 residues, see above). Despite these differences, the secondary structure composition of SH determined by NMR in DPC micelles and by FTIR in lipid bilayers is in reasonably good agreement.
Reconstruction of the Pentameric Model of SH Protein Using NMR Data
In the first stage, the relative orientations of N-terminal helix, TM helix, and C-terminal β-turn were defined using 51 RDCs in the program MODULE (30). In all models fitted, the N-terminal α-helix was found to be positioned orthogonal to the TM α-helix axis (see supplemental Fig. S3). The C-terminal β-turn structure was also found to be orthogonal to the TM-helix, and it may be lying on the surface of the micelle (Fig. 7, A–C).
FIGURE 7.

Overall structure of the SH pentamer. SH pentamer structure with the lowest HADDOCK score depicting the side view (A) and top and bottom views (B and C). Flexible regions Met1–Thr4 and Arg59–Thr64 were removed to avoid fraying effects. D, lumen of the pentameric channel (α-helices are omitted for clarity), with the most constricted region (green). E, radius of the lumen calculated by HOLE. F, electrostatic surface of the assembly showing the mostly hydrophobic central lumen. The SH assembly spans the entire bilayer with an overall length of about 45 Å.
The multibody HADDOCK algorithm (36) was used to reconstruct the pentameric structure of SH protein. To identify inter-helical NOEs, an exhaustive search was undertaken of all available NOEs that cannot be satisfied within the structures of monomers. After verification against a previously obtained low resolution model (18), the inter-monomeric contacts were implemented as enforced restraints for the C5-symmetrical input in HADDOCK. In this way, seven unique NOEs stemming from regions between Ser5 and Trp15 were found between the “head” of the N-terminal helix of a monomer and the “tail” of an adjacent N-terminal helix, and another eight unique intermonomeric NOEs were found between side chains of the TM α-helical domain (supplemental Table S1). To improve convergence, residues with negative HNOE values (Met1–Thr4 and Arg59–Thr64, Fig. 5C) were removed from the calculations. Residues comprising the “link” (Tyr17–Leu20) between the N-terminal and TM α-helices were defined as fully flexible, whereas the two helices and the C-terminal β-turn (segments Ser5–Trp15 and Ile22–Pro61) were defined as semi-flexible. Thus, RDCs, center-of-mass restraints, C5-symmetry restraints, noncrystallographic restraints, and five copies of the truncated monomers were used to build the pentameric SH assembly (Fig. 7, A–C).
The convergent pentameric model of SH shows a funnel-like pore of about 45 Å in length (Fig. 7, D–F), spanning residues belonging to the N-terminal helix, TM helix, and C-terminal β-turn. At its narrowest, the pore diameter is about 3.5 Å, formed by side chains of Ile36. The N-terminal side (cytoplasmic) of the pentamer is very wide due to the ring of the N-terminal helices (up to 18 Å in diameter), whereas at the C-terminal side (extracellular/lumenal) the pore is more constricted, to a maximum of about 8 Å in diameter.
To validate the general topology of the pentameric structure obtained from HADDOCK, we obtained paramagnetic induced relaxation enhancement data of cross-peaks in TROSY-HSQC spectra. The following reagents were used: (i) 5-doxyl stearic acid (5-DSA) to probe the vicinity of the phosphate headgroup of the DPC detergent; (ii) 16-doxyl stearic acid (16-DSA) to probe the central hydrophobic core of the DPC micelles, and (iii) water-soluble Gd(DOTA) to probe 1HN in residues that are water-exposed were measured.
In the presence of 1 mm 5-DSA (Fig. 8A), intensities of the N-terminal helix, C- and N-ends of the TM domain, and parts of the C-terminal β-turn decreased by more than 25%, whereas the TM region was not affected. This broadening effect indicates that the N-terminal helices and the C-terminal β-turn are either lying on or slightly embedded into the surface of the micelles. In the presence of 1 mm 16-DSA (Fig. 8B), residues of the N-terminal helix and C-terminal β-turn also showed some attenuation; however, the most affected residues were found in the TM domain, confirming their location at the center of the micelle hydrophobic core.
FIGURE 8.

Interaction with paramagnetic reagents in DPC micelles. Intensity retention upon addition of 5-DSA (A) and 16-DSA (B) to the sample. For clarity, residues with intensity below an arbitrary cutoff are shown in red. P represents proline residues, and Wε represents the side chain of Trp15. C and D, intensity retention upon addition of excess paramagnetic agent Gd(DOTA). C, 1H-15N-TROSY-HSQC of SH in 99.9% D2O-based buffer. D, spectrum after acquisition of overnight 1H-15N-TROSY-HSQC in the presence of 100 mm showing regions of intensity retention.
To delineate water-exposed residues, we employed a water-soluble probe Gd(DOTA). We found that at low concentrations (1–3 mm) of Gd(DOTA), the longitudinal proton relaxation of solvent-exposed residues was mostly enhanced, resulting in higher cross-peak volumes observed for the flexible regions at the protein termini (data not shown). However, in the presence of 100 mm Gd(DOTA), it was found that residues expected to be exposed to water are in fact broadened. The resonances still visible in the HSQC spectrum (Fig. 8D) closely correspond to residues protected from H/D exchange (Fig. 8C), thus confirming the position and topology of SH pentamer in the DPC micelles. The spectral regions in Fig. 8, C and D, are directly super-imposable, although slightly shifted due to the paramagnetic effect.
Patch Clamp of HEK 293 Cells Expressing SH Protein
To test the channel activity of SH protein, full-length protein was transiently expressed in HEK 293 cells. At neutral pH, no significant difference was observed between currents mediated by vector alone (Fig. 9A, left panel) and SH protein (Fig. 9A, middle panel) (unpaired t test, p = 0.1928, for currents evoked at 70 mV). However, a more acidic pH led to a significant increase in current density by almost 5-fold (unpaired t test, p = 0.0013, for currents evoked at 70 mV) (Fig. 9A, right panel).
FIGURE 9.

Current-density through HEK 293 cells expressing SH protein. A, traces of currents evoked in HEK 293 cells transfected with vector alone or with a vector containing wild type (WT) SH protein when placed in a pH 7.4 or pH 5.5 bath solution. The cells were held at voltages ranging from −100 to 70 mV at 10-mV intervals. B, current-density versus voltage curves for WT SH protein. Whole-cell peak currents, I (pA), were normalized against whole-cell membrane capacitance, Cm (pF). The curves are fits with the Boltzmann relationship. *, p < 0.01; **, p < 0.001 (compared with vector, unpaired t test). C, current versus voltage curves for H51A, where * < 0.05; **< 0.01 (compared with vector, unpaired t test). The p value was calculated using an unpaired t test. D, I/V plot for mutant H22A, where *, p < 0.1; **, p < 0.01. E and F, I/V plots for mutants AA and FF, respectively. G, comparison at the same scale of the average curves obtained for H22A (green), WT (blue), H51A (red), and FF/AA (black) in B–F. The label is next to the trace at low pH, in each case.
A rectifier behavior, i.e. more outward conductance (positive ions going out) than inward (positive ions going in), is observed at this lower pH (Fig. 9B). The bath solution used in this experiment, artificial cerebrospinal fluid, contained a high concentration of NaCl (124 mm), whereas the internal solution contained a high concentration of potassium ions (145 mm), to mimic physiological conditions. Thus, the estimated equilibrium potentials for sodium and potassium, ENa and EK, were +65 and −87 mV, respectively. In case of a strong selectivity for sodium or potassium, the reversal potential (potential at which no net current is observed) should be present near the respective equilibrium potential of each of these ions. The observed value of reversal potential of HEK 293 cells expressing SH protein was ∼0 mV (Fig. 9B), indicating no selectivity of SH channel for sodium or potassium.
We reasoned that the pH dependence observed in the pH range 5.5–7.4 may be related to the protonatable His residues His22 and His51 (pKa ∼6.5) (49). His22 is located at the interface between the TM domain and the cytoplasmic side (see sequence in Fig. 6B), and in our previous low resolution model for the SH-TM pentamer (18) it had a lumenal orientation. This is reminiscent of His37 in the TM domain of influenza A M2, M2A, which is lumenally oriented and involved in pH-mediated gating (50), and it also has a stabilizing role (51). The other His residue in SH protein, His51, is located at the extracellular C-terminal side and should be readily protonated when the pH changes in the patch clamp experiment. Thus, the experiment was repeated with single mutants H51A, H22A, and double mutants H22A/H51A (AA) and H22F/H51F (FF) (Fig. 9, C–G).
Although none of the mutants showed channel activity at pH 7.4, mutant H51A showed activity and pH activation comparable with WT (Fig. 9C), which supports a role for His22 protonation in activation. Although His22 is near the cytoplasmic side of the TM domain, it still may be exposed to changes in pH at the extracellular side, at least when His51 is missing. Surprisingly, mutant H22A showed an even larger increase in current-density relative to either WT or H51A under acidic conditions (7-fold; p = 0.0049, un-paired t test) (Fig. 9D). The mutation at His22 may lead to unstable TM-TM interactions and a very leaky membrane when the pH is lowered, may be through His51 protonation. Finally, none of the double mutants, AA or FF, lacking His showed activation upon lowering the pH (Fig. 9, E and F). All the traces obtained at pH 5.5 were compared in Fig. 9G).
Overall, these results suggest that the pH-sensitive behavior observed is due to His protonation. It is difficult to delineate the relative importance of these two histidines because absence of one of them may affect the overall conformation of the protein, particularly His22, located within the TM domain. However, given the pH-sensitivity of both single His mutants, it is likely that both residues have some gating role.
SH Protein Localization and Topology in Cells Expressing SH Protein
Both WT (active) and FF mutant (inactive) were found to be expressed abundantly in HEK 293 cells, and the C-terminal FLAG tag was localized at the plasma membrane when cells were not permeabilized with Triton X-100, as shown by using an antibody against a C-terminally located FLAG tag (Fig. 10). These conclusions are in agreement with previous studies performed in infected cells (10, 13). However, similar experiments performed to localize an N-terminally located FLAG tag were not successful, with or without Triton X-100 permeabilization, which suggests that the N-terminal end of SH may be membrane-embedded or -truncated. In any case, these results show that the lack of channel activity in the double mutant FF cannot be attributed to deficient expression or anomalous localization.
FIGURE 10.

Membrane localization of SH protein in HEK 293 cells. HEK 293 cells were transiently transfected with pIRES-AcGFP1 vector alone (A–C), a vector containing SHwt-FLAG (D–F), or the double mutant SHFF-FLAG (G–I). Cells were fixed, and AcGFP (green, 1st column) was visualized to determine SH expression. Rabbit anti-FLAG antibody conjugated with Alexa Fluor 647 (red, middle column) was used to localize the C-terminally located FLAG tag. Images are overlaid in the last column. The white bar corresponds to 10 μm.
BN-PAGE of SH Mutants
To support that the lack of activity observed in mutant FF is not due to protein unfolding or defective oligomerization, we compared the mobility of mutant FF and WT SH protein in BN-PAGE (Fig. 11). Mutant FF produced a single band after solubilization in a variety of detergents, with a mobility similar to that of WT SH protein. In the case of C8E5 or C14 betaine, SH WT has been shown to form homopentamers (see Fig. 3); therefore, the similar mobility observed for WT and mutant FF in BN-PAGE suggests mutant FF in these detergents is also pentameric.
FIGURE 11.
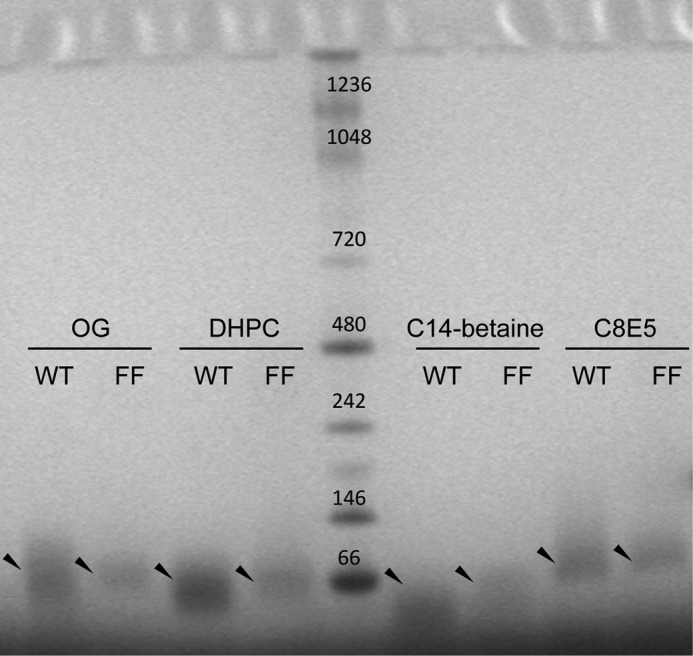
BN-PAGE analysis of SH WT and double mutant H22F/H51F (FF). Four different detergents were used: OG, DHPC, C14-betaine, and C8E5. The arrows indicate the position of the main band observed in each case for WT and FF mutant.
In contrast, species containing the mutation H22A, i.e. mutants H22A and double mutant AA, behaved most differently from species that have His22, i.e. WT or H51A. For example in SDS, H22A produced a distinct pattern with larger molecular weights, compared with WT and H51A, whereas double mutant AA had a tendency to aggregate before entering the gel (supplemental Fig. S4A).
Similar conclusions can be drawn from BN-PAGE experiments, where the sample was presolubilized in C14 betaine, DPC, or C8E5. For example, in C14 betaine (supplemental Fig. S4B), both H22A and AA ran faster than WT and H51A, and AA also showed a tendency to aggregate. In DPC (supplemental Fig. S4C), both H22A and AA produced smeared large aggregates, compared with WT and H51A. In C8E5 (supplemental Fig. S4D), both H22A and AA mutants aggregated before entering the gel, whereas WT and H51A produced almost identical patterns.
These results suggest that although His22 is critical for preserving TM interactions, it can be substituted for Phe but not for Ala. The latter is consistent with the patterns of conservation observed in multiple alignments (supplemental Fig. S6). Finally, although mutant H22A showed channel activity at low pH in patch clamp experiments, the AA mutant was inactive, suggesting that His51 also has an important functional role in activation.
pH Activation and Role of the Two Histidines
We have shown that the I/V plots obtained in the samples WT, H51A, and H22A are not equivalent (Fig. 9G); in order of decreasing conductance, they can be arranged as H22A > WT > H51A.
His51 is located extracellularly; therefore it should be readily protonated by changing the pH in the whole-cell patch clamp experiment.
Thus the activation of mutant H22A can be explained by protonation at His51, whereas the larger conductance than WT may be due to the disrupted native TM interactions mediated by H22A.
Activation in mutant H51A may be reduced relative to WT due to the more hidden location of His22, more difficult to protonate. Alternatively, the absence of His51 may make a conformational change mediated by His22 less efficient. Finally, mutants AA and FF cannot be activated because they both lack His side chains.
In summary, lack of channel activity correlates with predicted functional features of SH and not with loss of SH structural integrity or anomalous folding. The latter would be expected if the observed conductance was due to direct or indirect interaction with an endogenous channel.
In an attempt to localize the structural variations associated with pH changes, the pH of the SH protein sample in DPC micelles was increased from 4.4 to 6.5. Chemical shift changes were observed not only at His22 and His51 but also at other mostly polar residues as follows: Thr7, Phe10–Ser11, Lys13–Trp18, Thr19–His22, Thr25–Thr26, and residues located at the C-terminal end of the TM, Asn42–Leu44, and Arg56 and Leu57 (Fig. 12A). Fitting a 3.6 periodic sine wave to the N-terminal helix, maximal perturbation was observed in residues that are in phase: Thr7, Ser11, Phe14, which are oriented toward the lumen of the channel (Fig. 12B).
FIGURE 12.

Chemical shift perturbations in response to pH change. A, chemical shift perturbation when the pH of the solution was changed from 4.4 to 6.5. An arbitrary cutoff of Δδ = 0.05 is shown. P represents proline residues, Wε represents the side chain of Trp15, and * represents a nonobserved peak. B, SH pentameric assembly showing perturbations above the cutoff (in red) in A. One monomer has been removed for clarity.
Cu2+ Interaction with SH His Residues
For influenza A M2 proton channel, it has been shown that Cu2+ can inhibit channel activity by chelation to the histidine imidazole groups (52). To test if a similar effect could be observed in the SH protein, the bath solution was perfused with external buffer containing increasing concentrations of copper chloride, from 1 nm to 1 mm. A concentration of 1 μm Cu2+ successfully inhibited the current evoked through SH protein by a factor of 2 (0.53 ± 0.06) (data not shown). However, this inhibitory effect was also observed when using mutant H22A, which indicates that His51 is also involved in this regulation. At the same time, this suggests that His51 from different monomers may be in contact in biological membranes, i.e. the fragment from the TM to His51 may be in a different conformation in DPC micelles relative to biological membranes. This was also suggested by comparing H/D exchange in NMR and FTIR experiments (see above).
We then used TROSY-HSQC spectra to measure the chemical shift variation and intensity decrease of the 1H-15N backbone amide moieties when SH protein in DPC micelles was exposed to 1–50 μm of Cu2+. Although more localized to the interfacial regions of the pore, chemical shift changes were observed throughout the protein (supplemental Fig. S5), and neither His residue was particularly affected. The interaction of copper and SH protein in detergent micelles therefore is not specific to His residues.
SH Protein Is Not a Proton Channel
Inspired by the possible similarities between SH protein and the proton channel M2A, we tested if SH acts as a proton channel. However, we could not detect any proton transport when SH protein was reconstituted in E. coli liposomes or a mixture involving 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-l-serine], and cholesterol, an assay used to test proton channel activity in influenza A M2 (53) (results not shown) in any of the protein/lipid ratios tested.
DISCUSSION
RSV SH Protein Forms Pentameric Oligomers
In studies toward oligomeric size determination, the source of protein, i.e. synthetic or recombinant, presence of tags, mutations, and reconstitution environment may strongly affect the final outcome. In our hands, the electrophoretic mobility pattern of synthetic SH protein was always slightly different from recombinant SH protein,5 possibly due to some side chain modifications during synthesis. Therefore, in this study, we have used recombinant SH protein, where the extra amino acids are only three, N-terminally located to minimize any possible disruptive effect. In addition, the fusion construct used was expressed in the membranes and not in inclusion bodies. This not only suggests that MBP has a chaperone-like effect but indicates that in our work the state of SH protein when expressed should be close to the native conformation.
The results presented show that in a variety of the following environments, SH protein forms a single oligomer that is pentameric: (i) in PFO detergent; (ii) in a BN-PAGE system, and (iii) in sedimentation equilibrium experiments in C14 betaine, DPC, and C8E5, which is consistent with results obtained previously for peptide SH-TM (18). This confirms the importance of the TM domain in the pentamerization of SH protein.
In a recent electron microscopy study on full-length SH protein in liposomes or DHPC detergent micelles (17), very large (∼9 nm) structures were interpreted as pentameric or hexameric forms, with a central pore diameter of 19 and 26 Å, respectively. The authors proposed a hexameric model, and not a pentameric one, although a rationale for that choice was not made explicit (17). In that study, however, SH protein was in fact an 86-residue construct, i.e. SH was prolonged by 19 additional N-terminal amino acids, plus 3 C-terminal ones. This means that the construct was almost one-third longer than SH protein itself, and this fusion protein appeared in inclusion bodies when expressed. We note that a central pore of 19–26 Å is not compatible with the cation-selective ion channel activity observed by patch clamp. In addition, it is questionable how such a large pore could be formed with only 5–6 TM helices (one per SH monomer). The model we report herein, may offer an alternative explanation to a pentameric arrangement of TM domains; indeed, a ring formed by the N-terminal helices connected via a flexible linker to the TM domain would be compatible with the dimensions reported by electron microscopy.
However, we also note that although our AUC results indicate that WT SH monomer-monomer interactions are sufficiently strong, the resulting oligomerization state may still be sensitive to small changes in the sequence. This underlines the problems reported in the literature regarding oligomeric state determination of viroporins, especially when mutants or artificial constructs are used, as these may disrupt native interactions (54–57).
Secondary Structure and H/D Exchange of RSV SH Protein
Overall, the NMR results, combined with FTIR results, indicate that SH protein has one TM α-helix, an N-terminal extramembrane helix, and a C-terminal hairpin-like region. The N-terminal region is linked to the TM by an extended linker peptide formed by residues 17–21. An intriguing structural feature of SH protein is the observation of conformational heterogeneity manifested as doubling of cross-peaks, starting from the N terminus and propagating to residue 19, and also in the C terminus (supplemental Fig. S2). The N-terminal region corresponds to the ring of tightly packed short N-terminal helices, a feature reminiscent of the C-terminal extramembrane helix found in influenza A M2 (fragment 18–60) (58). Because of the significantly different RDCs observed for the 1H-15N indole group of Trp15 in minor and major conformations, with −17 and 0.4 Hz, respectively, one may conclude that the heterocycle ring of Trp15 can be aligned along or orthogonal to the membrane surface. This variable orientations of the indole ring of Trp15, and the pH activation likely mediated by histidine side chains, are also reminiscent of similar features encountered in influenza A M2, which has a “Trp gating” mechanism (58). Further studies are required to assess the physiological relevance of the minor alternative conformation.
We note that, although NMR and FTIR results are remarkably consistent despite the different environments used, there are several differences in terms of secondary structure content and in H/D exchange. We determined a higher α-helical content for SH protein using FTIR; therefore, it may be possible that the α-helical structure extends from the TM domain on both sides in lipid bilayers, rather than forming an extended structure.
These differences may be caused by the following: (i) the well known curvature effects of micelles used in NMR relative to the planar lipid bilayers used in FTIR; (ii) the different hydration between FTIR and NMR samples (higher hydration in NMR); and (iii) the molar ratio protein/detergent, which is significantly lower in NMR (1:200) relative to FTIR (1:50), in addition to inaccuracies of determination in the two techniques, especially in the case of FTIR.
SH Protein Forms Cation-selective Channels in Transfected Mammalian Cells
The fact that SH protein is found in plasma membrane (13) allowed the study of SH channel activity using whole-cell patch clamp. SH protein transiently expressed in mammalian cells produced nonselective cation channel activity, with passage of Na+ or K+. Poor selectivity has also been observed in other viroporins (60, 61), and it may be a hallmark of incomplete specialization of these channels. Indeed, in vitro studies show that SH protein is not essential, although it is important for effective viral infection in vivo, an importance also suggested by its strong sequence conservation.
These patch clamp studies also show a striking pH dependence in channel activity. Involvement of His as a sensor has been observed in other channels, e.g. influenza A M2 (58, 62), KcsA (His25) (63), ROMK1(64), TASK1 (65), and TREK-1 and TREK-2 (66).
In SH protein, only His22 is close to the TM domain and is facing the lumen of the channel (Fig. 12B) (18). However, inspection of sequence alignments shows that SH proteins in human RSV, PIV5, or mumps virus always have at least one His residue near the equivalent position of His51 in RSV SH protein, i.e. the extracellular/lumenal side (highlighted in yellow, supplemental Fig. S6), but the equivalent residue to His22 is substituted in some cases, for example in bovine RSV to Phe. This suggests His to Phe at this position is a conservative mutation for this class of proteins, consistent with our results, because mutant FF forms pentamers like the WT. It would therefore appear that the His51 residue in RSV is even more important than His22. Overall, these considerations suggest that a mutant H22F would form oligomers like WT and FF and would be activated by low pH.
It has been suggested that ion leakage may lead to dissipation of membrane potential and disruption of cell homeostasis, a common sign of apoptosis (67), leading to cell damage as infection progresses. In fact, the viroporins of Sindbis virus 6K, murine hepatitis virus E protein, influenza A M2 protein, HCV p7 protein, and poliovirus 2b and 3A protein have all been reported to affect apoptosis in infected cells (68–70). In contrast, the SH protein encoded by members of Paramyxoviridae family simian virus 5 (SV5) (71), parainfluenza virus 5 (PIV 5) (72), mump virus (8), and hRSV (72) all inhibit apoptosis in several mammalian cell lines. Although promotion of apoptosis may help with virus release, inhibition of apoptosis during infection may give an advantage to the virus to replicate.
For both RSV SH and another member of the Paramyxoviridae family, parainfluenza virus 5 (PIV5), it has been observed (71) that SH protein is necessary for the inhibition of tumor necrosis factor-α (TNF-α)-induced apoptosis (59, 72). The fact that the absence of SH can cause cell death in RSVΔSH-infected cells, compared with wild type RSV, and the fact that this is the case even in A549 cells, which are insensitive to TNF-α-induced cell death (72), suggest that this toxicity is not uniquely mediated by a TNF-α pathway. Also, in MDBK and L929 cells, SH from PIV5 itself or from RSV A or B subgroups has a protective role against the cytopathic effect produced by PIV5 (8, 59, 71). Similarly, SH from PIV5 could be substituted by SH from mumps virus (8), even though these two SH proteins have no sequence homology. These data argue against a mechanism mediated by a specific protein-protein interaction with an unknown protein. We postulate that the protective effect of SH may be mediated by the protein itself, for example via membrane permeabilization using the channel structure reported herein. Further studies on hRSV-infected cells should gain insight into the significance of SH viroporin activity in the hRSV life cycle.
Supplementary Material
Acknowledgments
The pTBMalE plasmid was a gift from Ian Brett (from the Steven Smith laboratory, Stony Brook University, New York). We also thank Alexander Bonvin for assistance on the HADDOCK webserver.
This work was supported by National Research Foundation Grant NRF-CRP4-2008-02 (to J. T.).

This article contains supplemental Figs. S1–S7, and Table S1, and an additional reference.
S. W. Gan, unpublished results.
- SH
- small hydrophobic
- hRSV
- human respiratory syncytial virus
- ATR
- attenuated total reflection
- TM
- transmembrane
- PFO
- perfluorooctanoic acid
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- MBP
- maltose-binding protein
- Ni2+-NTA
- nickel-nitrilotriacetic acid
- TEV
- tobacco etch virus
- AUC ES
- analytical ultracentrifugation equilibrium sedimentation
- H/D
- hydrogen/deuterium
- DHPC
- dihexanoyl phosphocholine
- DMPC
- dimyristoylphosphocholine
- HNOE
- heteronuclear NOE
- RDC
- residual dipolar coupling
- BN
- Blue Native
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- DSA
- doxyl stearic acid
- DPC
- dodecylphosphocholine
- Gd-DOTA
- gadolinium-tetraazacyclododecanetetraacetic acid.
REFERENCES
- 1. Dowell S. F., Anderson L. J., Gary H. E., Jr., Erdman D. D., Plouffe J. F., File T. M., Jr., Marston B. J., Breiman R. F. (1996) Respiratory syncytial virus is an important cause of community-acquired lower respiratory infection among hospitalized adults. J. Infect. Dis. 174, 456–462 [DOI] [PubMed] [Google Scholar]
- 2. Blount R. E., Jr., Morris J. A., Savage R. E. (1956) Recovery of cytopathogenic agent from chimpanzees with coryza. Proc. Soc. Exp. Biol. Med. 92, 544–549 [DOI] [PubMed] [Google Scholar]
- 3. Krusat T., Streckert H. J. (1997) Heparin-dependent attachment of respiratory syncytial virus (RSV) to host cells. Arch. Virol. 142, 1247–1254 [DOI] [PubMed] [Google Scholar]
- 4. Lamb R. A. (1993) Paramyxovirus fusion. Hypothesis for changes. Virology 197, 1–11 [DOI] [PubMed] [Google Scholar]
- 5. Techaarpornkul S., Barretto N., Peeples M. E. (2001) Functional analysis of recombinant respiratory syncytial virus deletion mutants lacking the small hydrophobic and/or attachment glycoprotein gene. J. Virol. 75, 6825–6834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bukreyev A., Whitehead S. S., Murphy B. R., Collins P. L. (1997) Recombinant respiratory syncytial virus from which the entire SH gene has been deleted grows efficiently in cell culture and exhibits site-specific attenuation in the respiratory tract of the mouse. J. Virol. 71, 8973–8982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Karron R. A., Buonagurio D. A., Georgiu A. F., Whitehead S. S., Adamus J. E., Clements-Mann M. L., Harris D. O., Randolph V. B., Udem S. A., Murphy B. R., Sidhu M. S. (1997) Respiratory syncytial virus (RSV) SH and G proteins are not essential for viral replication in vitro. Clinical evaluation and molecular characterization of a cold-passaged, attenuated RSV subgroup B mutant. Proc. Natl. Acad. Sci. U.S.A. 94, 13961–13966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wilson R. L., Fuentes S. M., Wang P., Taddeo E. C., Klatt A., Henderson A. J., He B. (2006) Function of small hydrophobic proteins of paramyxovirus. J. Virol. 80, 1700–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Whitehead S. S., Bukreyev A., Teng M. N., Firestone C. Y., St Claire M., Elkins W. R., Collins P. L., Murphy B. R. (1999) Recombinant respiratory syncytial virus bearing a deletion of either the NS2 or SH gene is attenuated in chimpanzees. J. Virol. 73, 3438–3442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Collins P. L., Mottet G. (1993) Membrane orientation and oligomerization of the small hydrophobic protein of human respiratory syncytial virus. J. Gen. Virol. 74, 1445–1450 [DOI] [PubMed] [Google Scholar]
- 11. Collins P. L., Olmsted R. A., Johnson P. R. (1990) The small hydrophobic protein of human respiratory syncytial virus. Comparison between antigenic subgroups A and B. J. Gen. Virol. 71, 1571–1576 [DOI] [PubMed] [Google Scholar]
- 12. Chen M. D., Vazquez M., Buonocore L., Kahn J. S. (2000) Conservation of the respiratory syncytial virus SH gene. J. Infect. Dis. 182, 1228–1233 [DOI] [PubMed] [Google Scholar]
- 13. Rixon H. W., Brown G., Aitken J., McDonald T., Graham S., Sugrue R. J. (2004) The small hydrophobic (SH) protein accumulates within lipid-raft structures of the Golgi complex during respiratory syncytial virus infection. J. Gen. Virol. 85, 1153–1165 [DOI] [PubMed] [Google Scholar]
- 14. Olmsted R. A., Collins P. L. (1989) The 1A protein of respiratory syncytial virus is an integral membrane protein present as multiple, structurally distinct species. J. Virol. 63, 2019–2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rixon H. W., Brown G., Murray J. T., Sugrue R. J. (2005) The respiratory syncytial virus small hydrophobic protein is phosphorylated via a mitogen-activated protein kinase p38-dependent tyrosine kinase activity during virus infection. J. Gen. Virol. 86, 375–384 [DOI] [PubMed] [Google Scholar]
- 16. Perez M., García-Barreno B., Melero J. A., Carrasco L., Guinea R. (1997) Membrane permeability changes induced in Escherichia coli by the SH protein of human respiratory syncytial virus. Virology 235, 342–351 [DOI] [PubMed] [Google Scholar]
- 17. Carter S. D., Dent K. C., Atkins E., Foster T. L., Verow M., Gorny P., Harris M., Hiscox J. A., Ranson N. A., Griffin S., Barr J. N. (2010) Direct visualization of the small hydrophobic protein of human respiratory syncytial virus reveals the structural basis for membrane permeability. FEBS Lett. 584, 2786–2790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gan S. W., Ng L., Lin X., Gong X., Torres J. (2008) Structure and ion channel activity of the human respiratory syncytial virus (hRSV) small hydrophobic protein transmembrane domain. Protein Sci. 17, 813–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Arkin I. T., MacKenzie K. R., Brunger A. T. (1997) Site-directed dichroism as a method for obtaining rotational and orientational constraints for oriented polymers. J. Am. Chem. Soc. 119, 8973–8980 [Google Scholar]
- 20. Torres J., Kukol A., Goodman J. M., Arkin I. T. (2001) Site-specific examination of secondary structure and orientation determination in membrane proteins: the peptidic (13)C=(18)O group as a novel infrared probe. Biopolymers 59, 396–401 [DOI] [PubMed] [Google Scholar]
- 21. Briggs J. A., Torres J., Arkin I. T. (2001) A new method to model membrane protein structure based on silent amino acid substitutions. Proteins Struct. Funct. Genet. 44, 370–375 [DOI] [PubMed] [Google Scholar]
- 22. Adams P. D., Arkin I. T., Engelman D. M., Brünger A. T. (1995) Computational searching and mutagenesis suggest a structure for the pentameric transmembrane domain of phospholamban. Nat. Struct. Biol. 2, 154–162 [DOI] [PubMed] [Google Scholar]
- 23. Tolley K. P., Marriott A. C., Simpson A., Plows D. J., Matthews D. A., Longhurst S. J., Evans J. E., Johnson J. L., Cane P. A., Randolph V. B., Easton A. J., Pringle C. R. (1996) Identification of mutations contributing to the reduced virulence of a modified strain of respiratory syncytial virus. Vaccine 14, 1637–1646 [DOI] [PubMed] [Google Scholar]
- 24. Qin H., Hu J., Hua Y., Challa S. V., Cross T. A., Gao F. P. (2008) Construction of a series of vectors for high throughput cloning and expression screening of membrane proteins from Mycobacterium tuberculosis. BMC Biotechnol. 8, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marley J., Lu M., Bracken C. (2001) A method for efficient isotopic labeling of recombinant proteins. J. Biomol. NMR 20, 71–75 [DOI] [PubMed] [Google Scholar]
- 26. McGhie E. J., Hume P. J., Hayward R. D., Torres J., Koronakis V. (2002) Topology of the Salmonella invasion protein SipB in a model bilayer. Mol. Microbiol. 44, 1309–1321 [DOI] [PubMed] [Google Scholar]
- 27. Marshall J., Molloy R., Moss G. W., Howe J. R., Hughes T. E. (1995) The jellyfish green fluorescent protein. A new tool for studying ion channel expression and function. Neuron 14, 211–215 [DOI] [PubMed] [Google Scholar]
- 28. Peterson B. Z., DeMaria C. D., Adelman J. P., Yue D. T. (1999) Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron 22, 549–558 [DOI] [PubMed] [Google Scholar]
- 29. Cavanagh J., Fairbrother W. J., Palmer A. G., Skelton N. J., Rance M. (2006) Protein NMR Spectroscopy: Principles and Practice, 2 Ed., Elsevier Academic Press, New York [Google Scholar]
- 30. Dosset P., Hus J. C., Marion D., Blackledge M. (2001) A novel interactive tool for rigid-body modeling of multidomain macromolecules using residual dipolar couplings. J. Biomol. NMR 20, 223–231 [DOI] [PubMed] [Google Scholar]
- 31. Herrmann T., Güntert P., Wüthrich K. (2002) Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 319, 209–227 [DOI] [PubMed] [Google Scholar]
- 32. Güntert P. (2004) Automated NMR structure calculation with CYANA. Methods Mol. Biol. 278, 353–378 [DOI] [PubMed] [Google Scholar]
- 33. Berjanskii M. V., Neal S., Wishart D. S. (2006) PREDITOR. A web server for predicting protein torsion angle restraints. Nucleic Acids Res. 34, W63–W69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bhattacharya A., Tejero R., Montelione G. T. (2007) Evaluating protein structures determined by structural genomics consortia. Proteins 66, 778–795 [DOI] [PubMed] [Google Scholar]
- 35. Brunger A. T. (2007) Version 1.2 of the crystallography and NMR system. Nat. Protoc. 2, 2728–2733 [DOI] [PubMed] [Google Scholar]
- 36. de Vries S. J., van Dijk M., Bonvin A. M. (2010) The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 5, 883–897 [DOI] [PubMed] [Google Scholar]
- 37. Clore G. M., Gronenborn A. M., Tjandra N. (1998) Direct structure refinement against residual dipolar couplings in the presence of rhombicity of unknown magnitude. J. Magn. Reson. 131, 159–162 [DOI] [PubMed] [Google Scholar]
- 38. Smart O. S., Neduvelil J. G., Wang X., Wallace B. A., Sansom M. S. (1996) HOLE. A program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 14, 354–360 [DOI] [PubMed] [Google Scholar]
- 39. Humphrey W., Dalke A., Schulten K. (1996) VMD, visual molecular dynamics. J. Mol. Graph. 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 40. Farmer B. T., Constantine K. L., Goldfarb V., Friedrichs M. S., Wittekind M., Yanchunas J., Robertson J. G., Mueller L. (1996) Localizing the NADP+-binding site on the MurB enzyme by NMR. Nat. Struct. Mol. Biol. 3, 995–997 [DOI] [PubMed] [Google Scholar]
- 41. Hu J., Qin H., Li C., Sharma M., Cross T. A., Gao F. P. (2007) Structural biology of transmembrane domains. Efficient production and characterization of transmembrane peptides by NMR. Protein Sci. 16, 2153–2165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wittig I., Beckhaus T., Wumaier Z., Karas M., Schägger H. (2010) Mass estimation of native proteins by blue native electrophoresis: principles and practical hints. Mol. Cell. Proteomics 9, 2149–2161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gan S. W., Vararattanavech A., Nordin N., Eshaghi S., Torres J. (2011) A cost-effective method for simultaneous homo-oligomeric size determination and monodispersity conditions for membrane proteins. Anal. Biochem. 416, 100–106 [DOI] [PubMed] [Google Scholar]
- 44. Fleming K. G. (2002) Standardizing the free energy change of transmembrane helix-helix interactions. J. Mol. Biol. 323, 563–571 [DOI] [PubMed] [Google Scholar]
- 45. Käll L., Krogh A., Sonnhammer E. L. (2004) A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 338, 1027–1036 [DOI] [PubMed] [Google Scholar]
- 46. Sonnhammer E. L., von Heijne G., Krogh A. (1998) A hidden Markov model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 6, 175–182 [PubMed] [Google Scholar]
- 47. Tamm L. K., Tatulian S. A. (1997) Infrared spectroscopy of proteins and peptides in lipid bilayers. Q. Rev. Biophys. 30, 365–429 [DOI] [PubMed] [Google Scholar]
- 48. Kay L. E., Torchia D. A., Bax A. (1989) Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy. Application to staphylococcal nuclease. Biochemistry 28, 8972–8979 [DOI] [PubMed] [Google Scholar]
- 49. Cymes G. D., Ni Y., Grosman C. (2005) Probing ion-channel pores one proton at a time. Nature 438, 975–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang C., Lamb R. A., Pinto L. H. (1995) Activation of the M2 ion channel of influenza virus. A role for the transmembrane domain histidine residue. Biophys. J. 69, 1363–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Howard K. P., Lear J. D., DeGrado W. F. (2002) Sequence determinants of the energetics of folding of a transmembrane four-helix-bundle protein. Proc. Natl. Acad. Sci. U.S.A. 99, 8568–8572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gandhi C. S., Shuck K., Lear J. D., Dieckmann G. R., DeGrado W. F., Lamb R. A., Pinto L. H. (1999) Cu(II) inhibition of the proton translocation machinery of the influenza A virus M2 protein. J. Biol. Chem. 274, 5474–5482 [DOI] [PubMed] [Google Scholar]
- 53. Pielak R. M., Schnell J. R., Chou J. J. (2009) Mechanism of drug inhibition and drug resistance of influenza A M2 channel. Proc. Natl. Acad. Sci. U.S.A. 106, 7379–7384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Griffin S. D., Beales L. P., Clarke D. S., Worsfold O., Evans S. D., Jaeger J., Harris M. P., Rowlands D. J. (2003) The p7 protein of hepatitis C virus forms an ion channel that is blocked by the antiviral drug, Amantadine. FEBS Lett. 535, 34–38 [DOI] [PubMed] [Google Scholar]
- 55. Clarke D., Griffin S., Beales L., Gelais C. S., Burgess S., Harris M., Rowlands D. (2006) Evidence for the formation of a heptameric ion channel complex by the hepatitis C virus p7 protein in vitro. J. Biol. Chem. 281, 37057–37068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Luik P., Chew C., Aittoniemi J., Chang J., Wentworth P., Jr., Dwek R. A., Biggin P. C., Vénien-Bryan C., Zitzmann N. (2009) The three-dimensional structure of a hepatitis C virus p7 ion channel by electron microscopy. Proc. Natl. Acad. Sci. U.S.A. 106, 12712–12716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Parthasarathy K., Ng L., Lin X., Liu D. X., Pervushin K., Gong X., Torres J. (2008) Structural flexibility of the pentameric SARS coronavirus envelope protein ion channel. Biophys. J. 95, L39–L41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schnell J. R., Chou J. J. (2008) Structure and mechanism of the M2 proton channel of influenza A virus. Nature 451, 591–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lin Y., Bright A. C., Rothermel T. A., He B. (2003) Induction of apoptosis by paramyxovirus simian virus 5 lacking a small hydrophobic gene. J. Virol. 77, 3371–3383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pervushin K., Tan E., Parthasarathy K., Lin X., Jiang F. L., Yu D., Vararattanavech A., Soong T. W., Liu D. X., Torres J. (2009) Structure and inhibition of the SARS coronavirus envelope protein ion channel. PLoS Pathog. 5, e1000511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wilson L., McKinlay C., Gage P., Ewart G. (2004) SARS coronavirus E protein forms cation-selective ion channels. Virology 330, 322–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Stouffer A. L., Acharya R., Salom D., Levine A. S., Di Costanzo L., Soto C. S., Tereshko V., Nanda V., Stayrook S., DeGrado W. F. (2008) Structural basis for the function and inhibition of an influenza virus proton channel. Nature 451, 596–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Takeuchi K., Takahashi H., Kawano S., Shimada I. (2007) Identification and characterization of the slowly exchanging pH-dependent conformational rearrangement in KcsA. J. Biol. Chem. 282, 15179–15186 [DOI] [PubMed] [Google Scholar]
- 64. Chanchevalap S., Yang Z., Cui N., Qu Z., Zhu G., Liu C., Giwa L. R., Abdulkadir L., Jiang C. (2000) Involvement of histidine residues in proton sensing of ROMK1 channel. J. Biol. Chem. 275, 7811–7817 [DOI] [PubMed] [Google Scholar]
- 65. Yuill K. H., Stansfeld P. J., Ashmole I., Sutcliffe M. J., Stanfield P. R. (2007) The selectivity, voltage dependence, and acid sensitivity of the tandem pore potassium channel TASK-1. Contributions of the pore domains. Pflugers Arch. 455, 333–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sandoz G., Douguet D., Chatelain F., Lazdunski M., Lesage F. (2009) Extracellular acidification exerts opposite actions on TREK1 and TREK2 potassium channels via a single conserved histidine residue. Proc. Natl. Acad. Sci. U.S.A. 106, 14628–14633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Burg E. D., Remillard C. V., Yuan J. X. (2006) K+ channels in apoptosis. J. Membr. Biol. 209, 3–20 [DOI] [PubMed] [Google Scholar]
- 68. Neznanov N., Kondratova A., Chumakov K. M., Angres B., Zhumabayeva B., Agol V. I., Gudkov A. V. (2001) Poliovirus protein 3A inhibits tumor necrosis factor (TNF)-induced apoptosis by eliminating the TNF receptor from the cell surface. J. Virol. 75, 10409–10420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Campanella M., de Jong A. S., Lanke K. W., Melchers W. J., Willems P. H., Pinton P., Rizzuto R., van Kuppeveld F. J. (2004) The coxsackievirus 2B protein suppresses apoptotic host cell responses by manipulating intracellular Ca2+ homeostasis. J. Biol. Chem. 279, 18440–18450 [DOI] [PubMed] [Google Scholar]
- 70. Madan V., Castelló A., Carrasco L. (2008) Viroporins from RNA viruses induce caspase-dependent apoptosis. Cell. Microbiol. 10, 437–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. He B., Lin G. Y., Durbin J. E., Durbin R. K., Lamb R. A. (2001) The SH integral membrane protein of the paramyxovirus simian virus 5 is required to block apoptosis in MDBK cells. J. Virol. 75, 4068–4079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Fuentes S., Tran K. C., Luthra P., Teng M. N., He B. (2007) Function of the respiratory syncytial virus small hydrophobic protein. J. Virol. 81, 8361–8366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chou J. J., Gaemers S., Howder B., Louis J. M., Bax A. (2001) A simple apparatus for generating stretched polyacrylamide gels, yielding uniform alignment of proteins and detergent micelles. J. Biomol. NMR 21, 377–382 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.