

Background: Heme oxygenase-1 (HO-1) is constitutively expressed in many cancers which are highly resistant to apoptosis.
Results: CO, a product of HO-1, inhibits K+ channels in the medulloblastoma cell line DAOY and protects against apoptosis.
Conclusion: HO-1 increases resistance to apoptosis in cancer cells via CO generation.
Significance: targeting HO-1 expression may increase the effectiveness of cancer therapies.
Keywords: Apoptosis, Cancer Biology, Carbon Monoxide, Heme Oxygenase, Patch Clamp Electrophysiology, Potassium Channels
Abstract
Tumor cell survival and proliferation is attributable in part to suppression of apoptotic pathways, yet the mechanisms by which cancer cells resist apoptosis are not fully understood. Many cancer cells constitutively express heme oxygenase-1 (HO-1), which catabolizes heme to generate biliverdin, Fe2+, and carbon monoxide (CO). These breakdown products may play a role in the ability of cancer cells to suppress apoptotic signals. K+ channels also play a crucial role in apoptosis, permitting K+ efflux which is required to initiate caspase activation. Here, we demonstrate that HO-1 is constitutively expressed in human medulloblastoma tissue, and can be induced in the medulloblastoma cell line DAOY either chemically or by hypoxia. Induction of HO-1 markedly increases the resistance of DAOY cells to oxidant-induced apoptosis. This effect was mimicked by exogenous application of the heme degradation product CO. Furthermore we demonstrate the presence of the pro-apoptotic K+ channel, Kv2.1, in both human medulloblastoma tissue and DAOY cells. CO inhibited the voltage-gated K+ currents in DAOY cells, and largely reversed the oxidant-induced increase in K+ channel activity. p38 MAPK inhibition prevented the oxidant-induced increase of K+ channel activity in DAOY cells, and enhanced their resistance to apoptosis. Our findings suggest that CO-mediated inhibition of K+ channels represents an important mechanism by which HO-1 can increase the resistance to apoptosis of medulloblastoma cells, and support the idea that HO-1 inhibition may enhance the effectiveness of current chemo- and radiotherapies.
Introduction
One defining feature of cancer is the resistance of tumor cells to apoptosis (1, 2). When normal cell removal through apoptosis is inhibited, tumor growth is promoted through neoplastic transformation and poorly restrained cellular proliferation (1, 3, 4). Intracellular K+ levels exert an important influence on different aspects of the complex process of apoptosis: K+ regulates caspase activation, mitochondrial membrane potential and volume and, through its abundance, osmolarity, and cell volume (5). K+ efflux via K+ channels triggers the apoptotic cascade, in part by initiating the early apoptotic volume decrease, but also by relieving inhibition of key components of the apoptotic cascade, including pro-caspase 3 (5–7). In addition, since K+ channels are fundamental to the regulation of membrane potential, their activity also regulates Ca2+ influx and excessive intracellular Ca2+ levels can also trigger apoptosis (7). For these reasons, suppression of K+ efflux via K+ channels can protect against apoptosis stimulated by a variety of insults, including oxidative stress (8). Indeed, K+ channels play key roles in both proliferation and apoptosis, and so are regarded as potentially important targets in the treatment of cancers (reviewed in e.g. Ref. 9).
Several different K+ channel types have been proposed to have important roles in apoptosis, including large conductance, Ca2+-sensitive K+ channels, tandem P domain (“leak” or K2P) K+ channels, and voltage-gated K+ channels (7, 10). Of these, one particular K+ channel, the voltage-gated, delayed rectifier K+ channel Kv2.1, has received much attention due to its role in neuronal apoptosis. For example, neurones transfected with a dominant negative Kv2.1 containing construct (which consequently lack functional Kv2.1 channels) were protected against oxidative stress-induced apoptosis (11). Furthermore, expression of Kv2.1 in CHO (11) or HEK293 cells (12) increased their susceptibility to apoptosis. Pro-apoptotic agents trigger K+ loss from cells by causing a rapid increase in the surface expression of Kv2.1 channels; this process requires p38 MAP kinase phosphorylation of Ser-800 in the intracellular C-terminal of the channel protein and additional phosphorylation of an N-terminal tyrosine (Y124)-regulated via Src kinase (13).
Heme oxygenase-1 (HO-1,2 also known as heat shock protein 32) is an inducible enzyme in non-cancerous tissues; its expression is stimulated in response to numerous factors including hypoxia, UV radiation, nitric oxide, and oxidants (14). In contrast, HO-1 is constitutively active in many tumor types (15). HO-1 breaks down heme to liberate biliverdin (a powerful antioxidant), ferrous iron (Fe2+) and carbon monoxide (CO). The antioxidant actions of HO-1 have been well documented, and its constitutive activity in cancerous tissue has been proposed to contribute to apoptotic resistance, thereby supporting hyperplasia. Indeed, tumor growth often requires HO-1 (14, 15), and experimental down-regulation of HO-1 inhibits growth of various cancer types as well as increasing their sensitivity to radiotherapy and chemotherapy (16). Although there is undoubtedly a role for biliverdin in this regard, increasing attention has been given to CO as a signaling molecule in cancer progression since it can strongly influence proliferation and apoptosis (17, 18). However, the mechanisms underlying the role of CO in these processes remain to be determined. We have recently demonstrated that CO can provide protection for central neurones in the face of oxidative stress by inhibiting Kv2.1, thereby suppressing the pro-apoptotic loss of intracellular K+ (12). In the present study we have investigated whether such a mechanistic pathway exists in tumor cells, focusing on the central nervous system medulloblastoma tumor, prevalent in childhood and from which the experimentally amenable DAOY cell line is derived (19, 20).
EXPERIMENTAL PROCEDURES
Immunohistochemical Analysis of Medulloblastoma Tissue
Cases of medulloblastoma were retrieved from the histopathology files of the archives of the Department of Histopathology, Leeds Teaching Hospitals (Leeds, UK) with ethical permission. Representative blocks of formalin fixed paraffin embedded tissue were chosen for each case (total of 5). Sections were cut at four microns and mounted on 3-aminopropyltriethoxysilane-coated slides. Sections were serially dewaxed with two exposures to Histoclear (5 min followed by 3 min in fresh solution), followed by rinses in absolute alcohol (3 min), 90% alcohol (2 min), 70% alcohol (1 min) followed by tap water (30 s). Sections were then blocked in PBS containing 10% goat serum (1 h). Primary antibodies (anti-HO-1 at 1:200 and anti-Kv2.1 at 1:1000) were then applied for 3 h at room temperature, sections washed (3 × 10 min) with PBS-Tween (PBST) and secondary antibodies applied (1:2000 for 1 h, room temperature in the dark). Mounted sections were then washed again (3 × 10 min) with PBST prior to dropwise addition of Vectashield mounting media (Vector Labs H-1200, with DAPI nuclear stain incorporated) to the slides, and a coverslip applied and sealed with nail varnish.
DAOY Medulloblastoma Cell Line Culture
The DAOY human medulloblastoma cell line (a kind gift from Dr MD Glitsch, University of Oxford) was propagated in Dulbecco's Modified Eagle's Medium (DMEM; Invitrogen, UK) supplemented with 10% (v/v) fetal calf serum, 1% penicillin-streptomycin and 1 mm glutamine. Cells were cultured at 37 °C in a humidified atmosphere containing 95% air and 5% CO2, and passaged when 80–90% confluent.
Membrane Preparation and Immunoprecipitation
Cells were grown to ∼90% confluence in 125 cm2 flasks, washed with PBS and trypsinized (3 min at 37 °C). Cells were collected in 10 ml of PBS, briefly centrifuged (200 rpm), and lysed in 500 μl of M-PER mammalian protein extraction reagent (Thermo Scientific, Essex, UK) containing Complete Mini protease inhibitors (Roche Diagnostics UK, Lewes, East Sussex, UK) and 0.2% Triton X-100 for membrane protein solubilization. This extraction solution was then incubated at 4 °C (30 min) with gentle agitation, the lysate subsequently collected and cleared by centrifugation (14,000 rpm. 15 min). Supernatants were transferred to fresh tubes and pre-cleared using protein G beads (20 μl, 30 min at 4 °C) to prevent nonspecific binding. This mixture was spun down, the pre-cleared lysate transferred to a fresh tube and incubated with anti-Kv2.1 antibody at 4 °C (4 h) with gentle rotation. Fresh protein G beads (50 μl) were then added to the antibody-protein mixture and incubated for 4 h at 4 °C with gentle rotation to allow antibody-antigen complexes to form. The complex was pelleted (3000 rpm for 5 min) and then washed (×3) in 1 ml of ice-cold PBS-containing 0.2% Triton X-100 and Complete Mini protease inhibitors. Proteins were eluted from the solid support using 1× SDS sample buffer, and analyzed via Western blotting as described below. Exactly the same protocol was followed for the negative control experiment (Fig. 3A) but with the omission of the anti-Kv2.1 antibody.
FIGURE 3.

DAOY cells express functional Kv2.1 channels. A, Western blots showing the presence of Kv2.1 channel protein in DAOY total cell lysate. The left-hand blot shows detection of Kv2.1 using an anti-Kv2.1 antibody in total DAOY cell lysate without immunoprecipitation (no IP); the right-hand blot is from lysate following immunoprecipitation with anti- Kv2.1 antibody (IP; E, elution). The lane labeled negative control was performed exactly as the IP but omitting the anti-Kv2.1 antibody. B, left: example voltage-gated K+ currents evoked in DAOY cells by step depolarizations to +50 mV before (control) during (TEA) and after (wash) exposure to 5 mm TEA. Right: example K+ currents evoked by step depolarizations to +50 mV before (control) during (4-AP) and after (wash) exposure to 10 mm 4-AP. C, normalized mean time courses (± S.E. bars) of K+ current amplitudes evoked by successive step depolarizations from −90 to +50 mV (100 ms). Cells were dialyzed with a pipette solution containing either no antibody (control, solid circles, n = 6 cells), an anti-Kv2.1 antibody (0.5 μg/ml, open circles, n = 6 cells) or an anti-Kv4.3 antibody (0.5 μg/ml, open triangles, n = 6 cells). Currents became significantly reduced (p < 0.05, unpaired t test) in the presence of anti-Kv2.1 antibody as compared with controls after 200 s dialysis. The inset shows an example of a family of currents evoked in response to voltage steps from −100 to +80 mV in 20 mV increments.
Western Blotting
Protein concentrations in cleared cell lysates (200 μl) were determined using a BCA assay kit according to manufacturers' instructions (Pierce). Four volumes of cell lysate sample were added to one volume of 5× sample buffer (60 mm Tris-Cl, pH 6.8, 2% SDS, 10% glycerol, 5% 2-mercaptoethanol, 0.01% bromphenol blue). Samples (20–30 μg protein) were loaded on to polyacrylamide-SDS gels (0.75-mm thick, 7% for high MW proteins or 12.5% for mid range MW proteins) and separated at 200 V (45 min) before being transferred on to PVDF membranes (30 V, overnight). Membranes were blocked with 5% nonfat milk protein in TBS-Tween (TBST, 0.05%) for 1 h and immunostained with the appropriate antibodies (1:500 dilution; anti-HO-1, Santa Cruz Biotechnology, UK, or anti-Kv2.1 antibody, Neuromab, Davis, CA) in 1% nonfat milk protein in TBST (3 h at room temperature). Membranes were then washed (3 × 5 min in TBST) and afterward incubated (1 h at room temperature) with secondary antibody conjugated to horseradish peroxidase (anti-rabbit Ig for HO-1 or anti-mouse Ig for anti-Kv2.1, both 1:2000 dilution; Amersham Biosciences UK). Finally, membranes were again washed in TBST (3 × 5 min) and immunoreactive bands visualized using the enhanced chemiluminescence (ECL) detection system and hyperfilm ECL according to manufacturers' instructions (GE Healthcare, UK). Band intensities were measured using Scion Image analysis software.
Electrophysiology
Fragments of coverslip with attached cells were transferred to a perfused (3–5 ml/min) recording chamber mounted on the stage of a Nikon T1-SM inverted microscope (Nikon, Tokyo, Japan). K+ currents were monitored by whole-cell voltage-clamp recordings. The standard perfusate (pH 7.4, 22 ± 1 °C) was composed of (mm): 140 NaCl, 4 KCl, 1 MgCl2, 10 HEPES, 2 CaCl2, and 10 glucose. Patch pipette resistance was 4–6 MΩ and series resistance was compensated (60–80%) after breaking into the whole-cell configuration. The pipette solution (pH 7.2) consisted of (mm): 140 KCl, 5 EGTA, 2 MgCl2, 1 CaCl2, 10 HEPES, and 10 glucose. Two voltage protocols were adopted: 1) a series of depolarizing steps from −100 to +80 mV in 10 mV increments for 500 ms; and 2) a repeated step to +50 mV from −80 mV for 100 ms. Signals were sampled at 10 kHz and low-pass filtered at 2 kHz. Voltage-clamp and analysis protocols were performed using an Axopatch 200A amplifier/Digidata 1200 interface (Axon Instruments, Foster City, CA) controlled by Clampex 9.0 software (Molecular Devices, Foster City, CA). Offline analysis was performed using Clampfit 9.0 (Molecular Devices). Current densities were calculated by measuring current amplitudes over the last 10 ms of a depolarizing step, when they were observed to have reached a steady-state value, then dividing these measured currents by each cell's capacitance as determined from calibrated analog offset controls.
In some experiments, apoptosis of coverslip-attached cells was induced by oxidative stress following a 5 min exposure (at 37 °C, 95% air, and 5% CO2) to either diamide (200 μm) or DTDP (100 μm). Following this insult, cells were washed with PBS and returned to fresh media containing 10 μm 1,3-boc-aspartyl-(ome)-fluoromethylketone (BAF; a caspase inhibitor) alone or, in some cases, together with the p38 inhibitor SB203580 (10 μm; Ascent scientific, Cambridge, UK). Inclusion of BAF allowed cells to remain viable for electrophysiological recordings, which were made 1–3 h after the initial oxidant exposure as previously described (12). To confirm the functional activity of Kv2.1 in DAOY cells, an anti-Kv2.1 antibody (Neuromab, Davis, CA) was added to the intracellular solution to give a final concentration of 0.5 μg/ml. An anti-Kv4.3 antibody was employed as control at the same concentration. CO was applied using the commercially-available CO-donor CORM-2 (tricarbonyldichlororuthenium (II) dimer, Sigma-Aldrich), freshly prepared in dimethyl sulfoxide (DMSO) to obtain a 30 mm stock and further diluted in bath perfusate to give a final concentration of 30 μm. The inactive form of the compound (iCORM) was used as a negative control as previously described (12).
Induction of HO-1 in DAOY Cells
HO-1 expression was induced in DAOY cells by three different means; hemin (Fe protoporphyrin IX; Merck Chemicals), cobalt protoporphyrin IX (CoPPIX; Sigma-Aldrich) and hypoxia. For studies using either hemin or CoPPIX, cells were initially seeded in 6-well plates for 24 h prior to HO-1 induction. Stock solutions for both chemicals (100 mm) were prepared in 0.1 n NaOH, and added to cells to achieve a working concentration of 1–200 μm. Following chemical exposure, cells were cultured for a further 24 h. For hypoxia-mediated HO-1 induction, cells were initially cultured to sub-confluence in 25 cm2 flasks and then transferred to a hypoxic work station (Ruskinn Life Sciences,) for an additional 3 to 24 h (0.5% O2:5% CO2:94.5% N2; 37 °C).
Generation of Inducible HO-1 shRNA Expression in DAOY Cells
The tetracycline-inducible Knock-outTM Single Vector Inducible RNAi System (Clontech), was employed to suppress HO-1 using shRNA, under the control of the U6 promoter. Oligonucleotides were custom-designed and HO-1-shRNA oligonucleotide generation was confirmed by ethidium bromide gel electrophoresis. Successful cloning of HO-1-shRNA into the pSingle-tTS-shRNA vector, via 5′-XhoI and 3′-HindIII restriction sites, was confirmed by restriction analysis and sequencing. Cells were transfected with the construct using the Genejammer transfection reagent (Stratagene) and stable DAOY/HO-1(shRNA) cell lines obtained by antibiotic selection (G-418, 1 mg/ml, Invitrogen, Paisley, UK). Individual colonies were picked and propagated. HO-1 expression was induced with CoPPIX (see Fig. 5B), either in the absence (control) or additional presence of doxycycline (Dox, 2 μg/ml) to induce HO-1-shRNA expression, and measured by Western blotting. G418 selection was maintained throughout the entire cloning process at 1 mg/ml then subsequently reduced to 400 μg/ml in all subsequent passages of cells once stable clones had been positively identified.
FIGURE 5.

HO-1 induction protects DAOY cells against oxidative stress. A, bar graph showing the mean (± S.E. bars) effect of oxidative stress (DTDP exposure, 0–500 μm) on DAOY cell viability. Cells were either untreated (control) or had HO-1 expression induced following incubation with hemin (200 μm, 24 h), CoPPIX (100 μm, 24 h), or hypoxia (0.5% O2, 16 h), as indicated. B, bar graph showing the mean effect on viability of diamide in DAOY cells stably expressing doxycycline-inducible HO-1 targeting shRNA. Cells were pre-exposed to hemin (200 μm, 48 h) with or without doxycycline (2 μg/ml), as indicated. The inset shows Western blot of HO-1 levels in control cells and in cells pre-exposed to hemin (200 μm, 48h) with or without doxycycline (2 μg/ml). C, bar graph showing the mean effect on viability of DAOY cells treated with diamide with and without the subsequent incubation of CORM-2 (100 μm, 3h) or iCORM (100 μm, 3 h). In A–C, each experiment was repeated three times in triplicate, and the mean number obtained each time was used for statistical analysis. (*, p < 0.05; **, p < 0.01, ***, p < 0.001; 2 way ANOVA with Bonferroni multiple-comparison test).
MTT Assay for Cell Viability
Cells were grown to confluence in 96-well plates and then exposed to varying concentrations of either diamide or 2,2′-dithiodipyridine (DTDP) for 10 min (37 °C, 95% air:5% CO2). This oxidative stress-inducing media was subsequently replaced with fresh medium (110 μl) containing thiazolyl blue tetrazolium bromide (MTT, 0.5 mg/ml), with or without additional drugs, and then left for a further 3 h in standard culture conditions. Following this incubation, an equal volume of acidified isopropanol solution (1 ml 1 m HCl in 24 ml isopropanol) was added to each well; the mixture then thoroughly triturated with the pipette to ensure all formazan crystals were dissolved. Finally, the plate was scanned using a spectrophotometer (wavelength of 570 nm).
Flow Cytometry
DAOY cells were cultured in 25 cm2 flasks for 24 h and then treated with various concentrations of either diamide or DTDP for 10 min. Following aspiration of this oxidative stress-inducing media, cells were then exposed to fresh media containing CORM-2 (100 μm) for an additional 3 h in standard culture conditions. Next, cells were washed twice with PBS, detached using 0.05% trypsin-EDTA (0.5 ml) and then resuspended in media (4.5 ml) prior to centrifugation (200 rpm for 2 min). Cell pellets were washed twice using PBS (2 ml) and apoptotic or necrotic cells were detected using the ApoScreen Annexin V Apoptosis Kit (Southern Biotech, UK) as instructed by the manufacturer. Specifically, cells were first resuspended in 1× binding buffer (100 μl) and then incubated in the dark (15 min) following the addition of annexin V-FITC (10 μl). Propidium iodide (PI, 10 μl) was added to this mixture, the stained cellular suspension then analyzed using the FACS Calibur system (BD Biosciences). Control cells were not exposed to oxidative stress. CellQuest software packages were used for cell analysis, cells were sorted into 4 groups: 1) healthy cells [FITC(−), PI(−)], 2) apoptotic cells [FITC(+), PI(−)], 3) necrotic cells (denoted as FITC(+), PI(+)), and 4) completely dead cells (FITC(−), PI(+)).
TUNEL Assay
As an additional assessment of apoptosis, TdT-mediated dUTP-biotin nick end labeling (TUNEL) assays were performed. DAOY cells were grown on coverslips in a 6-well plate (24 h) and then subjected to oxidative stress using either diamide or DTDP as above; culture medium was aspirated, replaced with fresh medium alone, or supplemented with either diamide (200 μm) or DTDP (100 μm), and then returned to a humidified incubator (37 °C, 95% air:5% CO2) for 5 min. This oxidative stress-inducing media was then replaced with fresh culture media and incubated for a further 1 h. Next, cells were gently washed with PBS, fixed with a 4% paraformaldehyde-PBS solution (10 min), washed again, and then permeabilized (20 min) with Cytonin (Trevigen, Gaithersburg, MD). The TUNEL assay was performed using a commercially available TACS 2 TdT-fluor in situ apoptosis detection kit (Trevigen) as per instructions. Coverslips were mounted on microscope slides using Vectashield containing DAPI (Vector Laboratories, Burlingame, CA), sealed with nail varnish, and then examined on a Zeiss LSM 510 confocal microscope (488- and 351-nm lasers). At least 10 separate fields of view, containing an average of 20 cells, were acquired at ×40 magnification for each coverslip examined. All TUNEL-positive cells (green fluorescence in the nucleus) were counted in each field of view and subsequently expressed as a percentage of the total number of nuclei in that same field.
Statistical Analysis
Data were analyzed using Origin (Northampton, MA) and Excel (Microsoft, UK) software. Data are presented as mean ± S.E. Statistical comparisons were made using paired Student's t tests or one-way ANOVA with Bonferroni's multiple-comparison test, as appropriate, with p < 0.05 taken as statistically significant in each case (Graphpad Prism).
RESULTS
To determine whether medulloblastoma tissue expressed the pro-apoptotic K+ channel Kv2.1, or the cytoprotective enzyme HO-1, we performed immunohistochemistry on fixed sections of medulloblastoma tissue (Fig. 1). Both proteins were clearly detectable in most if not all cells by this method and, as highlighted by the nuclear stain DAPI, HO-1, and Kv2.1 co-localized largely at the cell periphery. Identification of the presence of both Kv2.1 and HO-1 prompted us to consider further the possibility that Kv2.1 activity might influence apoptosis, and this in turn may be regulated by HO-1, in human medulloblastoma tissue.
FIGURE 1.

Medulloblastoma tissue expresses HO-1 and Kv2.1. Immunohistological images of a section of human medulloblastoma tissue which had been formalin-fixed and embedded in paraffin wax. In the upper four panels the section was immunostained using primary antibodies against HO-1 (green, top left image), Kv2.1 (red, top right image), and stained with DAPI to highlight nuclei (blue, bottom left image). All images were merged to form the bottom right image, which indicates co-localization (yellow) of HO-1 and Kv2.1. In the lower four panels, images were taken from a separate section under identical filter conditions, but primary antibodies against HO-1 and Kv2.1 were omitted.
To test this hypothesis mechanistically, we next investigated whether or not these two key proteins, HO-1 and Kv2.1, were also expressed in the human medulloblastoma cell line, DAOY. Our initial investigations consistently failed to detect HO-1 by Western blotting when cells were maintained under standard culture condition (Fig. 2, A–D). However, this enzyme could be induced by both hemin and CoPPIX, known inducers of HO-1 expression in other tissues, in a concentration-dependent manner (Fig. 2, A and B; note, however, that at the highest concentration (200 μm) CoPPIX was toxic to cells). One possible reason to account for the lack of constitutive expression of HO-1 in DAOY cells, yet clear expression in medulloblastoma tissue (Fig. 1), is the fact that hypoxia is a key feature of the tumor microenvironment, yet DAOY cells were cultured at high levels of ambient O2 levels (20%) typically encountered in tissue culture. To investigate this possibility further, we exposed cells to hypoxic conditions (0.5% O2, 3–24 h) typical of tumor microenvironments (21, 22) and found robust, time-dependent induction of HO-1 in these cells (Fig. 2C), suggesting that the hypoxic conditions, which are likely to occur within medulloblastoma tumors, may act as the signal for constitutive HO-1 expression. Fig. 2D compares the relative levels of expression of HO-1 induced by these 3 stimuli at concentrations employed in subsequent functional studies (see Fig. 5A).
FIGURE 2.

HO-1 induction in DAOY cells. A, example Western blot demonstrating induction of HO-1 expression (upper) following 24 h exposure to hemin at the concentrations indicated. C, control, NaOH, vehicle control. Below; bar graph showing relative mean induction (± S.E. bars, n = 3) from quantification of blots normalized to the induction produced by 200 μm hemin. B, as A, except HO-1 was induced by 24 h exposure to CoPPIX at the concentrations indicated. Below; bar graph showing relative mean induction (± S.E. bars, n = 3) from quantification of blots normalized to the induction produced by 100 μm CoPPIX. C, example Western blot showing induction of HO-1 following exposure of cells to hypoxia (0.5% O2) for the indicated periods of time. Also shown for comparison is the induction of HO-1 caused by 200 μm hemin. Below; bar graph showing relative mean induction (± S.E. bars, n = 3) caused by hypoxia from quantification of blots normalized to the induction produced by 200 μm hemin. D, example Western blot (upper) comparing HO-1 expression in DAOY cells in response to optimum levels of hypoxia, hemin, and CoPPIX used in subsequent functional studies. The bar graph plots the mean ± S.E. bars induction caused by hypoxia (16 h, n = 4), hemin (200 μm; n = 5), and CoPPIX (100 μm; n = 5) on HO-1 expression, normalized to the inductive effect of 100 μm CoPPIX. β-Actin loading controls are also shown for each panel.
Western blotting results suggested Kv2.1 was poorly expressed in DAOY cells (Fig. 3A). However, a clear band (MW ∼125 kDa) was detected following immunoprecipitation, suggesting this channel was indeed expressed in DAOY cells (the negative control followed exactly the same protocol but with the omission of the anti-Kv2.1 antibody). Furthermore, using whole-cell patch-clamp recordings, we identified outward, voltage-gated K+ currents in DAOY cells. These were small in amplitude (8.7 ± 0.7 pA/pF at +50mV, n = 30 cells, e.g. Fig. 3, B and C), but could be inhibited by both tetraethylammonium (TEA; 5 mm reversibly inhibited currents by 48.7 ± 5.6%, n = 12, p < 0.001, e.g. Fig. 3B, left) and 4-aminopyridine (4-AP; 10 mm 4-AP inhibited currents by 46.2 ± 5.4%, n = 10, p < 0.01, e.g. Fig. 3B, right). Sensitivity to these inhibitors is consistent with Kv2.1 contributing to these outward currents, but their lack of selectivity among different K+ channel types discounts them from defining the presence of Kv2.1. However, we found that intracellular dialysis with an anti-Kv2.1 antibody, previously shown to inhibit Kv2.1 selectively in hippocampal neurones (12) caused a time-dependent decrease in current amplitudes (∼50% of control levels, n = 6 cells, Fig. 3C), suggesting that Kv2.1 is indeed a major contributor to the total whole-cell K+ current seen in these cells. By contrast, intracellular dialysis with an anti-Kv4.3 antibody was without effect (Fig. 3C).
To investigate the possible role(s) of HO-1 and Kv2.1 in the vulnerability of DAOY cells to apoptosis, we next investigated the sensitivity of these cells to oxidative stress. Fig. 4A demonstrates the concentration-dependent effects of two established oxidants (diamide; left, and DTDP; right) on DAOY cell viability, determined using the MTT assay. Clearly, both agents were capable of causing cell death, but this assay does not identify the specific mechanism underlying oxidant-induced cell death. To probe this further, we treated cells with diamide (200 μm for 10min), double stained the cells with annexin V-FITC and PI, and then subjected them to FACS analysis. Diamide exposure markedly increased the proportion of cells that stained positively with both FITC and PI (Fig. 4B), indicating that a large fraction of cells were entering late apoptosis following such oxidant exposure. Finally, TUNEL analysis of both diamide and DTDP-treated cells showed a striking increase in the number of TUNEL-positive cells when compared with untreated cells (Fig. 4C). Collectively, these data indicate that oxidative stress leads to DAOY cell death via apoptosis.
FIGURE 4.

Oxidative stress induces apoptosis in DAOY cells. A, concentration-response relationships for the effects of diamide (left) and DTDP (right) on DAOY cells viability. Cells were treated for 10 min as described under “Experimental Procedures” and then assessed for viability using MTT assay. Each data point is the mean (± S.E. bars) taken from 3 measurements in each case. Data were fitted using Origin 7.5 software, yielding IC50 values of 256.8 ± 15.7 μm for diamide and 180.0 ± 28.2 μm for DTDP. B, flow cytometry analysis of DAOY cells under control conditions (left) and following oxidative stress (right; 100 μm DTDP for 10 min). Cells were stained with FITC-labeled annexin V and PI. Late apoptotic cells are located in the upper right quadrant of the two displays. C, TUNEL assay analysis of DAOY cells under control conditions (left, upper image) and during oxidative stress (200 μm diamide; lower image). Fluorescent images show DAPI-stained DAOY nuclei (blue) superimposed on specks of green, indicative of positive TUNEL staining for DNA fragmentation. Right: percentage of TUNEL-positive cells following treatment with DTDP (100 μm) or diamide (200 μm) versus untreated control cells (n = 10 fields of view for each coverslip; ***, p 0.001; 1-way ANOVA with Bonferroni multiple-comparison test).
Fig. 5A illustrates the effects of HO-1 induction on the sensitivity of DAOY cells to oxidant-induced death. Importantly, increased resistance to oxidant-induced death was observed following HO-1 induction regardless of the means by which the enzyme was induced (i.e. in cells treated with hemin, CoPPIX, or hypoxia). It is noteworthy that the greatest resistance to oxidative stress was observed in CoPPIX-treated cells (Fig. 5A), correlating with the most striking induction of HO-1 (Fig. 2). In further support of the idea that HO-1 affords protection against oxidative stress, we generated a line which stably expressed doxycycline-inducible shRNA directed against HO-1. HO-1 was induced by 48h exposure to hemin (200 μm). Induced expression of HO-1 was reduced (although not completely) in the presence of doxycycline (2 μg/ml), and this was associated with an increased vulnerability to oxidative stress (Fig. 5B).
These findings prompted us to investigate whether increased levels of CO, arising primarily from increased HO-1 expression, might account for the improved resistance to apoptosis. To do so, we exposed cells to the established CO-releasing molecule, CORM-2. As illustrated in Fig. 5C, cells were significantly more resistant to oxidative stress in the presence of CORM-2, but not in the presence of iCORM. These findings suggest the protective effect of HO-1 induction (Fig. 5A) may arise at least in part to the increased levels of CO resulting from increased HO-1 expression.
Whole-cell patch-clamp recordings revealed that voltage-gated K+ currents in DAOY cells (to which Kv2.1 is a major contributor; Fig. 3) were reversibly inhibited by the CO-donor CORM-2 (30 μm). An example time course of the effects of CORM-2 is plotted in Fig. 6A. In several experiments, we constructed current voltage relationships in cells just before application of CORM-2, and then after 3 min of exposure; mean data from such studies are plotted in Fig. 6B. An identical approach was used to test the effects of the inactive compound, iCORM, which was without effect (Fig. 6C). Exposure of cells to either diamide or DTDP (for 1–3 h, in the presence of caspase inhibitor; see “Experimental Procedures”) caused a striking increase in K+ current amplitudes, and this effect could be largely suppressed in the presence of CORM-2 (Fig. 6D, applied acutely as for control cells). Thus, oxidative stress induces an increase in outward K+ currents in DAOY cells which can be largely prevented by CO.
FIGURE 6.
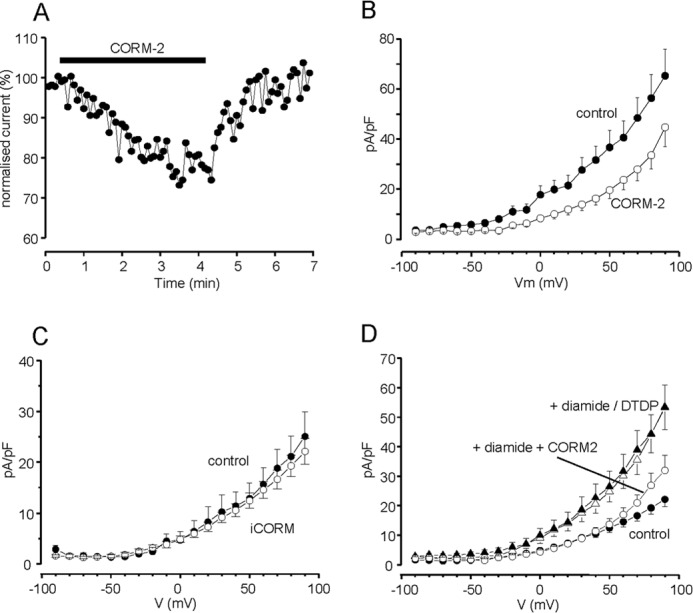
CO inhibits K+ currents and the oxidant-induced K+ current upsurge in DAOY cells. A, example time course of the reversible inhibitory effect of CORM-2. Currents were evoked by successive step depolarizations from −80 to +50 mV and CORM-2 (30 μm) was applied for the period indicated by the bar. Current amplitudes are normalized to those evoked by the first 10 depolarizations. B, mean (± S.E., n = 19 cells) current density versus voltage relationships in DAOY cells under control conditions (solid circles) and during exposure to CORM-2 (30 μm; open circles). CORM-2 caused significant reductions in current density (p < 0.05-p < 0.001, paired Student's t test) over the voltage range +10 to +90 mV. C, mean (± S.E., n = 10 cells) current density versus voltage relationships in DAOY cells under control conditions (solid circles) and during exposure to iCORM (30 μm; open circles). D, mean (± S.E.) current density versus voltage relationships determined in control cells (solid circles; n = 17), following exposure to 100 μm DTDP (solid triangles, n = 12) or 200 μm diamide (open triangles, n = 10) and following exposure to diamide in the presence of 30 μm CORM-2 (open circles, n = 8), as indicated. Effects of oxidants were significant (p < 0.05 - p < 0.01; unpaired t-tests), but diamide was without significant effect in the presence of CORM-2.
Given the significant presence of Kv2.1 in these cells, and the known role of Kv2.1 in apoptosis, these data are consistent with the hypothesis that CO may mediate the anti-apoptotic effects of HO-1 in these cells via inhibition of Kv2.1. To investigate this in more detail, we examined the ability of CORM-2 to inhibit residual K+ currents following dialysis with the anti-Kv2.1 antibody (as in Fig. 3C). Fig. 7 shows that, following the decrease in amplitude caused by intracellular anti-Kv2.1 antibody, extracellular application of 30 μm CORM-2 was without further significant effect, strongly suggesting that its inhibitory effect is selective among the voltage-gated K+ channels expressed in DAOY cells for Kv2.1.
FIGURE 7.
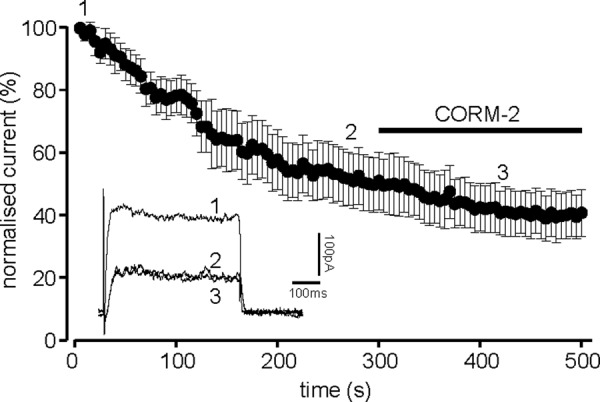
CO primarily inhibits Kv2.1 in DAOY cells. Normalized mean time course (± S.E. bars) of K+ current amplitudes evoked by successive step depolarizations from −90 to +50 mV (100 ms). Cells were dialyzed with a pipette solution containing an anti-Kv2.1 antibody (0.5 μg/ml, n = 6 cells). For the period indicated by the horizontal bar, the perfusate was switched for one containing 30 μm CORM-2. The inset shows example currents evoked in one of the dialyzed cells at the time points indicated by the numbers.
As further evidence to support this hypothesis, we investigated the involvement of p38 MAPK: phosphorylation of Kv2.1 at S800 by this kinase has previously been suggested to be an absolute requirement for the oxidant-induced increase in the plasma membrane localization of this channel (see Introduction). As illustrated in Fig. 8A, the dramatic diamide-induced increase in outward K+ current in DAOY cells was fully prevented in the presence of the p38 MAPK inhibitor, SB203580. This inhibitor was itself without direct effect on K+ currents in DAOY cells (Fig. 8B). Importantly, SB203580 provided significant protection against the oxidant-induced loss of cell viability (Fig. 8C), a finding which provides additional support for the idea that increased surface expression of Kv2.1 is an important early step in oxidative stress-induced apoptosis in DAOY cells.
FIGURE 8.
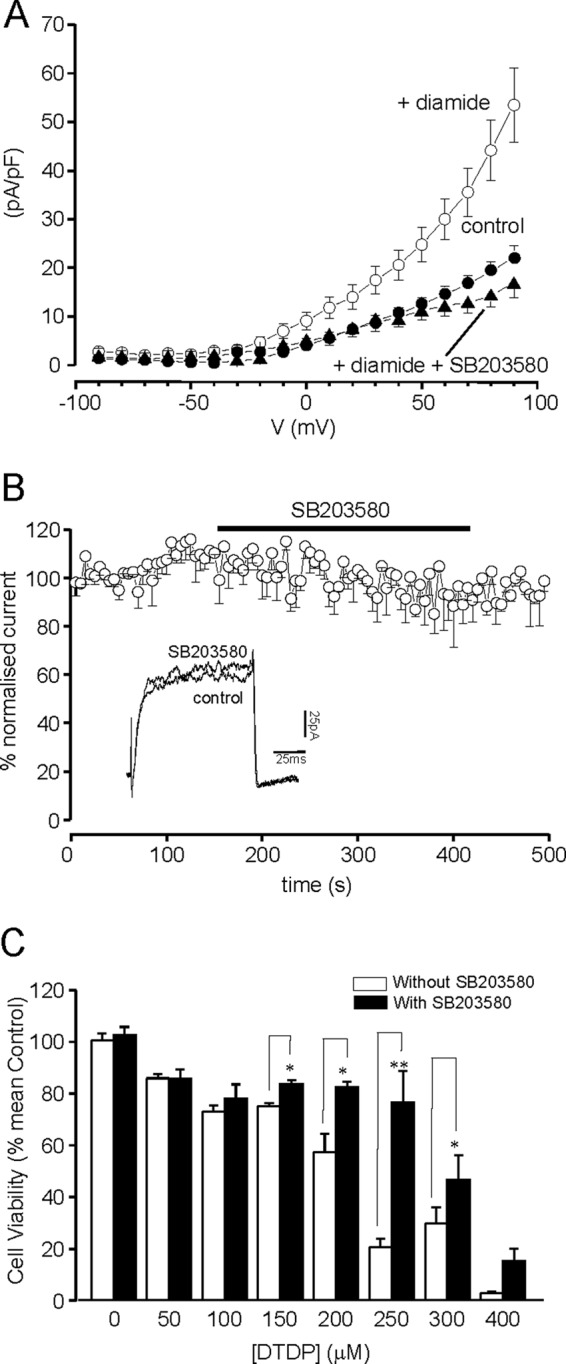
Inhibition of Kv2.1 phosphorylation with the selective inhibitor p38 MAPK inhibits the oxidant-induced upsurge of K+ current and protects DAOY cells against oxidative stress. A, mean (± S.E.) current density versus voltage relationships determined in control cells (solid circles, n = 10), following exposure to diamide (open circles; n = 10) and following exposure to diamide in the presence of p38 MAPK inhibitor (SB203580, 10 μm, closed triangles, n = 8), as indicated. Effects of SB203580 on the upsurge of K+ currents was significant (p < 0.05 to p < 0.001) over the voltage range +10 to +90 mV. B, normalized mean time course (± S.E. bars, n = 6) of K+ current amplitudes evoked by successive step depolarizations from −90 to +50 mV (100 ms). For the period indicated by the horizontal bar, the perfusate was switched for one containing 10 μm SB203580. C, bar graph showing the mean (± S.E. n = 3 experiments each performed in triplicate) cell viability under oxidative stress, with and without the p38 MAPK inhibitor (applied for 3 h). *, p < 0.05; **, p < 0.01; one way ANOVA with Bonferroni multiple-comparison test.
DISCUSSION
The present study identifies a Kv2.1-mediated apoptotic pathway in human medulloblastoma cells which can be regulated via the expression of HO-1, and therefore has the potential for beneficial therapeutic intervention. In central neurones the role of Kv2.1 as a pro-apoptotic K+ channel has developed in recent years and our current understanding is that oxidative stress causes a rapid increase in Kv2.1 channel insertion in the plasma membrane. This process is initiated by mobilization of intracellular Zn2+ (23) and requires p38 MAP kinase phosphorylation at Ser-800, as well as an additional Src kinase-mediated phosphorylation of an N-terminal tyrosine (Y124) (13). Our recent work indicated that CO could suppress this apoptotic pathway in central neurones by blocking Kv2.1 activity, an effect which may be important in certain pathological neurological conditions (such as neurodegenerative diseases), when HO-1 induction is apparent (12). Here, we provide a body of evidence to suggest strongly that CO can constitutively provide protection for medulloblastoma cells against Kv2.1-mediated apoptosis.
Our data show, for the first time, that human medulloblastoma tissue constitutively expresses HO-1 (Fig. 1). Thus, this tumor type can now be included in a growing list of cancer types known to express this enzyme constitutively (15, 24). Regulation of HO-1 expression in tumors is not fully understood, but in other tissues numerous stresses can induce the enzyme, and one likely factor is local oxygen availability: hypoxia is a key feature of the tumor microenvironment (21, 22, 25, 26). HO-1 expression in cancerous and non-cancerous tissue is controlled by the transcriptional regulator nuclear factor (erythroid-derived 2)-related factor 2 (Nrf2), the activity of which is increased in hypoxia by a seemingly paradoxical increase in oxidative stress (27, 28). Fig. 2 demonstrates that HO-1 is not expressed the medulloblastoma cell line DAOY under normal (relatively hyperoxic) tissue culture conditions, but its expression is dramatically induced by hypoxia. It is conceivable, therefore, that tumor hypoxia in vivo contributes to the constitutive expression of HO-1 in medulloblastoma tissue.
Several studies indicate that HO-1 provides cytoprotection and is anti-apoptotic in tumor cells. Increased HO-1 expression augments cell survival of numerous cancer types (15, 24). Furthermore, experimentally suppressed HO-1 expression reduces tumor cell viability (29, 30). This is of clinical significance as HO-1 inhibition has been shown to increase vulnerability to radiotherapy or chemotherapy (31, 32). Additional studies have also shown that the influence of HO-1 on cell viability and resistance to therapy arises in part from its anti-apoptotic capabilities (33–35). The present study is in agreement with such a role for HO-1 as a cytoprotective agent in DAOY cells. While we have not evaluated the influence of HO-1 acting as an antioxidant (via its action to degrade heme and generate biliverdin) (36), we have identified a clear, novel role for CO in this regard. Thus, the protective effects of HO-1 induction against oxidative stress were mimicked by exposing cells to the CO donor, CORM-2 (Fig. 5).
Previous studies have examined the electrophysiological properties of medulloblastoma cells and derived lines, including DAOY (37, 38). It is clear from these reports that DAOY cells express a variety of different K+ channel types, and in principle any of these could be involved in the early stages of apoptosis (7, 39, 40). Interestingly, Ernest et al. (38) identified an acid-sensitive 'leak' K+ current, termed TASK, in DAOY cells, and similar channels (TREK-1) have also been identified in prostate cancer cells. We sought evidence for the presence of Kv2.1 in DAOY cells, since it is most closely associated with apoptosis (see Introduction and earlier “Discussion”) and influences apoptosis in neurones in a manner which is sensitive to CO (12). To this end, we identified voltage-gated K+ currents in DAOY cells (as reported recently (38)), which were TEA and 4-AP-sensitive, consistent with the known properties of Kv2.1. Current density was low in these cells, so identification of the Kv2.1 protein was only achieved after immunoprecipitation (Fig. 3A). However, perhaps the most convincing demonstration of its functional presence was the fact that intracellular dialysis of a selective anti-Kv2.1 antibody (12) caused marked current inhibition, indicating that a large fraction of the outward whole-cell current in these cells arose due to K+ flux via Kv2.1 channels. Immunohistochemistry also identified the channel in medulloblastoma sections (Fig. 1).
As shown in Fig. 6, CO inhibited the whole-cell K+ current in DAOY cells. Given the dominant contribution of Kv2.1 to this whole-cell current, we propose that CO inhibits Kv2.1 in these cells. Furthermore, oxidative stress selectively increases the presence of these channels in the plasma membrane of neurones (8, 11, 12), and we found that oxidant-induced K+ current augmentation was largely reversed by CO (Fig. 6D), further supporting the idea that CO inhibits these channels in DAOY cells. Interestingly, CO does not modulate TASK channels, and actually augments TREK channels (41). Thus, although we cannot demonstrate complete selectivity for CO in its ability to inhibit Kv2.1, it is unlikely that modulation of these leak channels contributes to the observed effects of CO, and hence to its protective effects against apoptosis.
While we have demonstrated that CO inhibits Kv2.1 in DAOY cells, and that HO-1 induction or exogenous CO application provides protection against oxidative stress, these findings do not in themselves demonstrate directly that the protective effects of CO are mediated by its ability to inhibit K+ channels. However, results obtained with the p38 MAPK inhibitor SB203580 suggest that K+ channel inhibition is indeed responsible for such protective effects of CO. Thus, SB203580 almost completely inhibited the oxidant-induced increase in K+ current and also enhanced resistance to oxidative stress (Fig. 8). Since phosphorylation of Kv2.1 by p38 MAPK is required for its insertion into the membrane in response to oxidative stress (8), and inhibition of p38 was both anti-apoptotic and prevented K+ current increases, it is likely that the inhibition of Kv2.1 by CO in DAOY cells contributes significantly to the increased resistance to apoptosis attributable to HO-1.
In summary, our findings indicate that CO is anti-apoptotic in DAOY medulloblastoma cells via its ability to inhibit Kv2.1. These findings can account for the protective effect of increased HO-1 expression which is a common feature of many tumor types including (as we show here) medulloblastoma. Interestingly, HO-1 expression may be high in tumor cells in part because of the hypoxic microenvironment but can be increased further by the cellular stresses imposed by chemotherapeutic agents (reviewed by Ref. 15, 24). We suggest that resistance to therapy of some tumors may be due to increased endogenous CO production which prevents the early stages of apoptosis. Our findings support the growing concept that pharmacological targeting of HO-1 is likely to be beneficial in the treatment of some cancers (15, 16, 24), and may enhance the efficacy of current radiotherapy or chemotherapy. They also highlight a role for the Kv2.1 channel in apoptotic regulation of cancer cells and, consequently, a new target for therapeutic development.
This study was supported by Yorkshire Cancer Research, UK.
- HO-1
- heme oxygenase-1
- Dox
- doxycycline
- BAF
- 1,3-boc-aspartyl-(ome)-fluoromethylketone
- PI
- propidium iodide.
REFERENCES
- 1. Hanahan D., Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 2. Kelly G. L., Strasser A. (2011) The essential role of evasion from cell death in cancer. Adv. Cancer Res. 111, 39–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hanahan D., Weinberg R. A. (2000) The hallmarks of cancer. Cell 100, 57–70 [DOI] [PubMed] [Google Scholar]
- 4. Cory S., Adams J. M. (2002) The Bcl2 family: regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2, 647–656 [DOI] [PubMed] [Google Scholar]
- 5. Yu S. P., Canzoniero L. M., Choi D. W. (2001) Ion homeostasis and apoptosis. Curr. Opin. Cell Biol. 13, 405–411 [DOI] [PubMed] [Google Scholar]
- 6. Yu S. P. (2003) Regulation and critical role of potassium homeostasis in apoptosis. Prog. Neurobiol. 70, 363–386 [DOI] [PubMed] [Google Scholar]
- 7. Prevarskaya N., Skryma R., Shuba Y. (2010) Ion channels and the hallmarks of cancer. Trends Mol. Med 16, 107–121 [DOI] [PubMed] [Google Scholar]
- 8. Redman P. T., He K., Hartnett K. A., Jefferson B. S., Hu L., Rosenberg P. A., Levitan E. S., Aizenman E. (2007) Apoptotic surge of potassium currents is mediated by p38 phosphorylation of Kv2.1. Proc. Natl. Acad. Sci. U.S.A. 104, 3568–3573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang Z. (2004) Roles of K+ channels in regulating tumour cell proliferation and apoptosis. Pflugers Arch. 448, 274–286 [DOI] [PubMed] [Google Scholar]
- 10. Lehen'kyi V., Shapovalov G., Skryma R., Prevarskaya N. (2011) Ion channels in control of cancer and cell apoptosis. Am. J. Physiol. Cell Physiol. 301, 1281–1289 [DOI] [PubMed] [Google Scholar]
- 11. Pal S., Hartnett K. A., Nerbonne J. M., Levitan E. S., Aizenman E. (2003) Mediation of neuronal apoptosis by Kv2.1-encoded potassium channels. J. Neurosci. 23, 4798–4802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dallas M. L., Boyle J. P., Milligan C. J., Sayer R., Kerrigan T. L., McKinstry C., Lu P., Mankouri J., Harris M., Scragg J. L., Pearson H. A., Peers C. (2011) Carbon monoxide protects against oxidant-induced apoptosis via inhibition of Kv2.1. FASEB J. 25, 1519–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Redman P. T., Hartnett K. A., Aras M. A., Levitan E. S., Aizenman E. (2009) Regulation of apoptotic potassium currents by coordinated zinc-dependent signaling. J. Physiol. 587, 4393–4404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fang J., Akaike T., Maeda H. (2004) Antiapoptotic role of heme oxygenase (HO) and the potential of HO as a target in anticancer treatment. Apoptosis. 9, 27–35 [DOI] [PubMed] [Google Scholar]
- 15. Jozkowicz A., Was H., Dulak J. (2007) Heme oxygenase-1 in tumors: is it a false friend? Antioxid. Redox. Signal. 9, 2099–2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kinobe R. T., Dercho R. A., Nakatsu K. (2008) Inhibitors of the heme oxygenase - carbon monoxide system: on the doorstep of the clinic? Can. J. Physiol. Pharmacol. 86, 577–599 [DOI] [PubMed] [Google Scholar]
- 17. Wu L., Wang R. (2005) Carbon monoxide: endogenous production, physiological functions, and pharmacological applications. Pharmacol. Rev. 57, 585–630 [DOI] [PubMed] [Google Scholar]
- 18. Kim H. P., Ryter S. W., Choi A. M. (2006) CO as a cellular signaling molecule. Annu. Rev. Pharmacol. Toxicol. 46, 411–449 [DOI] [PubMed] [Google Scholar]
- 19. Taillandier L., Blonski M., Carrie C., Bernier V., Bonnetain F., Bourdeaut F., Thomas I. C., Chastagner P., Dhermain F., Doz F., Frappaz D., Grill J., Guillevin R., Idbaih A., Jouvet A., Kerr C., Donadey F. L., Padovani L., Pallud J., Sunyach M. P. (2011) Medulloblastomas: review. Rev. Neurol. 167, 431–448 [DOI] [PubMed] [Google Scholar]
- 20. Peña-Altamira E., Petazzi P., Contestabile A. (2010) Nitric oxide control of proliferation in nerve cells and in tumor cells of nervous origin. Curr. Pharm. Des. 16, 440–450 [DOI] [PubMed] [Google Scholar]
- 21. Vaupel P. (2004) Tumor microenvironmental physiology and its implications for radiation oncology. Semin. Radiat. Oncol. 14, 198–206 [DOI] [PubMed] [Google Scholar]
- 22. Brahimi-Horn M. C., Pouysségur J. (2007) Hypoxia in cancer cell metabolism and pH regulation. Essays Biochem. 43, 165–178 [DOI] [PubMed] [Google Scholar]
- 23. Sensi S. L., Paoletti P., Koh J. Y., Aizenman E., Bush A. I., Hershfinkel M. (2011) The Neurophysiology and pathology of brain zinc. J. Neurosci. 31, 16076–16085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Was H., Dulak J., Jozkowicz A. (2010) Heme oxygenase-1 in tumor biology and therapy. Curr. Drug Targets. 11, 1551–1570 [DOI] [PubMed] [Google Scholar]
- 25. Harris A. L. (2002) Hypoxia–a key regulatory factor in tumour growth. Nat. Rev. Cancer 2, 38–47 [DOI] [PubMed] [Google Scholar]
- 26. Wilson W. R., Hay M. P. (2011) Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 11, 393–410 [DOI] [PubMed] [Google Scholar]
- 27. Kim Y. J., Ahn J. Y., Liang P., Ip C., Zhang Y., Park Y. M. (2007) Human prx1 gene is a target of Nrf2 and is up-regulated by hypoxia/reoxygenation: implication to tumor biology. Cancer Res. 67, 546–554 [DOI] [PubMed] [Google Scholar]
- 28. Miyata T., Takizawa S., van de Ypersele S. C. (2011) Hypoxia. 1. Intracellular sensors for oxygen and oxidative stress: novel therapeutic targets. Am. J. Physiol. Cell Physiol. 300, C226–C231 [DOI] [PubMed] [Google Scholar]
- 29. Mayerhofer M., Florian S., Krauth M. T., Aichberger K. J., Bilban M., Marculescu R., Printz D., Fritsch G., Wagner O., Selzer E., Sperr W. R., Valent P., Sillaber C. (2004) Identification of heme oxygenase-1 as a novel BCR/ABL-dependent survival factor in chronic myeloid leukemia. Cancer Res. 64, 3148–3154 [DOI] [PubMed] [Google Scholar]
- 30. Busserolles J., Megías J., Terencio M. C., Alcaraz M. J. (2006) Heme oxygenase-1 inhibits apoptosis in Caco-2 cells via activation of Akt pathway. Int. J. Biochem. Cell Biol. 38, 1510–1517 [DOI] [PubMed] [Google Scholar]
- 31. Berberat P. O., Dambrauskas Z., Gulbinas A., Giese T., Giese N., Künzli B., Autschbach F., Meuer S., Büchler M. W., Friess H. (2005) Inhibition of heme oxygenase-1 increases responsiveness of pancreatic cancer cells to anticancer treatment. Clin. Cancer Res. 11, 3790–3798 [DOI] [PubMed] [Google Scholar]
- 32. Fang J., Sawa T., Akaike T., Greish K., Maeda H. (2004) Enhancement of chemotherapeutic response of tumor cells by a heme oxygenase inhibitor, pegylated zinc protoporphyrin. Int. J Cancer 109, 1–8 [DOI] [PubMed] [Google Scholar]
- 33. Chen G. G., Liu Z. M., Vlantis A. C., Tse G. M., Leung B. C., van Hasselt C. A. (2004) Heme oxygenase-1 protects against apoptosis induced by tumor necrosis factor-α and cycloheximide in papillary thyroid carcinoma cells. J. Cell Biochem. 92, 1246–1256 [DOI] [PubMed] [Google Scholar]
- 34. Liu Z. M., Chen G. G., Ng E. K., Leung W. K., Sung J. J., Chung S. C. (2004) Upregulation of heme oxygenase-1 and p21 confers resistance to apoptosis in human gastric cancer cells. Oncogene 23, 503–513 [DOI] [PubMed] [Google Scholar]
- 35. Tanaka S., Akaike T., Fang J., Beppu T., Ogawa M., Tamura F., Miyamoto Y., Maeda H. (2003) Antiapoptotic effect of haem oxygenase-1 induced by nitric oxide in experimental solid tumour. Br. J Cancer 88, 902–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gozzelino R., Jeney V., Soares M. P. (2010) Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 50, 323–354 [DOI] [PubMed] [Google Scholar]
- 37. Carignani C., Roncarati R., Rimini R., Terstappen G. C. (2002) Pharmacological and molecular characterisation of SK3 channels in the TE671 human medulloblastoma cell line. Brain Res. 939, 11–18 [DOI] [PubMed] [Google Scholar]
- 38. Ernest N. J., Logsdon N. J., McFerrin M. B., Sontheimer H., Spiller S. E. (2010) Biophysical properties of human medulloblastoma cells. J. Membr. Biol. 237, 59–69 [DOI] [PubMed] [Google Scholar]
- 39. Kunzelmann K. (2005) Ion channels and cancer. J. Membr. Biol. 205, 159–173 [DOI] [PubMed] [Google Scholar]
- 40. Voloshyna I., Besana A., Castillo M., Matos T., Weinstein I. B., Mansukhani M., Robinson R. B., Cordon-Cardo C., Feinmark S. J. (2008) TREK-1 is a novel molecular target in prostate cancer. Cancer Res. 68, 1197–1203 [DOI] [PubMed] [Google Scholar]
- 41. Dallas M. L., Scragg J. L., Peers C. (2008) Modulation of hTREK-1 by carbon monoxide. Neuroreport 19, 345–348 [DOI] [PubMed] [Google Scholar]