

Background: In ALS, FTD, and AD, TDP-43 is cleaved and mislocalized for unknown reasons.
Results: Cdc37 depletion causes TDP-43 clearance that can be blocked by tau over expression or beclin knockdown.
Conclusion: Cdc37 is essential for the stability of TDP-43 and can be affected by tau accumulation.
Significance: Normal TDP-43 turnover by the Cdc37/Hsp90 complex can be impaired by the emergence of tau co-pathology.
Keywords: Alzheimer Disease, Amyotropic Lateral Sclerosis (Lou Gehrig Disease), Autophagy, Chaperone Chaperonin, Tau
Abstract
The RNA-binding protein, trans-active response DNA-binding protein 43 (TDP-43), is normally found in the nucleus, but in amyotrophic lateral sclerosis, frontal temporal dementia, and some cases of Alzheimer disease it is cleaved and mislocalized to the cytosol, leading to accumulation. The mechanisms contributing to this are largely unknown. Here, we show that part of the normal clearance cascade for TDP-43 involves the Cdc37/Hsp90 complex. An Hsp90 inhibitor that disrupts the Cdc37/Hsp90 complex reduced TDP-43 levels to a greater extent than a standard Hsp90 ATPase inhibitor. When Cdc37 was depleted, TDP-43 underwent proteolytic clearance that was dependent on nuclear retrotranslocation and autophagic uptake. Accumulation of the microtubule-associated protein tau prevented the clearance of cleaved TDP-43, but not its production. This caused cleaved TDP-43 to accumulate, a feature observed in the brain of persons with Alzheimer disease. Clearance of cleaved TDP-43 was also prevented by knockdown of the autophagic inducer beclin1. Thus, in cells where TDP-43 clearance is normally needed, a system that employs manipulation of the Hsp90 complex and autophagy exists. But when tau accumulation is occurring, cleaved TDP-43 can no longer be cleared, perhaps explaining the emergence of these co-pathologies.
Introduction
TDP-434 has been linked to several neurodegenerative diseases including frontotemproral lobar degeneration (FTLD), amyotrophic lateral sclerosis (ALS), and Alzheimer disease (AD) (1–3). Specifically, it is the major component of ubiquinated inclusions in many cases of ALS and FTLD (2). Mutations in TDP-43 can cause neurodegeneration in ALS and FTLD (4, 5), but the extent of neurodegeneration caused by TDP-43 pathology in AD is not well understood. Normal function of TDP-43 is related to the binding of both mRNA and DNA, specifically regulating mRNA splicing, stability, translation, and transcription (6). In disease, TDP-43 abnormally aggregates, mislocalizes into the cytoplasm, and loses normal nuclear function (7, 8). One key feature of aggregated TDP-43 found in disease is that it is made up of truncated fragments (2, 7, 9). Cleavage of TDP-43 by caspase 3 or another protease may account for the mislocalization into the cytoplasm and eventual accumulation (1). In ALS and FTLD, TDP-43 pathology and its loss of function promote apoptosis, leading to neuronal toxicity (10). Part of this lost function could be due to its association with chaperone proteins. In fact, heat shock protein 90 (Hsp90) binds to TDP-43, suggesting that chaperones may contribute to the mislocalization and accumulation of TDP-43 in disease (11).
Hsp90 is a highly expressed molecular chaperone that regulates the maturation, activation, stability, and overall protein homeostasis of a variety of clients (12). Inhibitors of Hsp90 suspend the “opening and closing” of the chaperone thereby affecting client protein stabilization and activation (13–16). Hence, the client proteins are typically thought to undergo degradation by the ubiquitin-proteasome pathway (17, 18). Co-chaperones, such as Cdc37 and Aha1, help to regulate the chaperone cycle by assisting the recruitment of client proteins (14, 19–21). When Cdc37 or Aha1 is bound to the Hsp90 complex it can be considered a mature, folding complex that protects the functionality of the client; however, when Cdc37 or Aha1 is depleted, Hsp90 can triage clients for degradation (22, 23).
We speculated that altering the Hsp90 machinery could impact TDP-43 processing. Here, we show that disrupting the interaction of Hsp90 with Cdc37 facilitates caspase-mediated TDP-43 nuclear retrotranslocation, leading to TDP-43 autophagic clearance. This relationship was then investigated when tau levels were elevated or clearance mechanisms inhibited. Ultimately, these studies describe novel routes for TDP-43 clearance and pathogenesis that converge on a novel Cdc37/Hsp90 mechanism.
EXPERIMENTAL PROCEDURES
Antibodies, siRNAs, Plasmids, and Chemicals
PHF1 (anti-S396/S404 p-tau) was provided by P. Davies, Albert Einstein College of Medicine, Yeshiva University, New York. Cdc37 antibody was obtained from Cell Signaling (Danvers, MA). Anti-Tau (total tau) and Cdc37 (anti-mouse) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit anti-pS199/S202 tau was obtained from Anaspec (Fremont, CA). Anti-poly(ADP-ribose) polymerase, and anti-beclin1 antibodies were obtained from the Cell Signaling Apoptosis kit (Cell Signaling). Anti-GAPDH was obtained from Biodesign International (Saco, ME). Anti-Hsp90 was obtained from Stress Gen (Enzo Life Sciences, Farmingdale, NY). Anti-actin was obtained from Sigma-Aldrich. Secondary antibodies were obtained from Southern Biotech (Birmingham, AL). All antibodies were used at a 1:1,000 dilution with the exception of PHF1, which was used at a dilution of 1:200. Except the Cdc37 clone in pCMV6 vector, all other clones used were in pcDNA3.1 plasmid. All siRNAs were obtained from Qiagen, and target sequences are available upon request. siRNA efficiency for protein knockdown was validated by Western blotting. Celastrol and the caspase inhibitor were obtained from Calbiochem.
Cell Culture and Western Blotting
Regular HeLa cells and HeLa cells stably transfected with V5-tagged 4R0N tau were maintained as described previously (24). Transfections in cells were performed as described previously (25). Briefly, plasmid transfections were done utilizing the Lipofectamine 2000 reagent (Invitrogen). SiRNAs were transfected with SilentFect (Bio-Rad). Cells were harvested in MPER buffer (Pierce) containing 1× protease inhibitor mixture (Calbiochem), 1 mm phenylmethylsulfonyl fluoride (PMSF), and 1× phosphatase inhibitor I and II cocktails (Sigma). Measurements of tau levels in cell culture were performed by Western blot analysis.
Human Brain Tissue
Medial temporal gyrus from AD and normal (control) patients were homogenized and processed for co-immunoprecipitation with Cdc37 antibody as described previously (22). Samples were analyzed by Western blotting. Samples were provided by Dr. Tom Beach (Sun Health, Phoenix, AZ). Postmortem interval was between 2.5 and 3 h, and samples were gender- and age-matched.
Immunocytochemistry
M17 human neuroblastoma cells plated onto a chamber slide were transfected with Cdc37 or control siRNAs for 72 h. Immunocytochemistry was performed as described earlier (25). Primary antibodies, rabbit anti-TDP43 and mouse anti-Cdc37 (1:100), were used. Anti-rabbit (Alexa Fluor 488) and anti-mouse (Alexa Fluor 594) secondary antibodies (1:1,000) were added for detection. Imaging was performed with the Zeiss Imager AxioVision.
RESULTS
An Hsp90 Inhibitor That Disrupts the Cdc37/Hsp90 Interface More Potently Reduces TDP-43 Levels Than an Hsp90 ATPase Inhibitor
TDP-43 interacts with Hsp90 (11). We speculated that Hsp90 inhibition might affect TDP-43 stability because of this interaction. We incubated cells with the known Hsp90 inhibitors 17-AAG or celastrol for 24 h at 3 μm, and lysates were analyzed by Western blotting. Celastrol reduced both full-length and cleaved TDP-43 more effectively than 17-AAG (Fig. 1, A and B). Both drugs were more effective than siRNA, perhaps due to the ability of Hsp90 inhibitors to acutely target multiple Hsp90 isoforms unlike siRNAs. Because one of the functions of celastrol is to inhibit the interaction between Hsp90 and its co-chaperone Cdc37 (26), we speculated that TDP-43 might be a client of the Cdc37/Hsp90 complex. Indeed, co-immunoprecipitation studies showed that TDP-43 associates with both Hsp90 and Cdc37 in cells, guiding us to further pursue the effects of Cdc37 on TDP-43 processing (Fig. 1C).
FIGURE 1.

Allosteric Hsp90 inhibitor reduces TDP43 levels. A, Western blot of HeLa cell lysates treated with celastrol, 17-AAG, or vehicle control for 24 h. B, quantitation of Western blot by densitometry showing percentage levels of full-length (FL) and cleaved (C) TDP-43 following celastrol treatment (dark gray bars) or 17-AAG treatment (light gray bars) normalized to GAPDH relative to vehicle treated ± S.D. *, p < 0.05 by Student's t test. C, equal amounts of protein from HeLa cell lysates immunoprecipitated (IP) with rabbit anti-TDP-43 or control rabbit IgG antibodies. Cell lysates (inputs) and immunoprecipitates were analyzed by Western blotting for Hsp90, Cdc37, TDP-43, and GAPDH antibodies.
The effects of Cdc37 and Hsp90 depletion on TDP-43 processing were then evaluated in cells. Both total and caspase-cleaved TDP-43 levels were reduced in neuronal M17 cells transfected with Cdc37 siRNA (Fig. 2A). Conversely, the levels of both full-length and cleaved TDP-43 were preserved by Cdc37 overexpression (Fig. 2A). Whereas Cdc37 overexpression had no effect on TDP-43 localization (data not shown), Cdc37 siRNA caused TDP-43 to redistribute from the nucleus to the cytosol (Fig. 2B) in a manner similar to that observed in cells treated with the apoptosis inducer, staurosporine (7). Similar experiments in cells transfected with siRNA targeting Hsp90 or cDNA overexpressing Hsp90 showed that neither Hsp90 knockdown nor overexpression affected endogenous TDP-43 levels significantly (supplemental Fig. 1). This suggested that disrupting Cdc37 selectively could not only alter TDP-43 stability but also its proteolytic processing and localization.
FIGURE 2.

Modulation of Cdc37 levels regulates TDP-43 levels. A, Western blots of M17 neuronal cell lysates transfected with Cdc37 siRNA (Cdc37 K.D.), control siRNA, Cdc37 overexpression (O.E.) vector, or control vector. Densitometric quantitation of full-length (FL) and cleaved (C) TDP-43 levels after respective siRNA or DNA transfection is shown relative to GAPDH ± S.D. (error bars). **, p < 0.01 by Student's t test. B, M17 neuronal cells transfected with Cdc37 or control siRNAs for 72 h. Immunofluorescent staining of TDP-43 (red) and Cdc37 (green) in cells shows redistribution of TDP-43 from the nucleus to the cytosol.
Cdc37 Knockdown Stimulates Caspase-mediated Poly(ADP-ribose) Polymerase (PARP) Cleavage
Previous work showed that apoptotic activation could lead to TDP-43 cleavage (7). The impact of Cdc37 on this process was evaluated using the downstream apoptosis marker, PARP cleavage. Cells were transfected with Cdc37 siRNA for 72 h and treated with vehicle or a pancaspase inhibitor for 24 h. Depleting Cdc37 enhanced levels and cleavage of PARP relative to cells transfected with control siRNA (Fig. 3A). Caspase inhibition abrogated this process, whereas the caspase activator, staurosporine, facilitated it (Fig. 3). Levels of PARP and cleaved PARP were dramatically enhanced by Cdc37 siRNA, and this was enhanced by staurosporine (Fig. 3B). PARP is cleaved by caspases to become activated, at which point it consumes the cellular ATP to begin repairing DNA. Thus, increased levels of activated PARP caused by Cdc37 knockdown indicate that caspases are highly active in the cell.
FIGURE 3.

Cdc37 silencing stimulates PARP activation. A, HeLa cells transfected with Cdc37 or control siRNAs followed by caspase inhibitor treatment. Cell lysates analyzed by Western blotting. B, Western blots of HeLa cell lysates transfected with Cdc37 or control siRNAs and treated with staurosporine.
TDP-43 Clearance by Cdc37 Knockdown Is Caspase-dependent
Cdc37 knockdown triggered both destabilization of TDP-43 and apoptotic activation. This suggested that the Cdc37/Hsp90 complex might regulate TDP-43 in two ways. First, it typically stabilizes TDP-43, preventing it from degradation by an immature Hsp90 complex (27, 28); and second, it protects TDP-43 from cleavage leading to an alternative clearance pathway. Indeed, caspase inhibition completely prevented clearance of TDP-43 when Cdc37 levels were depleted by siRNA (Fig. 4). Staurosporine treatment enhanced cleavage of TDP-43, and clearance of full-length TDP-43 was accelerated, further supporting a mechanism whereby Cdc37 depletion facilitates TDP-43 removal from the nucleus by stimulating caspase-cleavage. Once in the cytosol, cleaved TDP-43 can be cleared.
FIGURE 4.
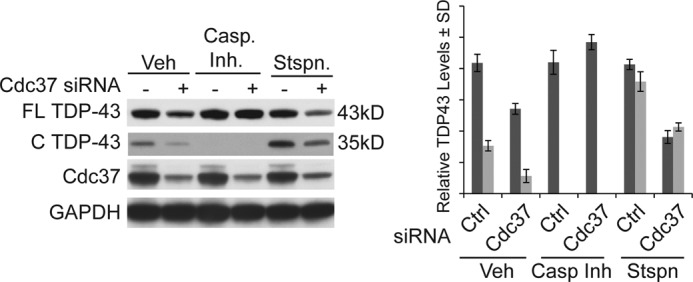
Clearance of TDP-43 by Cdc37 knockdown is dependent on caspase cleavage. Western blots of cell lysates transfected with Cdc37 or control siRNA and then treated with a pancaspase inhibitor (Casp. Inh.), staurosporine (Stspn.), or vehicle (Veh) for 24 h. Densitometric quantitation of full-length (FL) (dark gray bars) and cleaved (C) (light gray bars) TDP-43 levels after each treatment is shown relative to GAPDH ± S.D. (error bars).
Robust Accumulation of Tau Impairs Cdc37-dependent Clearance of TDP-43
Because TDP-43 and tau can both accumulate in AD and other diseases, we speculated that this clearance process might be affected by tau accumulation. Cells overexpressing TDP-43 were transfected with increasing amounts of tau and then subjected to control or Cdc37 siRNA for 72 h. Cdc37 siRNA facilitated tau clearance at all concentrations (Fig. 5A). At lower gene dosages of tau, TDP-43 clearance by Cdc37 knockdown was also observed; however, as tau levels increased, cleaved TDP-43 accumulated (Fig. 5, A and B), suggesting that tau could interfere with TDP-43 clearance pathways. Consistent with this idea, immunoprecipitation of Cdc37 in lysates from normal and AD brain tissue (n = 5 for both) showed that as phospho-tau levels were increased in the AD brain, so was the association of tau with Cdc37 (Fig. 5C and supplemental Fig. 2); however, this was at the expense of the TDP-43/Cdc37 interaction. Although TDP-43 interacted with Cdc37 in normal brain tissue, it was replaced in AD brain with phospho-tau. A corresponding conversion of full-length TDP-43 to cleaved TDP-43 was also observed in AD brain, suggesting altered clearance kinetics of TDP-43 in AD brain (Fig. 5, C and D).
FIGURE 5.

Tau overexpression displaces TDP-43 from Cdc37. A, Western blot of cell lysates transfected with and without Cdc37 siRNA and transfected with increasing amounts of tau cDNA (1, 2, or 3 μg). B, quantitation of full-length (FL) (dark gray bars) and cleaved (C) (light gray bars) normalized to GAPDH ± S.D. (error bars). C, human age matched normal and AD patient samples subjected to co-immunoprecipitation with Cdc37 antibody. Western blot analysis shows that FL TDP-43 associates with Cdc37 more in normal brain compared with AD brain. Increased interaction of Cdc37 with phospho-tau (pTau; detected with pS199/S202 Ab) coincides with a decreased interaction of Cdc37 with TDP-43 in AD brain and increased TDP-43 cleavage in the lysates (Inputs). D, quantitation of five samples (including data from supplemental Fig. 2) each from AD (light gray bars) and control brain tissue (dark gray bars). *, p < 0.05; **, p < 0.01.
Caspase-cleaved TDP-43 Clearance Is Mediated by Autophagy
The impaired clearance of TDP-43 by tau accumulation suggested that tau could be usurping the clearance machinery necessary for TDP-43 clearance stimulated by Cdc37 depletion. Because a major clearance pathway for tau is autophagic processing (29), we tested whether manipulating autophagy could similarly impair TDP-43 clearance following Cdc37 depletion. To disrupt autophagy, we used siRNA targeting beclin1. Beclin is a protein known to promote autophagy (30). Cells expressing TDP-43 were transfected with siRNA targeting beclin1 and either control or Cdc37 siRNA. Although beclin1 knockdown alone did not facilitate TDP-43 accumulation, it did abrogate clearance when Cdc37 levels were reduced (Fig. 6). Thus, Cdc37 knockdown triggers a cascade of events involving Hsp90 and caspases leading to clearance of TDP-43 by autophagy, a process that can be blocked by tau accumulation (Fig. 7).
FIGURE 6.
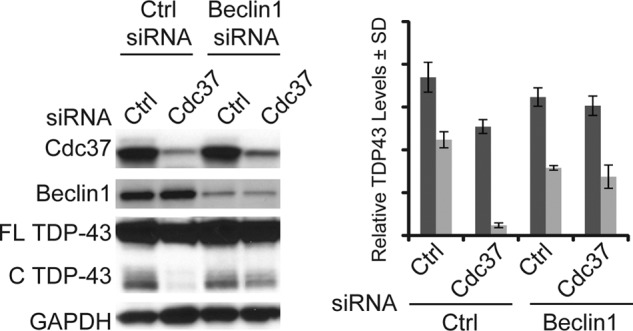
Cdc37 knockdown promotes beclin-dependent clearance of TDP-43. Cells transfected with beclin1 siRNA and either Cdc37 or control siRNA were subjected to Western blot analysis. Densitometric quantitation of full-length (FL) (dark gray bars) and cleaved (C) (light gray bars) TDP-43 levels after respective siRNA transfection is shown relative to GAPDH ± S.D. (error bars).
FIGURE 7.
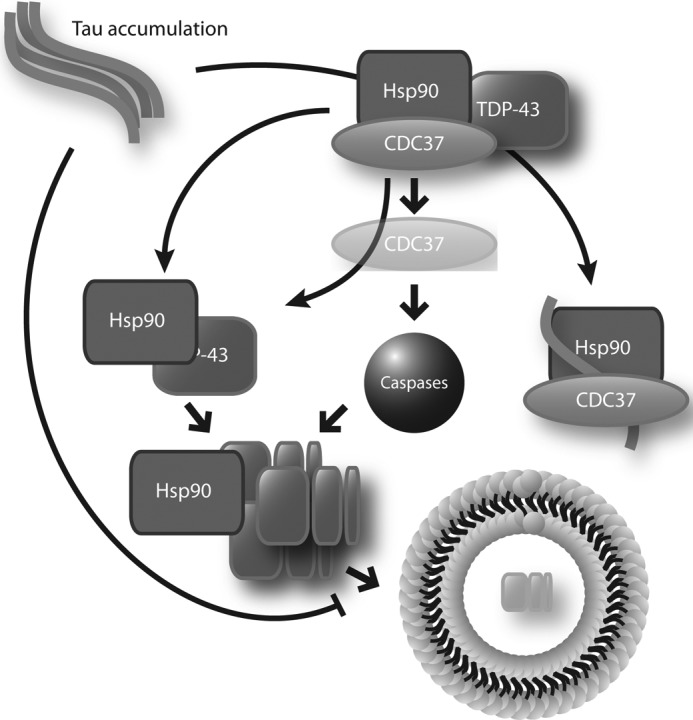
Cdc37 knockdown causes proteolytic clearance of TDP-43 by autophagy, but this can be abrogated by tau accumulation. The Cdc37/Hsp90 complex typically preserves TDP-43 in the nucleus. When Cdc37 is removed from the complex, TDP-43 is proteolytically cleaved and retrotranslocated to the cytosol. Here, it is cleared via beclin-dependent autophagy. Tau accumulation is able to interfere with the association of TDP-43 with the Cdc37/Hsp90 complex, causing cleaved TDP-43 to accumulate. This suggests that Hsp90 is involved in the autophagic clearance of cleaved TDP-43.
DISCUSSION
In this report, disrupting the Cdc37/Hsp90 interface reduced the stability of full-length and cleaved TDP-43 by altering its localization and facilitating beclin-dependent autophagic clearance. Cdc37 overexpression led to the accumulation of both TDP-43 species, whereas knockdown promoted caspase-dependent degradation. This is important because cleaved TDP-43 accumulates in ALS, FTLD, and AD (2, 3, 7, 9, 31). At first glance, this would suggest that therapies aimed at targeting Cdc37 and the Cdc37/Hsp90 interface could have efficacy in ALS and other diseases associated with TDP-43 accumulation. For example, celastrol disrupts the Cdc37/Hsp90 interface (26) and facilitated TDP-43 clearance. However, in an attempt to model disease, increasing the levels of tau in the cytosol slowed the clearance of cleaved TDP-43 following Cdc37 depletion. In this way, tau accumulation could prevent the clearance of cleaved TDP-43 triggered by changes in the Hsp90 complex. These findings could help to explain the emergence of tau and TDP-43 co-morbidities; as tau accumulates, it can physically block TDP-43 clearance dictated by chaperone proteins, leading to accumulation of cleaved TDP-43 pathology as well. It also suggests that strategies aimed at facilitating the clearance of TDP-43 based on chaperone manipulation may have adverse outcomes, depending on the abundance of tau in the neuron.
Caspase activation in neurons is a mystery. In the periphery, caspase activation is associated predominantly with cell death. But in neurons, caspase activation is common, and it is often observed in living neurons (32–34). This suggests that caspases may have a proteolytic function in long lived neurons that is beneficial in some cases. Indeed, neurons with high levels of caspase-cleaved substrates are thought to be longer lived than those without robust caspase cleavage (32). Moreover, recent work suggests that caspase cleavage can target proteins through distinct degradation pathways (35). One might envision this type of processing for TDP-43. Chaperone-guided caspase cleavage allows TDP-43 to localize to the cytosol for autophagic degradation in a retrotranslocation process (36). This would be similar to endoplasmic reticulum-associated degradation, where protein ubiquitination occurs in the endoplasmic reticulum lumen leading to retrotranslocation into the cytosol where proteasomal degradation occurs (37). In this way, caspases may have an integral role in dictating triage decisions of distinct proteins, particularly in the nucleus where Hsp90 and its co-chaperone repertoire are known to reside (38–40). Our results indicate that caspases consort with the Hsp90 complex to facilitate clearance. In general, this process may enhance the clearance of overly abundant clients by promoting proteolysis that engages the autophagy pathway. This process would also lose efficacy in aging because aging itself is associated with impaired protein clearance mechanisms and autophagy function (41–44). Therefore, when abnormal proteins become too abundant, chaperones recruit caspases to engage the autophagic system in their clearance, but with age, autophagy slows down and caspase-cleaved species begin to accumulate as is observed in postmortem tissues.
This study also points to a relationship between tau and TDP-43 that requires chaperones. The production of aberrant tau in the cytosol usurps the clearance machinery that is necessary for the clearance of TDP-43 from the nucleus when the Cdc37/Hsp90 interface is disrupted. The altered binding of TDP-43 to Cdc37 and Hsp90 in the AD brain suggests that tau may actually displace TDP-43 from the Hsp90 complex that is delivering TDP-43 for autophagic clearance. Thus, Hsp90 might actually be involved in the transport of TDP-43 to the autophagosome. These findings also begin to explain why neurons with tau accumulation do not always show TDP-43 pathology (2, 3). As tau becomes overabundant, only those neurons that are attempting to clear TDP-43 via the Hsp90 system exhibit accumulation of caspase-cleaved TDP-43 as well. Indeed, the abundance of TDP-43 and tau in the AD brain is disproportionate, such that tau accumulation is much more robust.
Ultimately, this study demonstrates that the clearance of TDP-43 from the nucleus requires orchestration of the Hsp90 machinery with caspases. It also points to a novel mechanism whereby Hsp90 and caspases can facilitate retrotranslocation of certain resident nuclear proteins leading to their clearance by autophagy. What precipitates this process remains to be determined. We show here that disrupting the association of Cdc37 from Hsp90 is essential to this degradation cascade. Given the relationship of Cdc37 with kinases, it is possible that traditional signaling plays an important role (27, 45). For example, the Cdc37/Hsp90 complex has been associated with the kinases Ulk1, mTOR, and AMPK, each of which is involved in autophagy (46, 47). It is possible that phosphorylation of these kinases regulates the interaction of Cdc37 with Hsp90 and can promote or prevent their interaction. It may also be more client-specific than that: TDP-43 is indeed phosphorylated, and it is possible that these phosphorylation events change the affinity of Cdc37 with the Hsp90/TDP-43 complex. Further exploration of this mechanism could provide new insights into cellular quality control as well as the emergence of tau and TDP-43 co-pathologies.
In conclusion, the studies here describe a chaperone-mediated autophagic mechanism through which TDP-43 clearance from the nucleus is controlled. In addition, these findings point to a mechanism by which TDP-43 pathogenesis arises sporadically in AD. It is possible that mutations in TDP-43 that cause ALS or FTLD may also affect this clearance pathway. New therapeutic strategies targeting this pathway could lead to treatments for ALS, FTLD, and AD.
Supplementary Material
Acknowledgments
We thank Dr. Peter Davies, Dr. Lester Binder, and Dr. Tom Beach for generously sharing their resources for our research.

This article contains supplemental Figs. 1 and 2.
- TDP-43
- trans-active response DNA-binding protein 43
- AD
- Alzheimer disease
- ALS
- amyotrophic lateral sclerosis
- FTLD
- frontotemproral lobar degeneration
- Hsp90
- 90-kDa heat shock protein
- PARP
- poly(ADP-ribose) polymerase
- 17AAG
- 17-(allylamino-17-demethoxygeldanamycin).
REFERENCES
- 1. Lee E. B., Lee V. M., Trojanowski J. Q. (2012) Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat. Rev. Neurosci. 13, 38–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Neumann M., Sampathu D. M., Kwong L. K., Truax A. C., Micsenyi M. C., Chou T. T., Bruce J., Schuck T., Grossman M., Clark C. M., McCluskey L. F., Miller B. L., Masliah E., Mackenzie I. R., Feldman H., Feiden W., Kretzschmar H. A., Trojanowski J. Q., Lee V. M. (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 [DOI] [PubMed] [Google Scholar]
- 3. Amador-Ortiz C., Lin W. L., Ahmed Z., Personett D., Davies P., Duara R., Graff-Radford N. R., Hutton M. L., Dickson D. W. (2007) TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann. Neurol. 61, 435–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sreedharan J., Blair I. P., Tripathi V. B., Hu X., Vance C., Rogelj B., Ackerley S., Durnall J. C., Williams K. L., Buratti E., Baralle F., de Belleroche J., Mitchell J. D., Leigh P. N., Al-Chalabi A., Miller C. C., Nicholson G., Shaw C. E. (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rutherford N. J., Zhang Y. J., Baker M., Gass J. M., Finch N. A., Xu Y. F., Stewart H., Kelley B. J., Kuntz K., Crook R. J., Sreedharan J., Vance C., Sorenson E., Lippa C., Bigio E. H., Geschwind D. H., Knopman D. S., Mitsumoto H., Petersen R. C., Cashman N. R., Hutton M., Shaw C. E., Boylan K. B., Boeve B., Graff-Radford N. R., Wszolek Z. K., Caselli R. J., Dickson D. W., Mackenzie I. R., Petrucelli L., Rademakers R. (2008) Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 4, e1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Buratti E., Dörk T., Zuccato E., Pagani F., Romano M., Baralle F. E. (2001) Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 20, 1774–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang Y. J., Xu Y. F., Dickey C. A., Buratti E., Baralle F., Bailey R., Pickering-Brown S., Dickson D., Petrucelli L. (2007) Progranulin mediates caspase-dependent cleavage of TAR DNA-binding protein-43. J. Neurosci. 27, 10530–10534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Winton M. J., Igaz L. M., Wong M. M., Kwong L. K., Trojanowski J. Q., Lee V. M. (2008) Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J. Biol. Chem. 283, 13302–13309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rohn T. T. (2008) Caspase-cleaved TAR DNA-binding protein-43 is a major pathological finding in Alzheimer's disease. Brain Res. 1228, 189–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cohen T. J., Lee V. M., Trojanowski J. Q. (2011) TDP-43 functions and pathogenic mechanisms implicated in TDP-43 proteinopathies. Trends Mol. Med. 17, 659–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang Y. J., Gendron T. F., Xu Y. F., Ko L. W., Yen S. H., Petrucelli L. (2010) Phosphorylation regulates proteasomal-mediated degradation and solubility of TAR DNA binding protein-43 C-terminal fragments. Mol. Neurodegener. 5, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Smith J. R., Workman P. (2009) Targeting CDC37: an alternative, kinase-directed strategy for disruption of oncogenic chaperoning. Cell Cycle 8, 362–372 [DOI] [PubMed] [Google Scholar]
- 13. Young J. C., Hartl F. U. (2000) Polypeptide release by Hsp90 involves ATP hydrolysis and is enhanced by the co-chaperone p23. EMBO J. 19, 5930–5940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Siligardi G., Panaretou B., Meyer P., Singh S., Woolfson D. N., Piper P. W., Pearl L. H., Prodromou C. (2002) Regulation of Hsp90 ATPase activity by the co-chaperone Cdc37p/p50cdc37. J. Biol. Chem. 277, 20151–20159 [DOI] [PubMed] [Google Scholar]
- 15. Young J. C., Moarefi I., Hartl F. U. (2001) Hsp90: a specialized but essential protein-folding tool. J. Cell Biol. 154, 267–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bukau B., Weissman J., Horwich A. (2006) Molecular chaperones and protein quality control. Cell 125, 443–451 [DOI] [PubMed] [Google Scholar]
- 17. Whitesell L., Cook P. (1996) Stable and specific binding of heat shock protein 90 by geldanamycin disrupts glucocorticoid receptor function in intact cells. Mol. Endocrinol. 10, 705–712 [DOI] [PubMed] [Google Scholar]
- 18. Dickey C. A., Kamal A., Lundgren K., Klosak N., Bailey R. M., Dunmore J., Ash P., Shoraka S., Zlatkovic J., Eckman C. B., Patterson C., Dickson D. W., Nahman N. S., Jr., Hutton M., Burrows F., Petrucelli L. (2007) The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J. Clin. Invest. 117, 648–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Harst A., Lin H., Obermann W. M. (2005) Aha1 competes with Hop, p50, and p23 for binding to the molecular chaperone Hsp90 and contributes to kinase and hormone receptor activation. Biochem. J. 387, 789–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mayer M. P., Nikolay R., Bukau B. (2002) Aha, another regulator for hsp90 chaperones. Mol. Cell 10, 1255–1256 [DOI] [PubMed] [Google Scholar]
- 21. Wang X., Venable J., LaPointe P., Hutt D. M., Koulov A. V., Coppinger J., Gurkan C., Kellner W., Matteson J., Plutner H., Riordan J. R., Kelly J. W., Yates J. R., 3rd, Balch W. E. (2006) Hsp90 co-chaperone Aha1 down-regulation rescues misfolding of CFTR in cystic fibrosis. Cell 127, 803–815 [DOI] [PubMed] [Google Scholar]
- 22. Jinwal U. K., Trotter J. H., Abisambra J. F., Koren J., 3rd, Lawson L. Y., Vestal G. D., O'Leary J. C., 3rd, Johnson A. G., Jin Y., Jones J. R., Li Q., Weeber E. J., Dickey C. A. (2011) The Hsp90 kinase co-chaperone Cdc37 regulates tau stability and phosphorylation dynamics. J. Biol. Chem. 286, 16976–16983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lin Y., Wu X., Deng Z., Wang J., Zhou S., Vrijmoed L. L., Jones E. B. (2002) The metabolites of the mangrove fungus Verruculina enalia no. 2606 from a salt lake in the Bahamas. Phytochemistry 59, 469–471 [DOI] [PubMed] [Google Scholar]
- 24. Jinwal U. K., O'Leary J. C., 3rd, Borysov S. I., Jones J. R., Li Q., Koren J., 3rd, Abisambra J. F., Vestal G. D., Lawson L. Y., Johnson A. G., Blair L. J., Jin Y., Miyata Y., Gestwicki J. E., Dickey C. A. (2010) Hsc70 rapidly engages tau after microtubule destabilization. J. Biol. Chem. 285, 16798–16805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jinwal U. K., Koren J., 3rd, Borysov S. I., Schmid A. B., Abisambra J. F., Blair L. J., Johnson A. G., Jones J. R., Shults C. L., O'Leary J. C., 3rd, Jin Y., Buchner J., Cox M. B., Dickey C. A. (2010) The Hsp90 cochaperone, FKBP51, increases tau stability and polymerizes microtubules. J. Neurosci. 30, 591–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang T., Hamza A., Cao X., Wang B., Yu S., Zhan C. G., Sun D. (2008) A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol. Cancer Ther. 7, 162–170 [DOI] [PubMed] [Google Scholar]
- 27. Stepanova L., Leng X., Parker S. B., Harper J. W. (1996) Mammalian p50Cdc37 is a protein kinase-targeting subunit of Hsp90 that binds and stabilizes Cdk4. Genes Dev. 10, 1491–1502 [DOI] [PubMed] [Google Scholar]
- 28. MacLean M., Picard D. (2003) Cdc37 goes beyond Hsp90 and kinases. Cell Stress Chaperones 8, 114–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang Y., Martinez-Vicente M., Krüger U., Kaushik S., Wong E., Mandelkow E. M., Cuervo A. M., Mandelkow E. (2009) Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum. Mol. Genet. 18, 4153–4170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liang X. H., Jackson S., Seaman M., Brown K., Kempkes B., Hibshoosh H., Levine B. (1999) Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402, 672–676 [DOI] [PubMed] [Google Scholar]
- 31. Kwong L. K., Neumann M., Sampathu D. M., Lee V. M., Trojanowski J. Q. (2007) TDP-43 proteinopathy: the neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron disease. Acta Neuropathol. 114, 63–70 [DOI] [PubMed] [Google Scholar]
- 32. de Calignon A., Fox L. M., Pitstick R., Carlson G. A., Bacskai B. J., Spires-Jones T. L., Hyman B. T. (2010) Caspase activation precedes and leads to tangles. Nature 464, 1201–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. D'Mello S. R., Kuan C. Y., Flavell R. A., Rakic P. (2000) Caspase-3 is required for apoptosis-associated DNA fragmentation but not for cell death in neurons deprived of potassium. J. Neurosci. Res. 59, 24–31 [PubMed] [Google Scholar]
- 34. Stadelmann C., Deckwerth T. L., Srinivasan A., Bancher C., Brück W., Jellinger K., Lassmann H. (1999) Activation of caspase-3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer's disease: evidence for apoptotic cell death. Am. J. Pathol. 155, 1459–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dolan P. J., Johnson G. V. (2010) A caspase cleaved form of tau is preferentially degraded through the autophagy pathway. J. Biol. Chem. 285, 21978–21987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rycyzyn M. A., Reilly S. C., O'Malley K., Clevenger C. V. (2000) Role of cyclophilin B in prolactin signal transduction and nuclear retrotranslocation. Mol. Endocrinol. 14, 1175–1186 [DOI] [PubMed] [Google Scholar]
- 37. Ye Y., Meyer H. H., Rapoport T. A. (2001) The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414, 652–656 [DOI] [PubMed] [Google Scholar]
- 38. Wilhelmsson A., Cuthill S., Denis M., Wikström A. C., Gustafsson J. A., Poellinger L. (1990) The specific DNA binding activity of the dioxin receptor is modulated by the 90-kDa heat shock protein. EMBO J. 9, 69–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gasc J. M., Renoir J. M., Faber L. E., Delahaye F., Baulieu E. E. (1990) Nuclear localization of two steroid receptor-associated proteins, hsp90 and p59. Exp. Cell Res. 186, 362–367 [DOI] [PubMed] [Google Scholar]
- 40. Pratt W. B. (1992) Control of steroid receptor function and cytoplasmic-nuclear transport by heat shock proteins. Bioessays 14, 841–848 [DOI] [PubMed] [Google Scholar]
- 41. Lu T., Pan Y., Kao S. Y., Li C., Kohane I., Chan J., Yankner B. A. (2004) Gene regulation and DNA damage in the ageing human brain. Nature 429, 883–891 [DOI] [PubMed] [Google Scholar]
- 42. Lipinski M. M., Zheng B., Lu T., Yan Z., Py B. F., Ng A., Xavier R. J., Li C., Yankner B. A., Scherzer C. R., Yuan J. (2010) Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 107, 14164–14169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cuervo A. M., Dice J. F. (2000) Age-related decline in chaperone-mediated autophagy. J. Biol. Chem. 275, 31505–31513 [DOI] [PubMed] [Google Scholar]
- 44. Cuervo A. M. (2003) Autophagy and aging: when “all you can eat” is yourself. SAGE KE 2003, pe25. [DOI] [PubMed] [Google Scholar]
- 45. Basso A. D., Solit D. B., Chiosis G., Giri B., Tsichlis P., Rosen N. (2002) Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J. Biol. Chem. 277, 39858–39866 [DOI] [PubMed] [Google Scholar]
- 46. Joo J. H., Dorsey F. C., Joshi A., Hennessy-Walters K. M., Rose K. L., McCastlain K., Zhang J., Iyengar R., Jung C. H., Suen D. F., Steeves M. A., Yang C. Y., Prater S. M., Kim D. H., Thompson C. B., Youle R. J., Ney P. A., Cleveland J. L., Kundu M. (2011) Hsp90-Cdc37 chaperone complex regulates Ulk1- and Atg13-mediated mitophagy. Mol. Cell 43, 572–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kim J., Kundu M., Viollet B., Guan K. L. (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.