

Abstract
Rhodium(II) dicarboxylate complexes were discovered to catalyze the intramolecular amination of unactivated primary-, secondary-, or tertiary aliphatic C–H bonds using aryl azides as the N-atom precursor. While a strong electron-withdrawing group on the nitrogen atom is typically required to achieve this reaction, we found that both electron-rich- and electron-poor aryl azides are efficient sources for the metal nitrene reactive intermediate.
The development of transition metal-catalyzed aliphatic C– H bond amination reactions that is stereoselective, uses readily accessible starting materials and catalysts, and is environmentally benign continues to inspire the efforts of research groups around the world.1,2 While considerable progress has been made, the current methods are still limited by the oxidative conditions to form the nitrene and the requirement for strong electron-withdrawing groups on the nitrene (Scheme 1). The use of azides as the source of the N-atom source would address these limitations because no oxidant would be required and the only by-product of the reaction would the environmentally benign N2-gas.3 While azides have been used for a variety of N-atom transfer reactions,2b,4 these transformations also require electron-withdrawing groups on the azide. We anticipated that a more general, complementary solution for aliphatic C–H bond amination might emerge if conditions were found to use electron-neutral aryl azides as the nitrogen-atom source. While we have reported a number of sp2-C–H bond amination reactions using aryl azides,5 our mechanism studies suggest that C–N bond formation occurs via a 4π-electron-5-atom electrocyclization.5b In contrast, aliphatic C–H bond amination requires metal-catalyzed C–H insertion or H-atom abstraction mechanisms—processes that have remained elusive to control using aryl azides as the N-atom source.6
Scheme 1.
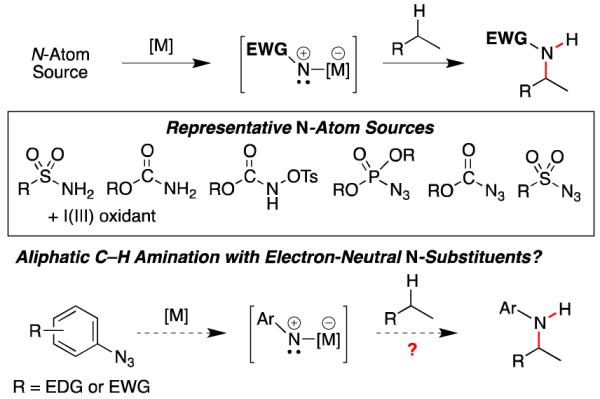
Nitrogen Substituent Requirements for Aliphatic C–H Bond Amination.
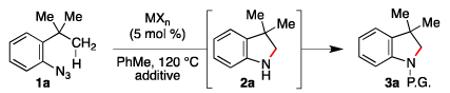
In search of the optimal conditions to achieve intramolecular aliphatic C–H bond amination using aryl azides, the reactivity of ortho-tert-butyl aryl azide 1a towards transition metal complexes was examined (Table 1).7 This aryl azide is relatively thermally robust with no reaction observed at 120 °C (entry 1).8 Exposure of 1a to commercially available transition metal complexes known to catalyze N-atom transfer reactions was met with limited success. Indoline 3a was not observed in the presence of iron,9 copper,10 cobalt,2b,4a-c ruthenium,2a,6,11 or iridium12 complexes (entries 2 – 7). Partial conversion to indoline 2 was observed when rhodium octanoate was used (entry 8). We anticipated that the partial conversions resulted from catalyst decomposition, and we found that using more thermally robust Rh2(esp) 132 improved both the conversion and yield of the process (entry 9). Examination of alternative solvents, concentration, and temperatures, however, did not increase the yield, and control experiments revealed that oxidative decomposition of the indoline occurred during purification.
Table 1.
Development of Optimal Conditions.
| entry | catalyst | additive | conv., %a |
yield, %b |
|---|---|---|---|---|
| 1 | none | n.a. | 0 | 0 |
| 2 | FeBr2 | n.a. | 0 | decc |
| 3 | CuBr | n.a. | 0 | 0 |
| 4 | CoTPP | n.a. | 0 | 0 |
| 5 | RuCl3•nOH2 | n.a. | 0 | 0 |
| 6 | [Ir(cod)OMe]2 | n.a. | 0 | 0 |
| 7 | [Rh(cod)OMe]2 | n.a. | 10 | 0c |
| 8 | Rh2(O2CC7H15)4 | n.a. | 35 | 35 |
| 9 | Rh2(esp)2 | n.a. | 99 | 75 |
| 10 | Rh2(esp)2 | Boc2O | 99 | 90 |
| 11 | Rh2(esp)2 | Ac2O | 99 | 83 |
| 12 | Rh2(esp)2 | Bz2O | 99 | aniline |
| 13 | Rh2(esp)2 | Tf2O | 99 | aniline |
As determined using 1H NMR spectroscopy.
Isolated after silica gel chromatography.
Aniline formed.
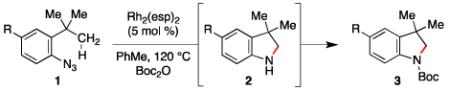
To improve the isolated yield of the amination reaction, in situ protection of the nitrogen atom was attempted. In line with our assumption, we found that the reaction yield was improved when the indoline was protected with either a Bocor Ac-group (entries 10 and 11). The reaction outcome appeared to correlate with the pKa of the acid by-product of the protection reaction: aniline was produced when the stronger benzoic- and triflic acids were produced (entries 12 and 13).14
Using these optimal conditions, the electronic- and steric constraints of the aliphatic C–H bond amination reaction was investigated (Table 2). In contrast to existing amination processes,2,4 our method does not require an electron-withdrawing group on the nitrogen: aryl azides bearing either para-electron-releasing, neutral or withdrawing groups were converted to indolines (entries 1 – 7). Illustrating the chemoselectivity of our process, olefins were tolerated as substituents (entry 5).
Table 2.
Scope and Limitations of Indoline Formation.
| entry | 1 | R | yield, %a |
|---|---|---|---|
| 1 | a | H | 84 |
| 2 | b | OMe | 63 |
| 3 | c | Me | 54 |
| 4 | d | CH2CH2Ph | 64 |
| 5 | e | CHCHPh | 70 |
| 6 | f | Ph | 54 |
| 7 | g | 3,5-OMe2C6H3 | 58 |
| 8 | h | Br | 73 |
Isolated after silica gel chromatography.
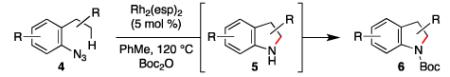
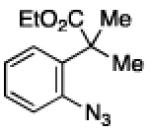
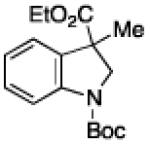
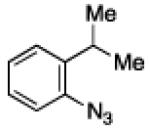
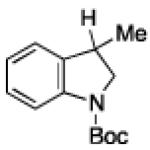
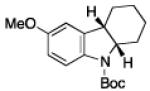
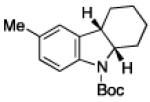
To further examine the scope of the transformation the identity of the ortho-alkyl substituent was varied (Table 3). While replacing one of the methyl groups with an ester group did not diminish the yield (entry 1), substitution with hydrogen reduced the reaction efficiency (entry 2). Amination can be achieved at tertiary- and secondary C–H bonds, although dehydrogenation of 5d occurred to afford indole. Dehydrogenation could be circumvented if an additional substituent was introduced at the benzylic position in of the aryl azide. Submission of 4e – 4k to reaction conditions produced indolines as single diastereomers (entries 5 – 11). In contrast, pyrolysis of aryl azide 4g was reported by Smolinsky to produce a 1:1 mixture of diastereomers.15 Although reaction with the methyl C–H bond in azide 4l could produce a six-membered ring, only amination of the methyl C–H bond was observed to afford indoline 5l as the sole product. While single diastereomers were obtained from substrates bearing orthocyclopentyl- or cyclohexyl-groups, diminished stereoselectivity was observed with ortho-cycloheptyl substituted aryl azide 4m (entry 13).
Table 3.
Scope and Limitations of Indoline Formation.
| entry | 4 | aryl azide | indoline | yield, %a |
|---|---|---|---|---|
| 1 | a |
|
|
70 |
| 2 | b |
|
|
20 |
| 3 | c |
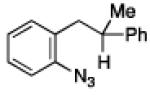
|
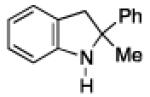
|
55b |
| 4 | d |
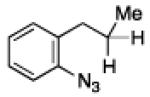
|
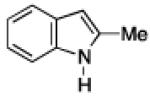
|
30c |
| 5 | e |
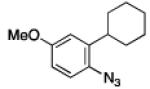
|
|
80 |
| 6 | f |
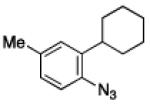
|
|
73 |
| 7 | g |
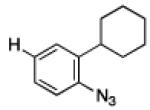
|
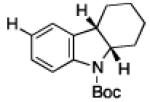
|
70 |
| 8 | h |
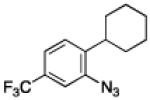
|
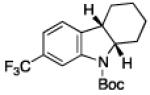
|
63 |
| 9 | i |
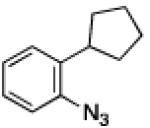
|
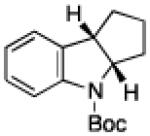
|
85 |
| 10 | j |
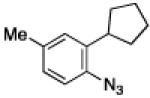
|
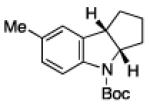
|
73 |
| 11 | k |
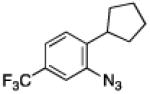
|
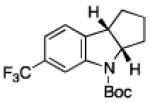
|
82 |
| 12 | l |
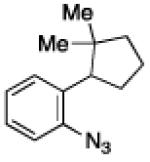
|
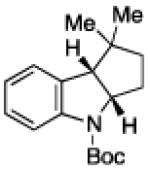
|
86 |
| 13 | m |
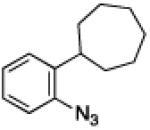
|
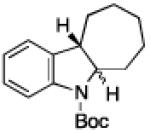
|
63 d.r. 82:18 |
Isolated after silica gel chromatography.
20% aniline observed.
30% aniline observed.
While indoline formation could occur through several different mechanisms,12a,16 our reactivity trends suggest that C–N bond formation occurs through N-atom transfer (Scheme 2). Coordination of the rhodium(II) carboxylate to either the α- or γ-nitrogen of the aryl azide produces 7.17 Extrusion of N2 then forms the rhodium nitrene 8.18 While rhodium could mediate a reversible one-electron oxidation to generate free nitrene,19 our current mechanistic hypothesis is that the nitrene remains metal bound for the C–H bond amination step since pyrolysis involving free nitrene is not diastereoselective.15 The mechanism of the amination step could be stepwise or concerted: hydride-20 or H-atom abstraction16a,21 (to form 9 or 10) followed by recombination produces the C–N bond; alternatively this bond could be formed through the concerted insertion16c,22 of the metal nitrene into the proximal C–H bond via transition state 11. Finally, the indoline is produced upon dissociation of the rhodium complex from 12.
Scheme 2.
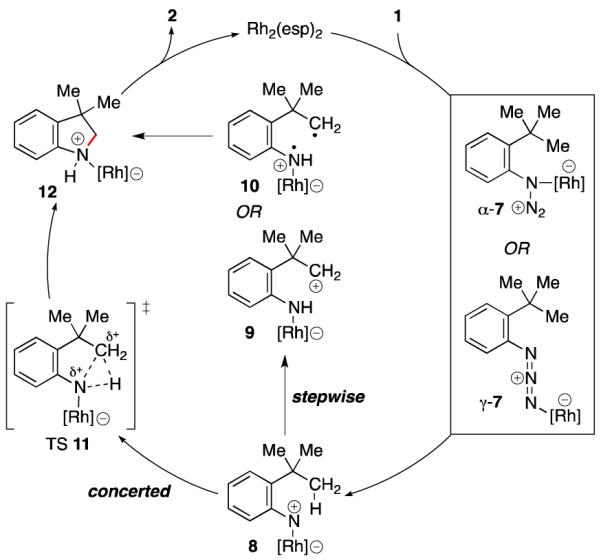
Possible Mechanisms for Intramolecular Aliphatic C–H Bond Amination from Aryl Azides.
Insight into the initial steps of the catalytic cycle was provided by the reactivity of 4-substituted aryl azides towards reaction conditions (eq 1). Underscoring the difference between our method for aliphatic C–H bond amination and others, we found that more electron-rich aryl azides (e.g. 1b) were more reactive toward reaction conditions. The increased reactivity of 1b relative to 1a could be due to either preferred coordination of 1b to Rh2(esp)2 or an accelerated N2 extrusion from the resulting azide-metal complex 7.
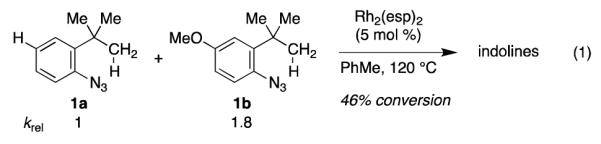 |
To examine the nature of the C–H bond cleavage step, two-labeled aryl azides were examined (Scheme 3). We anticipated that the number of indoline diastereomers from 14-d2 would reveal if the mechanism of C–H bond amination was stepwise or concerted. If the reaction was concerted, insertion into either the β-C–H or β-C–D bond would produce only two products. In contrast, if a radical (or cation) was formed at the β-carbon then scrambling of the C2-stereocenter could occur before recombination to form both 15-d2 and 16-d2. In support of a stepwise C–H bond amination, two diastereomers of 2-phenyl indoline (d.r. 50:50) and an intramolecular kinetic isotope effect of 6.7 were observed.23 The magnitude of this isotope effect is significantly smaller than reactions involving an H-atom abstraction by an aryl nitrene21 or an aryl metal nitrene6c (kH/kD 12 – 14), but larger than hydride shift reactions (kH/kD ~ 2 for Cannizzaro reaction and Meerwein–Ponndorf– Verley reduction).24-26 Smaller kinetic isotope effects were observed at lower reaction temperatures revealing that our amination reaction occurs above the isokinetic temperature and as a consequence is under entropic control.27,28 We found, however, that the spatial constraints of this reaction override these isotope effects: cyclopentanone-derived aryl azide 17-d1 reacted preferentially with the syn-C–D bond to form 18 exclusively.29
Scheme 3.
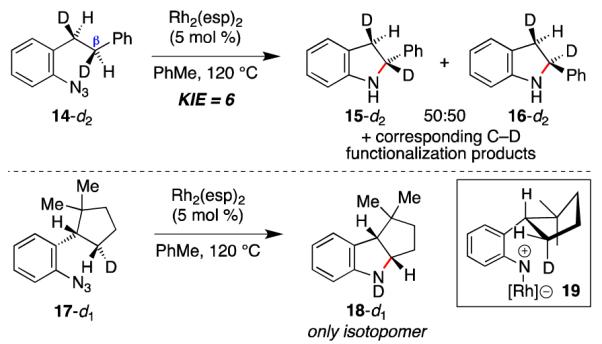
Isotope Labeling Studies.
In conclusion, we have developed an efficient and diastereoselective rhodium(II)-catalyzed aliphatic C–H bond amination reaction that uses an aryl azide as the nitrogen-atom source. Our method distinguishes itself from previously reported aliphatic C–H bond amination reactions by not requiring a strong electron-withdrawing group on the nitrogen atom. The reactivity of stereospecific labeled aryl azides revealed that the amination reaction occurred stepwise with the syn-C– H bond. Our current aims are to more deeply examine the nature of the catalytic intermediates in this C–H bond amination reaction and to extend this newfound reactivity of aryl azides to the stereoselective synthesis of complex, functionalized N-heterocycles
Supplementary Material
ACKNOWLEDGMENT
We are grateful to the National Institutes of Health NIGMS (R01GM084945) and the University of Illinois at Chicago for their generous financial support. We thank Mr. Furong Sun (UIUC) for high resolution mass spectrometry data.
Funding Sources National Institutes of Health NIGMS (R01GM084945) and the University of Illinois at Chicago
Footnotes
ASSOCIATED CONTENT Supporting Information. Experimental procedures, spectroscopic and analytical data for the products (PDF) are available free of charge via the Internet at http://pubs.acs.org.
Author Contributions The manuscript was written through contributions of all authors.
REFERENCES
- 1.For recent reviews, see: Du Bois J. Org. Process Res. Dev. 2011;15:758. doi: 10.1021/op200046v. Davies HML, Du Bois J, Yu J-Q. Chem. Soc. Rev. 2011;40:1855. doi: 10.1039/c1cs90010b. Stokes BJ, Driver TG. Eur. J. Org. Chem. 2011;4071 Lyons TW, Sanford MS. Chem. Rev. 2010;110:1147. doi: 10.1021/cr900184e. Collet F, Dodd RH, Dauban P. Chem. Commun. 2009;5061 doi: 10.1039/b905820f.
- 2.cf. Harvey ME, Musaev DG, Du Bois J. J. Am. Chem. Soc. 2011;133:17207. doi: 10.1021/ja203576p. Lu H, Tao J, Jones JE, Wojtas L, Zhang XP. Org. Lett. 2010;12:1248. doi: 10.1021/ol100110z. Kurokawa T, Kim M, Bois JD. Angew. Chem., Int. Ed. 2009;48:2777. doi: 10.1002/anie.200806192. Wasa M, Yu J-Q. J. Am. Chem. Soc. 2008;130:14058. doi: 10.1021/ja807129e. Huard K, Lebel H. Chem.– Eur. J. 2008;14:6222. doi: 10.1002/chem.200702027. Liang C, Collet F, Robert-Peillard F, Müller P, Dodd RH, Dauban P. J. Am. Chem. Soc. 2008;130:343. doi: 10.1021/ja076519d. Li Z, Capretto DA, Rahaman R, He C. Angew. Chem., Int. Ed. 2007;46:5184. doi: 10.1002/anie.200700760.
- 3.For reviews, see: Driver TG. Org. Biomol. Chem. 2010;8:3831. doi: 10.1039/c005219c. Cenini S, Gallo E, Caselli A, Ragaini F, Fantauzzi S, Piangiolino C. Coord. Chem. Rev. 2006;250:1234. doi: 10.1002/chem.200801148. Bräse S, Gil C, Knepper K, Zimmermann V. Angew. Chem., Int. Ed. 2005;44:5188. doi: 10.1002/anie.200400657. Katsuki T. Chem. Lett. 2005;34:1304.
- 4.cf. Subbarayan V, Ruppel JV, Zhu S, Perman JA, Zhang XP. Chem. Commun. 2009;4266 doi: 10.1039/b905727g. Gao G-Y, Lu H, Subbarayan V, Tao J, Zhang XP. Organometallics. 2009;29:389. Ruppel JV, Jones JE, Huff CA, Kamble RM, Chen Y, Zhang XP. Org. Lett. 2008;10:1995. doi: 10.1021/ol800588p. Jones JE, Ruppel JV, Gao G-Y, Moore TM, Zhang XP. J. Org. Chem. 2008;73:7260. doi: 10.1021/jo801151x. Kawabata H, Omura K, Uchida T, Katsuki T. Chem.— Asian J. 2007;2:248. doi: 10.1002/asia.200600363. Kawabata H, Omura K, Katsuki T. Tetrahedron Lett. 2006;47:1571.
- 5.Dong H, Latka RT, Driver TG. Org. Lett. 2011;13:2726. doi: 10.1021/ol2008268. Stokes BJ, Richert KJ, Driver TG. J. Org. Chem. 2009;74:6442. doi: 10.1021/jo901224k. Dong H, Shen M, Redford JE, Stokes BJ, Pumphrey AL, Driver TG. Org. Lett. 2007;9:5191. doi: 10.1021/ol702262f. Shen M, Leslie BE, Driver TG. Angew. Chem., Int. Ed. 2008;47:5056. doi: 10.1002/anie.200800689. Stokes BJ, Dong H, Leslie BE, Pumphrey AL, Driver TG. J. Am. Chem. Soc. 2007;129:7500. doi: 10.1021/ja072219k.
- 6.For descriptions of the competing reactions, see: Caselli A, Gallo E, Ragaini F, Ricatto F, Abbiati G, Cenini S. Inorg. Chim. Acta. 2006;359:2924. Ragaini F, Penoni A, Gallo E, Tollari S, Gotti CL, Lapadula M, Mangioni E, Cenini S. Chem.—Eur. J. 2003;9:249. doi: 10.1002/chem.200390018. Cenini S, Tollari S, Penoni A, Cereda C. J. Mol. Cat. A. 1999;137:135.
- 7.Refer to the Supporting Information for a comprehensive description of the reaction conditions examined.
- 8.Photolysis or pyrolysis of aryl azides with ortho-alkyl substituents affords a variety of products including tar, anilines, azepines, indoles, and indolines. For leading reports, see: Murata S, Yoshidome R, Satoh Y, Kato N, Tomioka H. J. Org. Chem. 1995;60:1428. Smolinsky G, Feuer BI. J. Org. Chem. 1964;29:3097.
- 9.For recent reports of iron-catalyzed aliphatic C–H bond amination from azides, see: King ER, Hennessy ET, Betley TA. J. Am. Chem. Soc. 2011;133:4917. doi: 10.1021/ja110066j. King ER, Betley TA. Inorg. Chem. 2009;48:2361. doi: 10.1021/ic900219b.
- 10.For recent reports of copper-catalyzed aliphatic C–H bond amination from azides, see: Badiei YM, Dinescu A, Dai X, Palomino RM, Heinemann FW, Cundari TR, Warren TH. Angew. Chem., Int. Ed. 2008;47:9961. doi: 10.1002/anie.200804304. Badiei YM, Krishnaswamy A, Melzer MM, Warren TH. J. Am. Chem. Soc. 2006;128:15056. doi: 10.1021/ja065299l.
- 11.For reports of Ru-catalyzed amination, see: Milczek E, Boudet N, Blakey S. Angew. Chem., Int. Ed. 2008;47:6825. doi: 10.1002/anie.200801445. Shou WG, Li J, Guo T, Lin Z, Jia G. Organometallics. 2009;28:6847.
- 12.Sun K, Sachwani R, Richert KJ, Driver TG. Org. Lett. 2009;11:3598. doi: 10.1021/ol901317j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Espino CG, Fiori KW, Kim M, Du Bois J. J. Am. Chem. Soc. 2004;126:15378. doi: 10.1021/ja0446294. [DOI] [PubMed] [Google Scholar]
- 14.Azepines, common aryl nitrene by-products, were never observed for any of the substrates investigated. In general, the only by-product observed in our Rh2(II)-catalyzed amination reaction was aniline.
- 15.Smolinsky G. J. Am. Chem. Soc. 1961;83:2489. [Google Scholar]
- 16.For leading reports on the mechanism of related metal-mediated C–H bond amination reactions, see: Lyaskovskyy V, Suarez AIO, Lu H, Jiang H, Zhang XP, de Bruin B. J. Am. Chem. Soc. 2011;133:12264. doi: 10.1021/ja204800a. Fiori KW, Espino CG, Brodsky BH, Du Bois J. Tetrahedron. 2009;65:3042. Fiori KW, Du Bois J. J. Am. Chem. Soc. 2007;129:562. doi: 10.1021/ja0650450.
- 17.For crystal structures of metal-azide complexes, see: Waterman R, Hillhouse GL. J. Am. Chem. Soc. 2008;130:12628. doi: 10.1021/ja805530z. Dias HVR, Polach SA, Goh S-K, Archibong EF, Marynick DS. Inorg. Chem. 2000;39:3894. doi: 10.1021/ic0004232. Fickes MG, Davis WM, Cummins CC. J. Am. Chem. Soc. 1995;117:6384. Proulx G, Bergman RG. J. Am. Chem. Soc. 1995;117:6382.
- 18.For computational studies on the mechanism of metal nitrenoid formation from azides, see: (a) refs. 12a and 16a. Cundari TR, Dinescu A, Kazi AB. Inorg. Chem. 2008;47:10067. doi: 10.1021/ic801337f.
- 19.Kornecki KP, Berry JF. Chem. Eur. J. 2011;17:5827. doi: 10.1002/chem.201100708. [DOI] [PubMed] [Google Scholar]
- 20.cf. Vadola PA, Sames D. J. Am. Chem. Soc. 2009;131:16525. doi: 10.1021/ja906480w. Murarka S, Deb I, Zhang C, Seidel D. J. Am. Chem. Soc. 2009;131:13226. doi: 10.1021/ja905213f.
- 21.Murata S, Tsubone Y, Kawai R, Eguchi D, Tomioka H. J. Phys. Org. Chem. 2005;18:9. [Google Scholar]
- 22.Lin X, Zhao C, Che C-M, Ke Z, Phillips DL. Chem.— Asian J. 2007;2:1101. doi: 10.1002/asia.200700068. Nägeli I, Baud C, Bernardinelli G, Jacquier Y, Moraon M, Müller P. Helv. Chim. Acta. 1997;80:1087.
- 23.The observed kinetic isotope effect is composed of both a primary-and an assumed small secondary isotope effect. We thank the reviewer for pointing out that this secondary effect is not necessarily the same for the two diastereomeric transition states that lead to 15-d2 and 16-d2.
- 24.Wiberg KB. J. Am. Chem. Soc. 1954;76:5371. Cohen R, Graves CR, Nguyen ST, Martin JML, Ratner MA. J. Am. Chem. Soc. 2004;126:14796. doi: 10.1021/ja047613m.
- 25.A kH/kD value of 5 was reported by Lebel and Huard for the Rh2(II)-catalyzed C–H bond amination of cyclohexane using N-tosyloxycarbamate as the N-atom source. See ref. 2e.
- 26.If C–H bond cleavage did occur via a hydride shift, formation of carbocation 9 would be expected to trigger the migration of one of the methyl groups. For alkyl group migrations in Rh2(II)-catalyzed reactions of aryl azides, see: Sun K, Liu S, Bec PM, Driver TG. Angew. Chem., Int. Ed. 2011;50:1702. doi: 10.1002/anie.201006917.
- 27.At 100 °C and 80 °C the kinetic isotope effect was determined to be 5.7 and 3.7. The linear correlation of these data with temperature revealed the isokinetic temperature to be 43 °C.
- 28.For leading discussions on the isokinetic temperature, see: Carpenter BK. Determination of Organic Reaction Mechanisms. Wiley; New York: 1984. pp. 149–150. Leffler JE. J. Org. Chem. 1955;20:1202.
- 29.In contrast, other aliphatic C–H bond functionalization reactions show a preference for equatorial C–H bonds, see: Chen K, Eschenmoser A, Baran PS. Angew. Chem., Int. Ed. 2009;48:9705. doi: 10.1002/anie.200904474. Chen MS, White MC. Science. 2007;318:783. doi: 10.1126/science.1148597. Wehn PM, Lee J, Du Bois J. Org. Lett. 2003;5:4823. doi: 10.1021/ol035776u.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.