

Abstract
Patients infected with human immunodeficiency virus type 1 (HIV-1) often display neurological complications in late stage disease and increased viral loads directly correlated with higher concentrations of extracellular HIV-1 viral protein r (Vpr) in the blood serum and cerebrospinal fluid. Additionally, HIV-1-infected patients with a low CD4+ T-lymphocyte count displayed lower concentrations of reduced glutathione (GSH), the main intracellular antioxidant molecule, and lower level of survival. To establish a correlation between increased concentrations of extracellular Vpr and an oxidative stress-induced phenotype, the U-87 MG astroglioma cell line has been used to determine the downstream effects induced by Vpr. Conditioned media obtained from the human endothelial kidney (HEK) 293T cell line transfected either in the absence or presence of HIV-1 Vpr contained free Vpr. Exposure of U-87 MG to this conditioned media decreased intracellular levels of both adenosine triphosphate (ATP) and GSH. These observations were recapitulated using purified recombinant HIV-1 Vpr both in U-87 MG and primary human fetal astrocytes in a dose- and time-dependent manner. Vpr-induced oxidative stress could be partly restored by co-treatment with the antioxidant molecule N-acetyl-cysteine (NAC). In addition, free Vpr augmented production of reactive oxygen species due to an increase in the level of oxidized glutathione (GSSG). This event was almost entirely suppressed by treatment with an anti-Vpr antibody or co-treatment with NAC. These studies confirm a role of extracellular Vpr in impairing astrocytic levels of intracellular ATP and GSH. Studies are underway to better understand the intricate correlation between reductions in ATP and GSH metabolites and how they affect neuronal survival in end-stage disease.
Keywords: ATP, glutathione, Vpr, oxidative stress, astrocytes, HIV-1-induced neuropathogenesis
1. Introduction
Infection with human immunodeficiency virus type 1 (HIV-1) leads to a spectrum of neurodegenerative diseases from mild neurocognitive disorders to more severe forms such as HIV-associated dementia or neuroAIDS (Kaul et al., 2001). HIV-1-induced neurological disease is a consequence of a cascade of events that in the most severe form includes a massive influx of HIV-1-infected monocytic cells across the blood-brain barrier once this barrier is compromised due to persistent insult during the course of disease (Gonzalez-Scarano and Martin-Garcia, 2005). Cells crossing the blood-brain barrier trigger a series of interrelated events with ensuing effects on central nervous system (CNS) homeostasis (Williams and Burdo, 2011). HIV-1 infects CNS-resident cells during the early phase of infection by infiltrating HIV-1-infected peripheral blood cells crossing the blood-brain barrier (An et al., 1999). Both peripheral blood HIV-1-infected monocytes and to a lesser extent CD4+ T cells play crucial roles in these processes after they penetrate the CNS. Among cells of the CNS, microglia and perivascular macrophages are the primary targets of productive HIV-1 infection (Gonzalez-Scarano and Martin-Garcia, 2005). Astrocytes have also been shown to be susceptible to infection although the outcome is nonproductive with little infectious virus released (Brack-Werner, 1999; Brack-Werner et al., 1992; Churchill et al., 2006; Churchill et al., 2009; Tornatore et al., 1994; Tornatore et al., 1991). Oligodendrocytes, although infected by HIV-1 (Albright et al., 1996), are not believed to contribute to any great extent to the overall production of virus within the CNS (Bissel and Wiley, 2004). In contrast, neurons are considered to be refractile to HIV-1 infection (Kaul et al., 2001), and their demise, a manifestation of disease progression, occurs predominantly through a number of indirect mechanisms.
HIV-1 infection of the astrocytic compartment has been shown to occur via a CD4-independent mechanism (Harouse et al., 1991) in the absence of significant virus production. Nonetheless, given their high abundance in the CNS, astrocytes could represent a major viral reservoir. More recent studies have reevaluated the role astrocytes might play in late stage disease by demonstrating a higher frequency of HIV-1-infected astrocytes located in the proximity of perivascular areas, correlating with increased severity of neurocognitive impairment (Churchill et al., 2009). Astroglial infection is characterized by an initial small burst in viral production followed by a state of persistent infection with the presence of multiply spliced short transcripts (encoding primarily the accessory Nef protein), inefficient translation of large-unit structural proteins (gag and env), and almost undetectable levels of viral genomic transcripts (Brack-Werner, 1999; Brack-Werner et al., 1992; Gorry et al., 1999). Restriction in astrocytic infection is attributed not only to limited viral entry (Li et al., 2007) but also to a post-transcriptional block in the export of viral transcripts from the nucleus (Kleinschmidt et al., 1994). During late stage HIV-1 disease, an increased frequency of infiltrating monocytes and CD4+ T cells may also deliver neurotoxic factors, such as chemokines and viral proteins (Tat, viral protein r [Vpr], gp120, and Nef) (Giulian et al., 1990), which stimulates astroglia to secrete an elevated amount of glutamate, increasing the overall level of excitotoxicity. This sequence of events could play a significant role in astrocytic and neuronal dysregulation, leading to mild to severe neurocognitive impairment. Another characteristic of patients infected with HIV-1 in late stage disease is diffuse intracellular oxidation in the form of decreased availability of glutathione (GSH), the main cellular antioxidant and redox buffer, and augmented lipid oxidation, which triggers a cascade of downstream signaling events (Herzenberg et al., 1997). GSH is a tripeptide, comprised of cysteine, glutamate, and glycine, which has been shown to be synthesized in two subsequent adenosine triphosphate (ATP)-dependent reactions. Initially, glutamate and cysteine, the rate-limiting amino acid, are combined by the action of the γ-glutamyl-cysteine synthetase, forming the dipeptide γ-glutamyl-cysteine, which has been shown to be used as a substrate by glutathione synthetase to produce the end product GSH. As a thiol, GSH has been shown to function as an electron donor, thus detoxifying reactive oxygen species (ROS), covalently binding to xenobiotics or other toxic compounds. A decline in reduced GSH could be the result of different processes, leading to augmented levels of oxidized glutathione (GSSG), and reduction of the GSH/GSSG ratio, which is maintained at an elevated level under physiological conditions. Within the CNS, astrocytes possess a much more efficient glutathione system of peroxide detoxification compared to neurons (Dringen et al., 1999) and secrete GSH into the extracellular environment, which is taken up by neurons to synthesize GSH intracellularly (Snyder et al., 2009). All cell types require not only GSH for their survival but also a fully efficient glutathione detoxifying system to promptly scavenge both exogenously applied peroxides or endogenous ROS generated continuously during oxidative metabolism. GSH deficits could be generally counteracted by supplementation with L-cysteine such as N-acetyl-cysteine (or NAC, its acetylated product, less prone to oxidation in the extracellular environment), providing more substrate for GSH synthesis.
ATP is the major chemical energy molecule for intracellular metabolism. It is generated through phosphorylation of adenosine monophosphate and cellular respiration and is used as a phosphate group donor to fuel a large number of intracellular processes. Previously published studies and evidence of the physiological alterations caused by extracellular Vpr to astrocytes led us to examine the role of Vpr in causing a decline in these two metabolites as a possible cause of the aggravation observed in patients infected with HIV-1 who are suffering from severe neurocognitive impairment in late stage HIV-1 disease. Indeed, mouse models wherein the Vpr protein was only expressed in brain monocytoid cells showed release of the protein and downstream effects in the highly controlled CNS compartment, including glial hyper-activation, secretion of neurotoxins and subsequent neuronal loss (Jones et al., 2007). Additionally, Vpr-induced effects were also noticeable in the peripheral nervous system, with increased calcium influx and action potential frequency. This resulted in dorsal root ganglion neuronal damage due to excessive interferon alpha production (Acharjee et al., 2010). In a further study by the same group, transgenic mice expressing Vpr in microglial cells displayed augmented astrocytic apoptosis due to excessive effector caspase activation (Noorbakhsh et al., 2010).
Our study delineates a correlation between exogenous Vpr and reduction in ATP and GSH in a dose-dependent manner, along with increased intracellular oxidation in the form of increased ROS production and decreased GSH/GSSG ratios in an astroglioma cell line. These lines of evidence not only led us to confirm previous studies performed in different cell types (thus suggesting a Vpr-induced cell-type independent phenotype) but also to advocate an appealing hypothesis with respect to the role played by Vpr in late stage disease when its levels increase in patients infected with HIV-1.
2. Material and methods
2.1. Materials and cell maintenance
The human endothelial kidney (HEK 293T) cell line (American Type Culture Collection, ATCC, CRL-11268, Manassas, VA) was grown in 1× Dulbecco’s modified Eagle’s medium (Cellgro, Mediatech, Manassas, VA) supplemented with 10% heat-inactivated fetal bovine serum (GemCell, Gemini Bio-Products, West Sacramento, CA), antibiotics (penicillin and streptomycin, at a concentration of 0.04 mg/ml each, Cellgro), glucose (4.5 g/L, Cellgro), sodium pyruvate (1 mM, Cellgro), and HEPES (10 mM, Cellgro). The human astroglioma cell line U-87 MG (ATCC, HTB-14) was grown in 1× Dulbecco’s modified Eagle’s medium supplemented with 10% heat-inactivated fetal bovine serum, penicillin, and streptomycin (at a concentration of 0.04 mg/ml each), 0.04 mg/ml kanamycin sulfate, and 7.5% sodium bicarbonate (final concentration 0.05%). Primary human fetal astrocytes (HFA) were purchased from ScienCell Research Laboratories (Carlsbad, CA) and maintained in astrocyte medium containing 1% astrocyte growth supplement, 2% FBS, and 1% penicillin/streptomycin as described by the manufacturer. All experiments using HFA were performed with cells passaged no more than twice because the astrocytic intracellular marker glial fibrillary acidic protein (GFAP) is gradually lost. All cells (primary or cell line) were maintained at 37°C in 5% CO2 at 90% relative humidity. NAC, BSO, and L-glutathione were obtained from Sigma-Aldrich (St. Louis, MO).
2.2. Plasmid construction
For all studies, the noninfectious HIV-1 pNL4-3R+E− molecular clone was used (obtained through the National Institutes of Health (NIH) AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH, obtained from Dr. Nathaniel Landau (Connor et al., 1995; He et al., 1995)). To construct the 6His-HA-Vpr plasmid, two single-stranded DNA sequences (sense and antisense) were initially synthesized (Integrated DNA Technologies, Coralville, Iowa): AGCTTGCCACC ATGGGACATCATCACC ATCACCATTACCCATACGATGTTCCAGATTACGCTT and the reverse complement 5 – CCGGAAGCGTAATCTGGAACATCGTATGGGTAATGGTGATGGTGATGATGTC CCATGGTGGCA. The sense sequence incorporates a consensus Kozak sequence prior to the initial ATG transcription start site (in bold). The sense sequence also contains 6His and HA coding sequences underlined and double underlined, respectively. The 5′ end of the antisense sequence also contains an AccIII cleavage site. The sense and antisense sequences were annealed in 1× saline-sodium citrate solution, boiled for 5 min, and incubated for 1 h at 45°C. The double-stranded DNA sequence was subsequently digested with the AccIII restriction endonuclease (Promega, Madison WI) for 1 h at 65°C. The Vpr coding sequence was amplified by a polymerase chain reaction (PCR) assay from the pNL4-3R+E− molecular clone using forward (5′ – CGCATCCGGAGAACAAG CCCCAGAAGACC) and reverse (5′ – GCAGCTCGAGCTAGGATCTACTGGCTCC) PCR primers, engineered to harbor an AccIII and an XhoI restriction endonuclease cleavage site, respectively (underlined). The PCR-amplified fragment was digested with AccIII at 65°C for 1 h. The double-stranded DNA sequence containing the 6His and HA tags along with the Vpr PCR-amplified fragment (both containing the compatible AccIII overhangs) was then ligated overnight at 4°C with T4 DNA ligase (Promega). The double-stranded 6His-HA-Vpr segment and the pcDNA3.1 vector were then digested with the respective restriction endonucleases HindIII and XhoI (Promega) and cloned by overnight ligation at 4°C with T4 DNA ligase (Promega). In cotransfection studies, the pNL4-3R−E− molecular clone (with a 4–base-pair insertion designed to knock out the Vpr coding sequence) was also used and obtained similarly to the aforementioned pNL4-3R+E−. The 3HA-Vpr plasmid, described previously (Xiao et al., 2008), contains three adjacent stretches of the HA tag at the 5′ end of the Vpr ATG transcription start site.
For recombinant Vpr purification, a GST-tagged Vpr construct was used (provided by Dr. Bassel Sawaya, Temple University, Philadelphia, PA (Deshmane et al., 2009; Rom et al., 2009)). The aforementioned 6His-HA-Vpr DNA sequence was cloned within the pGEX-4T-1 vector (GE Healthcare, Waukesha, WI) to obtain GST-6His-HA-Vpr, since the presence of the 6His stretch facilitated the purification process. Additionally, the presence of a thrombin cleavage site (Pro-Arg↓Gly-Ser) at the 3′ end of the GST DNA sequence and 5′ end of the initial Vpr ATG transcription start site separated the two proteins (GST from the Vpr), thus yielding a 6His-HA-Vpr protein for in vitro studies. The following PCR primers were utilized: 5′ – ATTCGGATCCATGGGACATCATCACC (forward) and 5′ – GGCTTCTAGACTAGGATC TACTGGCTCC (reverse), engineered to contain BamHI and XbaI cleavage sites (underlined), respectively. The cloning process was performed similar to that described for 6His-HA-Vpr, followed by restriction endonuclease digestion with BamHI and XbaI (Promega) and a final ligation overnight at 4°C with T4 DNA ligase (Promega).
2.3. Western immunoblot assays
Harvested cells or cell pellets were washed twice in phosphate-buffered saline (PBS), lysed in 0.5× radio-immunoprecipitation assay buffer supplemented with a protease and phosphatase inhibitor cocktail (Calbiochem, Merck, Darmstadt, Germany) and subjected to three freeze-thaw cycles. Lysed samples were then cleared of nucleic acids by centrifugation and the supernatant was collected. Protein concentrations were determined utilizing the Coomassie Plus (Bradford) Protein Assay (Pierce, Thermo Fisher Scientific, Rockville, IL), as described by the manufacturer. Equal amounts of whole cell lysates were separated using 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to an Immobilon-P polyvinylidene difluoride membrane, followed by blocking with 5% (w/v) nonfat dry milk in PBS. Membranes were incubated overnight at 4°C with primary antibody in 3% (w/v) nonfat dry milk in PBS. Specific protein bands were then detected using a horseradish peroxidase-conjugated secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA). Spotted antigen-antibody complexes were visualized using a chemiluminescent detection procedure (Pierce). For stripping, membranes were incubated for 1 h at room temperature using the Restore Western Blot Stripping Buffer (Thermo Fisher Scientific) and reblotted in 5% (w/v) nonfat dry milk in 1× PBS. Antibodies used in these studies were the anti-HA (Cell Signaling Technology, Danvers, MA), anti-β-actin (Sigma-Aldrich), and anti-p24 (Santa Cruz Biotechnology, Santa Cruz, CA), all diluted at 1:1000 except the anti-β-actin (1:5000).
2.4. Transfection and preparation of conditioned medium
The procedure was similar to that described previously, with a number of small modifications (Xiao et al., 2008). Briefly, exponentially growing HEK 293T cells (2 × 106) were seeded in 10 ml of growth medium in 10-cm Petri dishes. Transfection was performed using the classical calcium phosphate method, where a solution of CaCl2 (final concentration 60 mM) was slowly added to 15 μg of a DNA mixture composed of Vpr-containing or Vpr-deficient plasmids (5 μg) in trans with the Env- and Vpr-deficient molecular clone (10 μg of pNL4-3R−E−). The CaCl2–DNA mixture was then dispensed dropwise into a solution of 2× HEPES buffered saline, pH 7.0. Following a 30-min incubation, the CaCl2–DNA–2× HEPES buffered saline mixture was added dropwise onto the cell culture monolayer. The cell culture medium was changed after incubation for 5 h, and the cells were left unperturbed for 48 h, which allowed the cells to produce viral particles. Virus-containing conditioned medium was centrifuged and filtered through a 0.45-μm syringe filter. Cell-free virus-containing conditioned medium was slowly layered on top of a 20% sucrose gradient and subjected to centrifugation at 28,000 rpm (Beckman Coulter, Brea, CA) for 2 h at 4°C. CFVF conditioned medium (on top of the sucrose level) was collected and either used immediately with freshly cultured U-87 MG astrocytes or stored at −30°C for a brief period before use. Pelleted virions were resuspended in 80 μl of 1× PBS and maintained at 4°C overnight, after which time viral particles were lysed as indicated above to determined the level of Vpr incorporation.
The CFVF portion of cells cotransfected with the R−E− molecular clone, along with the 3HA-Vpr plasmid, was precipitated with ice-cold acetone. Samples were then incubated overnight at −80°C, washed in ice-cold acetone, and subjected to centrifugation to pellet and concentrate proteins within the supernatant. Pellets were then lysed in 0.5× radio-immunoprecipitation assay buffer and subjected to immunoprecipitation with an anti-HA antibody. Anti-HA immunoprecipitation samples were captured by protein G-agarose beads and then resolved using 12% SDS-PAGE. Spotted antigen-antibody complexes were visualized using a more sensitive SuperSignal ECL method (Pierce, Fisher Scientific).
2.5. Purification of recombinant Vpr
Recombinant Vpr was purified as reported previously, with minor modifications (Deshmane et al., 2009; Rom et al., 2009). GST-6His-HA-Vpr was expressed from BL21 DE3 RIPL (Stratagene, Agilent Technologies, Santa Clara, CA) bacteria by induction with isopropyl β-D-1-thiogalactopyranoside for 15 min at 28°C. Cultures were then pelleted and lysed in NETN lysis buffer, NaCl (100 mM), EDTA (1 mM), Tris pH 8.0 (20 mM), NP-40 (0.5%), phenylmethylsulfonyl fluoride (1 mM), and protease inhibitor cocktail (1:100). The lysate was sonicated until clear and subsequently purified by affinity chromatography using glutathione-sepharose beads (GE Healthcare) binding to the GST tag of the 6His-HA-Vpr protein. The retained protein was released in elution buffer (30 mM reduced GSH in 50 mM Tris·HCl buffer, pH 8) and cleaved with thrombin (10 U/mg of total protein) for 2 h. An additional affinity chromatography step was performed using a His SpinTrap nickel column (GE Healthcare) to concentrate 6His-HA-Vpr. Due to the presence of imidazole in the eluted protein, the preparation was dialyzed overnight at 4°C in 1× PBS, and the protein concentration was determined by the Bradford assay.
2.6. Immunocytochemical analyses
Adherent U-87 MG astroglioma cells were grown on polylysine-coated coverslips. Cells were washed twice with 1× PBS prior to fixation in 4% paraformaldehyde for 20 min at room temperature. Permeabilization of plasma membranes was performed with 0.2% Triton X-100 in 1× PBS for 5 min. Cells were washed with blocking buffer (5% goat serum, 1% bovine serum albumin in 1× PBS) and incubated overnight with primary antibody at 4°C. Staining with secondary Alexa Fluor 488 (green) or Alexa Fluor 595 (red)-conjugated antibody (Invitrogen, Carlsbad, CA) was performed for 1 h at room temperature in the dark. Cells were washed three times with 1× PBS between each step and incubated in 300 nM 4′,6-diamidino-2-phenylindole (DAPI)-containing 1× PBS to stain nuclei. Coverslips were then rinsed in ddH2O to remove saline residues, mounted on microscope slides with ProLong antifade reagent (Invitrogen), and immediately imaged with a deconvolution microscope (Olympus, Center Valley, PA). For all cell types, a minimum of 10 fields at 20× or 40× magnification was analyzed. For higher magnification, z-section images were captured (at least 20 different planes) at a 0.2-μm distance for each individual channel; planes were deconvolved using the nearest-neighbor algorithm; and volume images were obtained to confirm localization by planar view. The following primary antibodies were used for immunofluorescence microscopy studies: glial fibrillary acidic protein (Cell Signaling), xCT (Novus Biologicals, Littleton, CO), and 4F2hc (Abcam, Cambridge, MA), all at a dilution of 1:200.
To detect intracellular accumulation of ROS, the RedoxSensor Red CC-1 dye (Invitrogen) was utilized. Cells were fixed, permeabilized, and incubated with 2 μM RedoxSensor Red CC-1 dye in 1× PBS at 37°C for 10 min. Following three washes with 1× PBS, cells were stained with DAPI, mounted on slides, and processed as indicated above. All procedures were performed in dim light to avoid photo-oxidation of the dye. To block Vpr-induced downstream effects, an anti-Vpr antibody was kindly provided by Dr. Jeffrey Kopp (NIH, Kidney Disease Section).
2.7. Quantitation of ROS+ cells
To better estimate the downstream effects of extracellular Vpr on accumulation of ROS, the number of ROS+ cells (i.e. cells positively staining for the RedoxSensor Red CC-1 dye) was quantitated using two masks for the DAPI and FITC channels. DAPI+ cells with a fluorescent green staining within the cell membrane (as overlapped by phase contrast images) were considered FITC+ (or ROS+) and accounted for in the analysis. A minimum of 10 different fields of view each with at least 50 viable cells were captured and quantitated with this automated method. Results are then reported as percentage of DAPI+ ROS+ cells for the different treatments.
2.8. GSH and ATP assays
For all experiments involving measurement of intracellular ATP and reduced GSH concentrations, the CellTiter-Glo Luminescent Cell Viability Assay and the GSH-Glo Glutathione Assay (Promega), respectively, were used as described by the manufacturer with minor modifications. To determine the total protein concentration, the Bio-Rad Protein Assay (Bio-Rad Laboratories, Hercules, CA) was used as described by the manufacturer. Treated or untreated cells were collected by low speed centrifugation and 5% of each preparation was used for ATP and GSH determinations; the remainder of each preparation was used to normalize to total protein concentration.
Cells used for the ATP measurement were resuspended in 50 μl of 1× PBS and dispensed in an individual well of a 96-well opaque plate. An equal volume (50 μl) of CellTiter-Glo Reagent was added to both samples and standards; the contents were mixed and incubated at room temperature for 10 to 30 min. Luminescent signals were detected using a precalibrated Glomax luminometer (Promega) at different time points until each signal peaked to the maximum value. ATP concentrations were calculated after the noise (no cells) was subtracted from a standard curve. Cells used for the GSH measurement were instead resuspended in 25 μl of 1× PBS and dispensed in single wells of a 96-well plate; an equivalent amount of 2×GSH-Glo reagent (1:1:50 of Luciferin-NT substrate:GST:GSH-Glo reaction buffer) was added to each well and mixed. After incubation for 30 min at room temperature, 50 μl of reconstituted Luciferin Detection Reagent was added to each sample followed by an additional incubation for 15 min. GSH luminescent signals were recorded using a procedure similar to that for obtaining ATP measurements until values peaked; GSH concentrations were determined from the standard curve after background subtraction. For determination of total GSH and oxidized glutathione (GSSG) to calculate GSH/GSSG ratios, a similar approach was followed using the GSH/GSSG-Glo Glutathione Assay (Promega). Cells cultured in a 96-well tissue opaque culture plate (BD Bioscience, Sparks, MD) were harvested at each specific time point by removal of the cell culture medium followed by immediate lysis and assay for total and oxidized GSH as described by the manufacturer. To calculate GSH/GSSG ratios, the following formula was used: 2(total GSH/GSSG) − 1, since each molecule of oxidized glutathione gives rise to two molecules of reduced glutathione on breakage of the disulfide bond. All results were plotted as the ratio of treated to untreated cells.
2.9. Statistical analysis
The results were expressed as means ± standard error and analyzed statistically using the Student’s two-tailed t test. Differences between groups were considered significant if P values < 0.05 were obtained.
3. Results
3.1. Detection of HIV-1 Vpr as an extracellular protein
Soluble Vpr has been detected in vivo in the blood serum and the cerebrospinal fluid as a cell-free, virus-free (CFVF) protein of patients infected with HIV-1, and concentrations of extracellular Vpr were higher in patients in late stage disease compared to those who were asymptomatic or in the clinical latency phase of disease (Hoshino et al., 2007; Levy et al., 1994; Levy et al., 1995). A recent study also demonstrated that human endothelial kidney (HEK) 293T cells, Jurkat cell lines, as well as peripheral blood mononuclear cells, are able to release Vpr in the extracellular space in an vitro model system (Xiao et al., 2008). These observations underscored the ability of the virus to spread its detrimental effects not only within the infected cell population but also to uninfected bystander cells in a paracrine fashion. Little is known about the cellular sources of extracellular Vpr within the CNS and how they could affect HIV-1 disease progression as previously discussed (Ferrucci et al., 2011). The studies reported herein are aimed at examining the downstream effects of exogenous CFVF Vpr in an astroglioma cell line as a model of the cascade of events triggered by Vpr. In this regard, HEK 293T cells were used as a readily transfectable in vitro model cell line to verify the presence of Vpr in a CFVF supernatant. Intracellular expression of Vpr was examined after calcium phosphate transfection of HEK 293T cells. As expected, the intracellular expression of both the 6-histidine (6His)-hemagglutinin (HA)-Vpr and 3HA-Vpr was readily detectable by immunoblotting (Figure 1A, ~15 kDa and ~19 kDa, respectively), which showed successful transfection of HEK 293T cells. The p24 capsid protein was detected only in those samples transfected with the Vpr- and Env-defective proviral construct (NL4-3R−E−), which proved the presence of functionally structured virions. On the other hand, virion-incorporated 3HA-Vpr was found only in viral particles egressing from cells co-transfected with the NL4-3R−E− molecular clone and the 3HA-Vpr plasmid (Figure 1B). When the CFVF in the extracellular medium was analyzed by western immunoblotting, only the media derived from cells cotransfected with both 3HA-Vpr and the Vpr-deficient molecular clone resulted in the presence of extracellular Vpr (Figure 1C), as previously reported (Xiao et al., 2008). However, unlike Xiao and coworkers, the cleavage of Vpr was not detected in the CFVF extracellular medium, although a cleaved product could be observed in the cell lysate (Figure 1A, lane 3). However, the acetone treatment may not precipitate the cleavage product (found in the intracellular lysate compartment) when applied to CFVF medium, which could explain the different in the two results. Additionally, a slower-migrating complex around 35–40 kDa was detected (Figure 1C, lane 4), which might represent dimerization of Vpr. Collectively, these results show expression of Vpr intracellularly, within viral particles and in the CFVF extracellular medium only in those cells transfected with the molecular viral clone.
Figure 1. Viral protein r (Vpr) is expressed in different intra- and extracellular compartments.
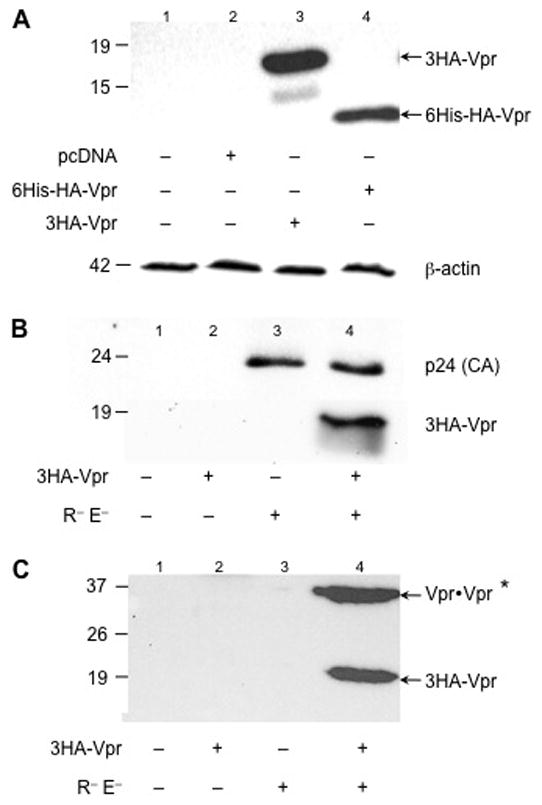
(A) Intracellular expression of two types of hemaglutinin (HA)-tagged Vpr proteins in HEK 293T cells. Whole cell lysates of untransfected 293T cells or 293 T cells transfected with either an empty vector (pcDNA) or with selected Vpr-containing plasmids were separated using 10% SDS-PAGE and probed with anti-HA antibody. In lanes 3 and 4, pcDNA-3HA-Vpr or the faster-migrating constructs (6His-HA-Vpr) have been transfected, respectively. As a loading control, membrane was stripped and probed with anti-β-actin antibody. (B) Expression of p24 capsid protein of virions collected 48 h post-transfection from the supernatant of 293T cells transfected with the constructs reported below. (C) After ultracentrifugation at 28,000 rpm, cell-free, virus-free supernatant was collected and precipitated with acetone. Subsequently, immunoprecipitation (with anti-HA antibody) was performed followed by western immunoblotting. Only the samples cotransfected with Vpr and the other viral proteins display Vpr in the CFVF. *Possible presence of a Vpr dimer. 6His: 6-histidine
3.2 Determining extracellular HIV-1 Vpr-induced effects on astroglioma metabolism
HIV-1 Vpr has been reported to be toxic when added to mixed neural cell cultures (Huang et al., 2000; Sabbah and Roques, 2005). To examine the downstream effects extracellular Vpr exerts upon astrocytes (the most abundant cell types in the CNS), we proceeded to establish an expression system using HEK 293T cells. In this regard, the production of Vpr was explored within the intra- and extracellular environments of transfected HEK 293T cell cultures. U-87 MG cells were exposed to different conditioned media obtained from either 293T cells in the absence or presence of transfection with the Vpr-deficient NL4-3R−E− molecular clone or the 3HA-Vpr plasmid in conjunction with the NL4-3R−E− molecular clone. A kinetic analysis was performed with respect to time and the level of intracellular GSH using a luciferase-based GSH detection assay (Figure 2A). A downward trend in reduced GSH was observed in U-87 MG cells exposed to the Vpr-containing conditioned medium (dark grey bar) starting at 12 h post-treatment (although not statistically significant) and continued for the duration of the treatment up to 48 h (Figure 2A). In contrast, HIV-1 conditioned medium devoid of Vpr does not induce a decrease in the levels of GSH and ATP in U-87 MG cells (light grey bars). As a positive control, U-87 MG cells were treated with buthionine sulfoximine (BSO), which has been shown to irreversibly inhibit γ-glutamylcysteine synthetase and GSH synthesis. U-87 MG cells treated with BSO showed an incremental decrease in the levels of GSH in a dose- and time-dependent manner (data not shown), displaying a tenfold decrease 24 h post-treatment (black bar). Because GSH has been shown to be synthesized in two ATP-dependent reactions from its constituent amino acids (cysteine, glutamate, and glycine) and because recent evidence demonstrates extracellular Vpr-induced ATP reduction in neurons (Kitayama et al., 2008; Rom et al., 2009), the levels of ATP were examined within U-87 MG exposed to Vpr-containing or Vpr-deficient conditioned media. At 24 h, Vpr-containing conditioned media (dark grey bars) showed a 30% reduction in the levels of ATP, which became statistically significant by 48 h post exposure (50% decrease, Figure 2B). These results confirm previously published studies (Rom et al., 2009), although the ATP decline was not as pronounced, most likely due to the low concentration of extracellular Vpr in tissue culture conditioned media.
Figure 2. HIV-1 conditioned medium containing Vpr induces a decrease in the levels of reduced GSH and ATP in U-87 MG cells and counteracted by anti-hemaglutinin (HA) antibody.
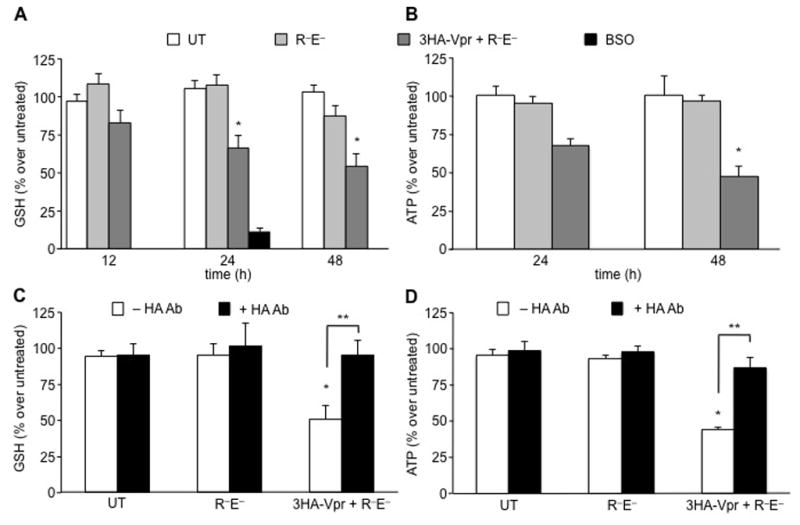
(A) To examine the temporal effect of extracellular Vpr on the levels of intracellular GSH, conditioned medium from HEK 293T cells transfected with the indicated constructs was applied to U-87 MG cells for 24 h and 48 h. Only cells cultured with extracellular Vpr-containing conditioned medium manifested a decrease in the levels GSH in a timely fashion (dark grey). On the contrary, untreated cells (white bars) or conditioned medium from Vpr-deficient transfected 293T cells (light grey) did not exhibit an altered level of GSH. As a positive control, U-87 MG cells treated with buthionine sulfoximine (BSO) (100 μM for 24 h), an irreversible inhibitor of γ-glutamylcysteine synthetase, induced a 90% decrease in the concentration of GSH (black bar). (B) The levels of ATP were shown to decrease only in conditioned medium containing Vpr at 24 h post-treatment. (C) Viral protein r (Vpr)-containing conditioned medium induced reduction in the level of GSH was almost entirely recovered after treatment with anti-HA antibody directed against the N-terminus tag of Vpr. Anti-HA antibody added to conditioned medium not containing Vpr did not induce any effects on GSH concentrations as shown by the black bars. (D) Similarly, Vpr-containing conditioned medium-treated U-87 MG displayed reduced levels of ATP compared with Vpr-deficient conditioned medium-treated cells. Addition of the anti-HA antibody resulted in almost complete recovery of initial ATP levels.
*P value < 0.01, **P value < 0.05 (Student paired t test). Values are determined as percent over control at 0 h time point (*) or between untreated (UT) and HA antibody (Ab) treated samples (**).
3.3. Replenishment of ATP and GSH by treatment with anti-HA antibody
We hypothesized that the observed decreases in GSH and ATP pool levels induced by Vpr-containing 293T conditioned medium could be attributed to the presence of extracellular Vpr. Hence, U-87 MG cells were exposed to Vpr-containing or Vpr-deficient 293T conditioned media in the absence or presence of anti-HA antibody, targeting the N-terminus HA tag of Vpr to neutralize or partially block Vpr present within the extracellular medium, thus not allowing downstream signaling leading to the observed decreases in the levels of GSH and ATP. The antibody directed against the N-terminal HA tag (used to detect extracellular Vpr as in Figure 1C) was used to block Vpr and prevent its translocation or downstream signaling effects, which caused the observed decreases in astrocytic metabolism. Indeed, U-87 MG cells exposed to Vpr-containing conditioned media in the presence of anti-HA antibody demonstrated almost a total recovery in the levels of GSH (Figure 2C) and ATP (Figure 2D). These observations point to a direct involvement of Vpr in the reduction of the level of intracellular GSH, either through translocation from the extracellular compartment or by signaling through membrane-associated receptors, which could generate a cascade of signaling events leading to the decreased levels of ATP and GSH. Overall, anti-HA antibody treatment of U-87 MG cells exposed to Vpr-containing conditioned medium showed an amelioration of Vpr-induced metabolic effects.
3.4. NAC counteracts extracellular Vpr-induced oxidative stress
Oxidative stress-inducing stimuli promote glutathione depletion and a cascade of signaling events leading to transcription of stress-response genes and DNA damage (Deshmane et al., 2009). The antioxidant compound NAC, a precursor of cysteine, may partially reverse GSH exhaustion. To evaluate whether Vpr-induced oxidative stress in astrocytes could be counteracted by treatment with NAC, the presence of system xc, primarily responsible for cysteine uptake, in U-87 MG astroglioma cells was verified. System xc, a heterodimeric protein complex composed of a catalytic unit (xCT) and a regulatory heavy chain (4F2hc), also referred to as CD98 (Sato et al., 1999), is the key mediator in the uptake of extracellular cystine (the oxidized form of cysteine) in exchange for intracellular glutamate and functions in a Na+- and ATP-independent manner. Because U-87 MG cells have been shown to express both subunits (Chung et al., 2005; Kim et al., 2001; Lyons et al., 2007) at the mRNA and protein levels, it was determined by immunofluorescence microscopy as to whether system xc was correctly localized at the cell membrane. In this regard, both the xCT (Figure 3A) and 4F2hc (Figure 3B) units were expressed at the plasma membrane and within the cytoplasm, the latter possibly due to constitutive expression of both units of the system xc, which are synthesized and transported to the cell membrane to meet cellular demands (Lyons et al., 2007). In addition, a magnified view (100×) of both subunits showed accumulation predominantly at the plasma membrane (Figure 3C).
Figure 3. N-acetyl-cysteine (NAC) partially rescues reduced glutathione (GSH)-depleted U-87 MG cells.
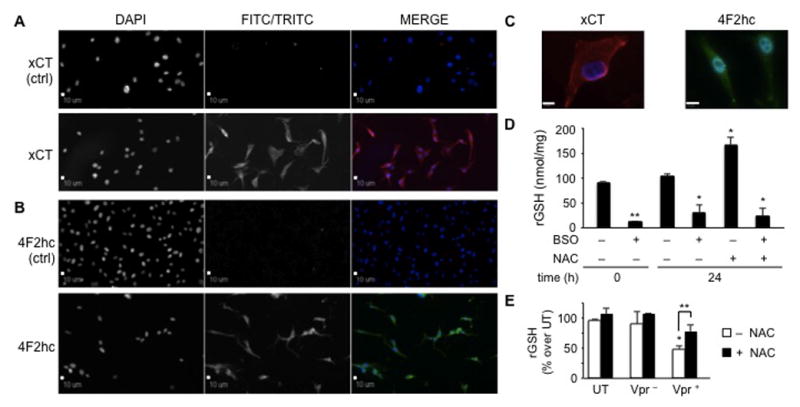
U-87 MG astroglioma cells possess the cystine/glutamate antiporter system, composed of a light subunit, catalytic unit (xCT) (A) and a regulatory heavy chain (4F2hc) (B), both of which localize predominantly at the plasma membrane. Astroglioma cells abundantly synthesized both subunits for continuous uptake of cystine in exchange for intracellular glutamate, thus giving rise to a clear intracellular staining pattern. The first column shows stained nuclei (DAPI), whereas the second column represents the TRITC/red (for xCT) or FITC/green (for 4F2hc) channel. These two channels are shown in black and white and in color when the results from the two channels are merged in the third column (magnification 20×). (C) Detailed images of the two subunits in U-87 MG cells are shown at magnification 100×, which revealed dispersed intracellular presence as well as presence at the plasma membrane. Scale bars = 10 μm. (D) The xc system is functional in U-87 MG. Cells were cultured in the absence or presence of known compounds capable of either decreasing (buthionine sulfoximine, BSO) or increasing (N-acetyl-cysteine, NAC) the intracellular concentration of reduced glutathione (GSH). Treatment with BSO (100 μM) induced an approximate tenfold decrease in GSH concentration. Cells treated for 24 h with NAC (5 mM) displayed a 70% to 80% increase in the level of intracellular GSH. On the other hand, the level of intracellular GSH in cells pretreated with BSO and subsequently with NAC was not rescued due to the irreversibility of the action of BSO on γ-glutamyl-cysteine synthetase. *P values < 0.05; **P value < 0.001 compared with untreated cells (Student paired t test). (E) U-87 MG cells treated with Vpr-deficient or Vpr-containing conditioned medium displayed an increase in intracellular GSH concentrations. U-87 MG cells exposed to extracellular Vpr-containing conditioned medium and treated with NAC for 24 h partially recovered cells from the induction of oxidative stress. *P value < 0.001 of untreated compared to Vpr-treated U-87 MG. **P value < 0.05 compared to U-87 MG exposed to Vpr-containing conditioned medium in the absence of NAC (Student paired t test). P-values are determined as percent over control at 0 h time point.
After demonstrating the detection of both subunits of the system xc, their functionality was examined with respect to taking up cystine for glutamate. Activity of the system xc could be quantitated indirectly by measuring the synthesis of the downstream product (glutathione), which relies primarily on high affinity cystine uptake by the system xc. To verify the functionality of the cysteine/glutamate antiporter system, U-87 MG astrocytes were treated with 5 mM NAC and the levels of GSH were measured 24 h postexposure. NAC-treated cells displayed a statistically significant increase of 80% of the original untreated levels of GSH (Figure 3D). This increase was specific and due to NAC uptake, because untreated cells did not exhibit altered levels of GSH. Functionality of the system xc has been reported previously for astroglioma cell lines. Gliomas are known to secrete abundant amounts of glutamate through the system xc (Lyons et al., 2007). As a result, they can expand and survive in an otherwise excitotoxic and neurotoxic environment. As a positive control, U-87 MG cells were also treated with the GSH-decreasing compound BSO, which has been shown to promote an irreversible decrease in GSH levels. Indeed, addition of NAC to the cell culture medium of U-87 MG cells previously treated with BSO did not rescue the intracellular levels of GSH (Figure 3D).
After the ability of U-87 MG cells to take up extracellular cysteine was verified, we sought to determine whether NAC treatment could counteract Vpr-induced intracellular oxidation. U-87 MG cells treated either with Vpr-deficient or Vpr-containing conditioned medium were then exposed to 5 mM NAC for 24 h prior to harvest. Cells assayed for GSH content displayed a statistically significant restoration of the GSH pool back to about 80% of initial values (Figure 3E). This observation underscores how the extracellular Vpr-induced decrease in GSH metabolism could be partially rescued after the addition of an antioxidant compound. Nevertheless, complete reversal of the oxidative phenotype could not be achieved.
3.5. Effects of recombinant HIV-1 Vpr on astrocytic metabolism and ROS production
To recapitulate our previous observations, we proceeded to use recombinant Vpr employing the 6His-HA-Vpr protein (as described in Materials and Methods). To further extend the previous studies using Vpr-containing HEK 293T conditioned medium, U-87 MG cells were exposed to recombinant HIV-1 Vpr and assayed for the intracellular levels of ATP and GSH. Vpr induced a statistically significant decrease in ATP concentrations as early as 24 h post-treatment (30% reduction), decreasing to 50% and to 20% of the initial values after 48 h and 72 h of exposure, respectively (Figure 4A, black line). Similarly, the level of GSH was decreased at all time points examined, with a statistically significant decrease of 40% of the initial values after 72 h (Figure 4A, dotted line). Different oxidative stress-inducing compounds affect not only intracellular levels of total GSH, but also, and more importantly, the concentration of reduced GSH. These events coincide with an increased concentration of oxidized GSSG and a decrease in the ratio of reduced to oxidized glutathione (GSH/GSSG), an overall indicator of intracellular health. Because extracellular Vpr induced a statistically significant decrease in reduced GSH (Figure 4A), we sought to determine whether this decrease was due to a reduction in the GSH/GSSG ratio as a consequence of increased formation of GSSG. In an experiment similar to the one shown in Figure 4A, U-87 MG astrocytic cells were exposed to Vpr, and the GSH/GSSG ratio was assayed at different time points. We found that, whereas untreated U-87 MG cells maintained a constant ratio (Figure 4B, black line), extracellular Vpr induced a significant decrease in the GSH/GSSG ratio due to the abundant production of oxidized glutathione (Figure 4B, dotted line). Additionally, exposure of U-87 MG to a nonspecific recombinant glutathione S-transferase (GST) protein decreased neither ATP nor GSH levels nor the GSH/GSSG ratio (data not shown), which confirmed the specificity of the observations by recombinant extracellular Vpr.
Figure 4. Extracellular rVpr induces a significant decline in intracellular ATP, reduced GSH, GSH/GSSG ratio, and accumulation of ROS in U-87 MG cells.
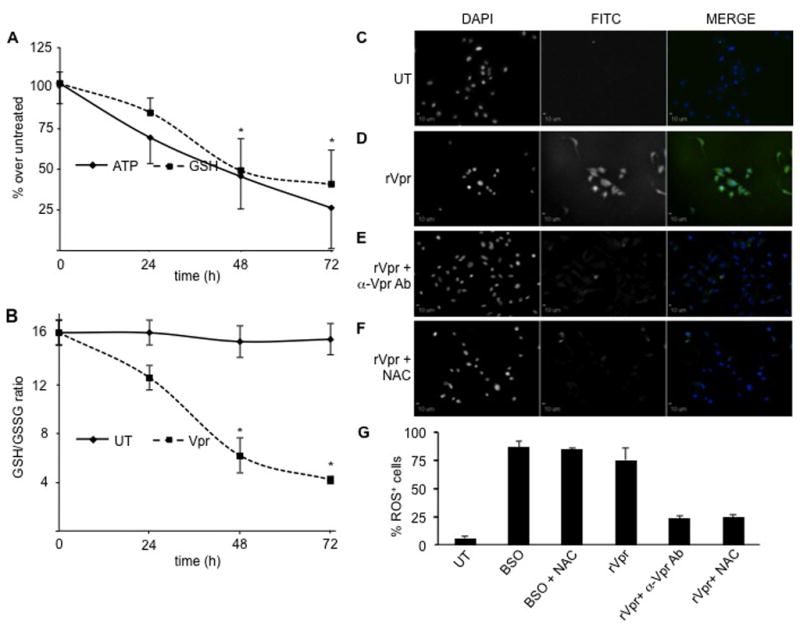
(A) U-87 MG astroglioma cells exposed to 5 μg/ml of extracellular recombinant Vpr showed a time-dependent decrease in ATP levels up to fourfold compared to untreated cells after 72 h exposure (black line). In a similar time-dependent manner, the concentration of GSH decreased about 2.5-fold after 72 h (dotted line). *P values < 0.05 compared to untreated cells (Student paired t test). (B) The intracellular levels of reduced and oxidized glutathione and the GSH/GSSG ratios were determined after incubation of U-87 MG astroglioma cells in either the absence or the presence of 5 μg/ml of recombinant Vpr for the indicated periods of time. Whereas untreated cells maintained constant levels of reduced and oxidized glutathione (GSH/GSSG stable), Vpr-treated cells exhibited a time-dependent decrease in the GSH/GSSG ratio. While untreated U-87 MG cells displayed no staining with the ROS-sensitive RedoxSensor Red CC-1 dye (C), exposure to extracellular recombinant Vpr (5 μg/ml) induced abundant accumulation of ROS (D), which was partially blocked by treatment with either anti-Vpr antibody (E) or addition of 5 mM NAC (F) for 24 h. In all images, the first column shows stained nuclei (DAPI), whereas the second column represents the FITC/green (RedoxSensor Red CC-1 dye) channel. These two channels are shown in black and white and in color when the results from the two channels are merged in the third column. All magnifications are 20×. Scale bars = 10 μm.
To further demonstrate the extracellular ability of Vpr to impair intracellular antioxidant defense mechanisms, we determined whether exposure to extracellular Vpr induced formation of ROS. ROS products are natural by-products of intracellular metabolism; however, if not disposed of properly by the cellular machinery in a timely manner, they accumulate within the mitochondria and lysosomes, thereby initiating intracellular signaling with damaging effects. Using a photo-oxidizable molecule (RedoxSensor Red CC-1), U-87 MG cells were treated with different compounds and examined for the intracellular accumulation of ROS. Untreated cells did not display any photo-oxidation of the ROS-susceptible dye (Figure 4C); on the other hand, BSO-treated U-87 MG cells exhibited abundant accumulation of ROS (data not shown), which is symptomatic of an excessive state of intracellular oxidation. As shown previously (Figure 3D), treatment with NAC was not capable of rescuing BSO-induced ROS buildup (data not shown) because this process was irreversible. When cells were exposed to recombinant Vpr, they displayed an increased accumulation of ROS, which resulted in intense intracellular staining (Figure 4D). However, treatment of U-87 MG with an anti-Vpr antibody abrogated Vpr-induced formation of ROS (Figure 4E). Similarly, addition of NAC (5 mM) for 24 h rescued U-87 MG astrocytic cells from induction of oxidative stress (Figure 4F).
To further expand our understanding concerning the effects of recombinant Vpr on astrocytic function and confirm our gain of knowledge in the astrocytic cell line, primary human fetal astrocytes (HFA) were used to study the downstream effects of Vpr treatment. To this end, a time- and dose-dependent analysis of the levels of ATP and GSH showed a significant decrease in both metabolites with all concentrations of Vpr employed at all time points, starting from 24 h postexposure (Figure 5A and B). Concentration values of ATP and GSH decreased almost in parallel compared with those from untreated HFA. This kinetic analysis confirmed results previously shown in the astroglioma U-87 MG cell line, wherein levels of ATP and GSH decreased synchronously with time (Figures 2A and B and 4A and B). In addition, exposure to recombinant Vpr disrupted the morphology of the cells, in that the finely regulated astrocytic architecture visible in untreated cells (Figure 5C) was lost in Vpr-treated HFA, which appeared more round-shaped with long thin processes and a condensed intracellular cytoplasm (Figure 5D).
Figure 5. Primary human fetal astrocytes (HFA) exposed to extracellular Vpr manifest a dose- and time-dependent decrease in concentrations of ATP and GSH.
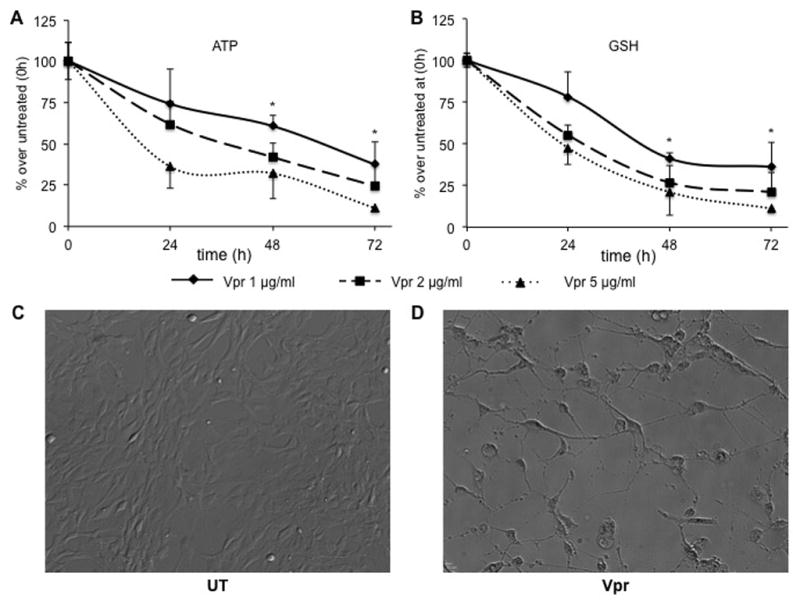
(A) Kinetic analysis of intracellular ATP concentration in HFA treated with different concentrations of extracellular Vpr demonstrated that ATP decreased to half of the initial levels in astrocytes 48 h post-treatment and decreased tenfold at 72 h after exposure. (B) A similar decrease was observed for GSH, with a slightly slower kinetic response than ATP. Phase contrast images of untreated (UT) HFA (C) and Vpr-treated (D) HFA (48 h post-treatment) show a detrimental loss of architecture and cell-cell communication. *P value < 0.05 between primary HFA exposed to any concentration of Vpr and untreated HFA (Student paired t test).
4. Discussion
The studies reported herein have been driven by the increasing evidence accumulated over the past decade that HIV-1 Vpr is present as a soluble protein within the blood serum and the CSF of patients infected with HIV-1 (Hoshino et al., 2010; Hoshino et al., 2007; Levy et al., 1994; Levy et al., 1995) and accumulates within these compartments to increasing concentrations as disease progresses toward the later stages. As an extracellular protein, HIV-1 Vpr has been shown to negatively affect the survival of brain-resident cells, especially neurons and astrocytes, which are the cell types most sensitive to local insult; they become dysfunctional and are gradually lost with disease progression, especially due to the lack of highly effective penetration of anti-retroviral treatment into the CNS. As a result, the studies were performed to improve overall insight into the role of HIV-1 Vpr in neurodegeneration and more specifically to investigate the impact of Vpr on the most abundant brain-resident cell population, the astrocyte. The impact of extracellular Vpr was first investigated within the context of conditioned medium derived from HEK 293T cells transfected with plasmids expressing Vpr and other HIV-1 proteins. The effects of Vpr-containing conditioned medium were first determined using an astroglioma cell line (U-87 MG). To this end, these cells were exposed to selected conditioned media in the absence or presence of HIV-1 Vpr. Reductions in ATP and GSH concentrations were blocked by treatment with anti-Vpr antibody, which led us to attribute these effects directly to the presence of extracellular Vpr present in the conditioned medium. To further rule out any effect of HIV-1 Vpr being due to the presence of possible Vpr-induced secreted cytokines and/or chemokines, we used different preparations of purified recombinant HIV-1 Vpr protein to determine that extracellular HIV-1 Vpr was capable of decreasing the level of the intracellular ATP pool. Since there is no published report that documents the levels of extracellular HIV-1 Vpr within the CNS, we employed concentration values as reported in previous in vitro studies (Acharjee et al., 2010; Noorbakhsh et al., 2010; Piller et al., 1999; Piller et al., 1998; Sabbah and Roques, 2005). In addition, we showed that recombinant HIV-1 Vpr protein similarly induced a reduction in the concentration of GSH, the main intracellular antioxidant molecule and essential to scavenge and reduce oxidant compounds produced during cellular metabolism. It was also shown that the concentrations of ATP and GSH were reduced by half after 48 h exposure to Vpr; by 72 h the decrease was about 20% and 40% for ATP and GSH, respectively. Additionally, a partial recovery from these effects was obtained on addition of the antioxidant compound NAC. The lack of complete recovery could be due to the presence of other Vpr-induced factors within the conditioned medium or to the time of NAC treatment (24 h post-exposure to recombinant Vpr and 24 h prior to harvest), because exogenous Vpr could have initiated a series of downstream effects that are not readily counteracted by NAC. In addition, NAC could also be partially oxidized in the extracellular milieu, so that not all NAC molecules gain access to the intracellular environment. The absence of a complete recovery is also evident intracellularly from the presence of few cells stained by the ROS-sensitive dye. Our results were also confirmed in primary human fetal astrocytes (a model more similar to in vivo human adult astrocytes than the astroglioma cell line); wherein similar (almost synchronously) decreases in both ATP and GSH were visible in a time and dose-dependent fashion. The direct effects of extracellular Vpr are particularly noticeable with respect to the morphology displayed by primary astrocytes upon long exposure to the viral protein, with severe loss of inter-astrocytic network and increased cell death and shrinkage. Current studies are underway in human fetal astrocytes to better examine the causality between Vpr exposure and induction of programmed cell death, wherein we noticed a caspase-dependent induction of apoptosis (Ferrucci et al., manuscript in preparation).
Because the concentrations of both ATP and GSH decreased during treatment with recombinant HIV-1 Vpr, two scenarios are possible from the results obtained. In one scenario, the GSH decrease could be a consequence of a decrease in the level of ATP, since GSH is synthesized in two subsequent ATP-dependent reactions. In this case, extracellular HIV-1 Vpr could enter into astrocytic cells and target the mitochondrial adenine nucleotide translocator protein, which has been shown to be involved in exchanging ATP molecules newly formed within the matrix for cytoplasmic ADP. Indeed, it was shown that the C-terminus of Vpr has a high binding affinity for a loop of the adenine nucleotide translocator in the intermembrane space of the mitochondria (Jacotot et al., 2001; Sabbah et al., 2006; Vieira et al., 2000). This high affinity binding would lead to a blockage in the passage of ATP to the cytoplasm, thus hampering ATP-dependent reactions such as GSH synthesis. It was reported previously that extracellular HIV-1 Vpr enters several different cell types, probably through a receptor-independent mechanism, and that bloodborne cells (primarily monocytes and CD4+ T cells) as well as neurons internalize extracellular Vpr when treated with preparations of recombinant Vpr (Jacotot et al., 2000; Rom et al., 2009; Sherman et al., 2002). This process eventually leads to the incorporation of soluble extracellular Vpr into the intracellular environment, possibly resulting in effects similar to those of intracellularly synthesized Vpr in infected cells. This evidence would be of particular interest in the uninfected bystander cells because Vpr, known to block cell cycle progression and induce apoptosis, could represent one of the causes of the massive cell death among uninfected bystander cells. These effects could be the result of Vpr-mediated recruitment of the DDB1-CUL4A-VprBP E3 ligase complex in nuclear foci, which leads to ataxia telangiectasia and Rad-3-related activation and G2 arrest (Belzile et al., 2010). In addition, a recent study evaluated how the C-terminus domain of Vpr is effectively internalized in HeLa cells and showed that several different pathways are involved, such as transmembrane passage, clathrin- and caveolae-mediated endocytosis, and macropinocytosis (Greiner et al., 2011).
In a second scenario, the decreases in the levels of ATP and GSH would be uncoupled. Although external stimuli causing a decrease in ATP may potentially affect several intracellular processes, the cellular machinery tends to preserve basic metabolic mechanisms, thus rerouting a sufficient amount of ATP to support these reactions. For instance, the ATP-dependent Na+/K+ pump maintains its normal activity even in circumstances where ATP levels decrease, although below a certain threshold the pump does not function properly (Lahti et al., 2003). Treatment of cells with the chemotherapeutic compound doxorubicin has also been shown to decrease levels of both ATP and GSH (Cui et al., 2006). Although in this study no direct correlation was made between the decreases in the levels of ATP and GSH, it was shown that the decrease in the level of ATP was due to decreased activity of glyceraldehyde 3-phosphate dehydrogenase, which inhibited glycolysis and thus ATP production, whereas GSH reduction was correlated with the inhibition of glucose-6-phosphate dehydrogenase, which caused a decrease in nicotinamide adenine dinucleotide phosphate, responsible for GSH recycling. In the latter case, it could be expected that the effects of Vpr might be similar to those of doxorubicin. Indeed, impairment of glyceraldehyde 3-phosphate dehydrogenase activity and glycolysis would lead to decreased levels of ATP and an increased accumulation of ROS, which may not be disposed of in a timely manner, thereby inducing a decrease in GSH concentrations. Another chemotherapeutic compound (iodoacetate) has been shown to decrease ATP but not GSH (Verity et al., 1991), pointing to a drug-dependent mechanism of action insofar as ATP and GSH reductions are concerned. While, our studies within the U-87 MG astroglioma cell line could represent an in vitro limitation with respect to our findings although in line with preliminary data in primary human fetal astrocytes, further studies are currently under way to determine the intricate relationship between the reductions in ATP and GSH in order to determine which of the two aforementioned scenarios are caused by recombinant Vpr. While measurements of ATP and GSH declines are usually employed to assess cell viability and redox state respectively, these quantitation methods are often performed without taking into account the cell number or the protein content. Our findings showing decreases of both metabolites normalized over the milligram cellular content is much more remarkable as (late) apoptotic/necrotic cells are not accounted for. Our studies reveal promising results for determining the intrinsic, downstream effects triggered by exogenous Vpr and how these may relate to end-stage outcomes in patients infected with HIV-1.
5. Conclusions
The importance of extracellular HIV-1 Vpr in the CNS during the course of HIV-1-associated neurological disease is likely linked to abnormalities generated in the monocytic, microglial cell, astrocytic, and neuronal compartments. We have found that astrocytic metabolism is negatively affected by the presence of extracellular HIV-1 Vpr, in that two of the most important intracellular metabolites (ATP and GSH) are both reduced to greater extents in the presence of Vpr. The results with recombinant Vpr validated our observations using HEK 293T Vpr-containing conditioned media. Exogenous Vpr has been shown to induce an oxidative intracellular environment that is characterized by excessive accumulation of ROS and an altered balance of reduced, overoxidized glutathione. Overall, these observations indicate that Vpr triggers intracellular signaling events that affect astrocytic cell energy and antioxidative defense storage in a dose-dependent manner. The study of these effects on astrocytes is fundamental to understanding the fate of the neuronal population because their survival strongly depends on several astrocytic factors that may be altered detrimentally on exposure to Vpr.
Highlights.
Extracellular HIV-1 Vpr induces a decline in astrocytic ATP and GSH levels
Exogenous HIV-1 Vpr promotes accumulation of reactive oxygen species
N-acetyl-cysteine treatment ameliorates HIV-1 Vpr-induced oxidative stress
Exposure of astrocytes to HIV-1 Vpr increases levels of oxidized glutathione
Treatment with HIV-1 Vpr disrupts primary astrocytes architectural structure
Acknowledgments
We are thankful to Dr. Nathaniel Landau and Dr. Bassel Sawaya for providing the constructs employed in our studies and to Dr. Jeffrey Kopp for the anti-Vpr antibody. This work was supported in part by funds from the Public Health Service, National Institutes of Health through grants from the National Institute of Neurological Disorders and Stroke [NS32092 to B.W.] and the National Institute of Drug Abuse [DA19807 to B.W.]. Dr. Michael Nonnemacher was also supported by faculty development funds provided by the Department of Microbiology and Immunology and the Institute for Molecular Medicine and Infectious Disease.
Abbreviations
- ATP
adenosine triphosphate
- BSO
buthionine sulfoximine
- CFVF
cell-free, virus-free
- CNS
central nervous system
- DAPI
4′,6-diamidino-2-phenylindole
- 4F2hc
regulatory heavy chain
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- GST
glutathione S-transferase
- HA
hemaglutinin
- HEK
human endothelial kidney
- HIV-1
human immunodeficiency virus type 1
- NAC
N-acetyl-cysteine
- NETN
NaCl, EDTA, Tris, NP-40
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- ROS
reactive oxygen species
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- Vpr
vial protein r
- xCT
catalytic unit
Footnotes
Conflict of interest statement
The authors declare that they have no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Adriano Ferrucci, Email: adriano.ferrucci@drexel.edu.
Michael R. Nonnemacher, Email: michael.nonnemacher@drexelmed.edu.
Éric A. Cohen, Email: eric.cohen@ircm.qc.ca.
Brian Wigdahl, Email: brian.wigdahl@drexelmed.edu.
References
- Acharjee S, Noorbakhsh F, Stemkowski PL, Olechowski C, Cohen EA, Ballanyi K, Kerr B, Pardo C, Smith PA, Power C. HIV-1 viral protein R causes peripheral nervous system injury associated with in vivo neuropathic pain. The FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2010;24:4343–4353. doi: 10.1096/fj.10-162313. [DOI] [PubMed] [Google Scholar]
- Albright AV, Strizki J, Harouse JM, Lavi E, O’Connor M, Gonzalez-Scarano F. HIV-1 infection of cultured human adult oligodendrocytes. Virology. 1996;217:211–219. doi: 10.1006/viro.1996.0108. [DOI] [PubMed] [Google Scholar]
- An SF, Groves M, Gray F, Scaravilli F. Early entry and widespread cellular involvement of HIV-1 DNA in brains of HIV-1 positive asymptomatic individuals. J Neuropathol Exp Neurol. 1999;58:1156–1162. doi: 10.1097/00005072-199911000-00005. [DOI] [PubMed] [Google Scholar]
- Belzile JP, Abrahamyan LG, Gerard FC, Rougeau N, Cohen EA. Formation of mobile chromatin-associated nuclear foci containing HIV-1 Vpr and VPRBP is critical for the induction of G2 cell cycle arrest. PLoS Pathog. 2010;6:e1001080. doi: 10.1371/journal.ppat.1001080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissel SJ, Wiley CA. Human immunodeficiency virus infection of the brain: pitfalls in evaluating infected/affected cell populations. Brain Pathol. 2004;14:97–108. doi: 10.1111/j.1750-3639.2004.tb00503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brack-Werner R. Astrocytes: HIV cellular reservoirs and important participants in neuropathogenesis. AIDS. 1999;13:1–22. doi: 10.1097/00002030-199901140-00003. [DOI] [PubMed] [Google Scholar]
- Brack-Werner R, Kleinschmidt A, Ludvigsen A, Mellert W, Neumann M, Herrmann R, Khim MC, Burny A, Muller-Lantzsch N, Stavrou D, et al. Infection of human brain cells by HIV-1: restricted virus production in chronically infected human glial cell lines. AIDS. 1992;6:273–285. [PubMed] [Google Scholar]
- Chung WJ, Lyons SA, Nelson GM, Hamza H, Gladson CL, Gillespie GY, Sontheimer H. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J Neurosci. 2005;25:7101–7110. doi: 10.1523/JNEUROSCI.5258-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill MJ, Gorry PR, Cowley D, Lal L, Sonza S, Purcell DF, Thompson KA, Gabuzda D, McArthur JC, Pardo CA, Wesselingh SL. Use of laser capture microdissection to detect integrated HIV-1 DNA in macrophages and astrocytes from autopsy brain tissues. J Neurovirol. 2006;12:146–152. doi: 10.1080/13550280600748946. [DOI] [PubMed] [Google Scholar]
- Churchill MJ, Wesselingh SL, Cowley D, Pardo CA, McArthur JC, Brew BJ, Gorry PR. Extensive astrocyte infection is prominent in human immunodeficiency virus-associated dementia. Ann Neurol. 2009;66:253–258. doi: 10.1002/ana.21697. [DOI] [PubMed] [Google Scholar]
- Connor RI, Chen BK, Choe S, Landau NR. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology. 1995;206:935–944. doi: 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
- Cui J, Tungaturthi PK, Ayyavoo V, Ghafouri M, Ariga H, Khalili K, Srinivasan A, Amini S, Sawaya BE. The role of Vpr in the regulation of HIV-1 gene expression. Cell Cycle. 2006;5:2626–2638. doi: 10.4161/cc.5.22.3442. [DOI] [PubMed] [Google Scholar]
- Deshmane SL, Mukerjee R, Fan S, Del Valle L, Michiels C, Sweet T, Rom I, Khalili K, Rappaport J, Amini S, Sawaya BE. Activation of the oxidative stress pathway by HIV-1 Vpr leads to induction of hypoxia-inducible factor 1alpha expression. J Biol Chem. 2009;284:11364–11373. doi: 10.1074/jbc.M809266200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dringen R, Kussmaul L, Gutterer JM, Hirrlinger J, Hamprecht B. The glutathione system of peroxide detoxification is less efficient in neurons than in astroglial cells. J Neurochem. 1999;72:2523–2530. doi: 10.1046/j.1471-4159.1999.0722523.x. [DOI] [PubMed] [Google Scholar]
- Ferrucci A, Nonnemacher MR, Wigdahl B. Human immunodeficiency virus viral protein R as an extracellular protein in neuropathogenesis. Adv Virus Res. 2011;81:165–199. doi: 10.1016/B978-0-12-385885-6.00010-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulian D, Vaca K, Noonan CA. Secretion of neurotoxins by mononuclear phagocytes infected with HIV-1. Science. 1990;250:1593–1596. doi: 10.1126/science.2148832. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5:69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- Gorry PR, Howard JL, Churchill MJ, Anderson JL, Cunningham A, Adrian D, McPhee DA, Purcell DF. Diminished production of human immunodeficiency virus type 1 in astrocytes results from inefficient translation of gag, env, and nef mRNAs despite efficient expression of Tat and Rev. J Virol. 1999;73:352–361. doi: 10.1128/jvi.73.1.352-361.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greiner VJ, Shvadchak V, Fritz J, Arntz Y, Didier P, Frisch B, Boudier C, Mely Y, de Rocquigny H. Characterization of the mechanisms of HIV-1 Vpr(52–96) internalization in cells. Biochimie. 2011 doi: 10.1016/j.biochi.2011.05.033. [DOI] [PubMed] [Google Scholar]
- Harouse JM, Laughlin MA, Pletcher C, Friedman HM, Gonzalez-Scarano F. Entry of human immunodeficiency virus-1 into glial cells proceeds via an alternate, efficient pathway. J Leukoc Biol. 1991;49:605–609. doi: 10.1002/jlb.49.6.605. [DOI] [PubMed] [Google Scholar]
- He J, Choe S, Walker R, Di Marzio P, Morgan DO, Landau NR. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J Virol. 1995;69:6705–6711. doi: 10.1128/jvi.69.11.6705-6711.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzenberg LA, De Rosa SC, Dubs JG, Roederer M, Anderson MT, Ela SW, Deresinski SC. Glutathione deficiency is associated with impaired survival in HIV disease. Proc Natl Acad Sci U S A. 1997;94:1967–1972. doi: 10.1073/pnas.94.5.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino S, Konishi M, Mori M, Shimura M, Nishitani C, Kuroki Y, Koyanagi Y, Kano S, Itabe H, Ishizaka Y. HIV-1 Vpr induces TLR4/MyD88-mediated IL-6 production and reactivates viral production from latency. J Leukoc Biol. 2010;87:1133–1143. doi: 10.1189/jlb.0809547. [DOI] [PubMed] [Google Scholar]
- Hoshino S, Sun B, Konishi M, Shimura M, Segawa T, Hagiwara Y, Koyanagi Y, Iwamoto A, Mimaya J, Terunuma H, Kano S, Ishizaka Y. Vpr in plasma of HIV type 1-positive patients is correlated with the HIV type 1 RNA titers. AIDS Res Hum Retroviruses. 2007;23:391–397. doi: 10.1089/aid.2006.0124. [DOI] [PubMed] [Google Scholar]
- Huang MB, Weeks O, Zhao LJ, Saltarelli M, Bond VC. Effects of extracellular human immunodeficiency virus type 1 vpr protein in primary rat cortical cell cultures. J Neurovirol. 2000;6:202–220. doi: 10.3109/13550280009015823. [DOI] [PubMed] [Google Scholar]
- Jacotot E, Ferri KF, El Hamel C, Brenner C, Druillennec S, Hoebeke J, Rustin P, Metivier D, Lenoir C, Geuskens M, Vieira HL, Loeffler M, Belzacq AS, Briand JP, Zamzami N, Edelman L, Xie ZH, Reed JC, Roques BP, Kroemer G. Control of mitochondrial membrane permeabilization by adenine nucleotide translocator interacting with HIV-1 viral protein rR and Bcl-2. J Exp Med. 2001;193:509–519. doi: 10.1084/jem.193.4.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacotot E, Ravagnan L, Loeffler M, Ferri KF, Vieira HL, Zamzami N, Costantini P, Druillennec S, Hoebeke J, Briand JP, Irinopoulou T, Daugas E, Susin SA, Cointe D, Xie ZH, Reed JC, Roques BP, Kroemer G. The HIV-1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. J Exp Med. 2000;191:33–46. doi: 10.1084/jem.191.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones GJ, Barsby NL, Cohen EA, Holden J, Harris K, Dickie P, Jhamandas J, Power C. HIV-1 Vpr causes neuronal apoptosis and in vivo neurodegeneration. J Neurosci. 2007;27:3703–3711. doi: 10.1523/JNEUROSCI.5522-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410:988–994. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- Kim JY, Kanai Y, Chairoungdua A, Cha SH, Matsuo H, Kim DK, Inatomi J, Sawa H, Ida Y, Endou H. Human cystine/glutamate transporter: cDNA cloning and upregulation by oxidative stress in glioma cells. Biochim Biophys Acta. 2001;1512:335–344. doi: 10.1016/s0005-2736(01)00338-8. [DOI] [PubMed] [Google Scholar]
- Kitayama H, Miura Y, Ando Y, Hoshino S, Ishizaka Y, Koyanagi Y. Human immunodeficiency virus type 1 Vpr inhibits axonal outgrowth through induction of mitochondrial dysfunction. J Virol. 2008;82:2528–2542. doi: 10.1128/JVI.02094-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinschmidt A, Neumann M, Moller C, Erfle V, Brack-Werner R. Restricted expression of HIV1 in human astrocytes: molecular basis for viral persistence in the CNS. Res Virol. 1994;145:147–153. doi: 10.1016/s0923-2516(07)80016-1. [DOI] [PubMed] [Google Scholar]
- Lahti AL, Manninen A, Saksela K. Regulation of T cell activation by HIV-1 accessory proteins: Vpr acts via distinct mechanisms to cooperate with Nef in NFAT-directed gene expression and to promote transactivation by CREB. Virology. 2003;310:190–196. doi: 10.1016/s0042-6822(03)00164-8. [DOI] [PubMed] [Google Scholar]
- Levy DN, Refaeli Y, MacGregor RR, Weiner DB. Serum Vpr regulates productive infection and latency of human immunodeficiency virus type 1. Proc Natl Acad Sci U S A. 1994;91:10873–10877. doi: 10.1073/pnas.91.23.10873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DN, Refaeli Y, Weiner DB. Extracellular Vpr protein increases cellular permissiveness to human immunodeficiency virus replication and reactivates virus from latency. J Virol. 1995;69:1243–1252. doi: 10.1128/jvi.69.2.1243-1252.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Bentsman G, Potash MJ, Volsky DJ. Human immunodeficiency virus type 1 efficiently binds to human fetal astrocytes and induces neuroinflammatory responses independent of infection. BMC Neurosci. 2007;8:31. doi: 10.1186/1471-2202-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons SA, Chung WJ, Weaver AK, Ogunrinu T, Sontheimer H. Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res. 2007;67:9463–9471. doi: 10.1158/0008-5472.CAN-07-2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noorbakhsh F, Ramachandran R, Barsby N, Ellestad KK, LeBlanc A, Dickie P, Baker G, Hollenberg MD, Cohen EA, Power C. MicroRNA profiling reveals new aspects of HIV neurodegeneration: caspase-6 regulates astrocyte survival. FASEB J. 2010;24:1799–1812. doi: 10.1096/fj.09-147819. [DOI] [PubMed] [Google Scholar]
- Piller SC, Ewart GD, Jans DA, Gage PW, Cox GB. The amino-terminal region of Vpr from human immunodeficiency virus type 1 forms ion channels and kills neurons. Journal of virology. 1999;73:4230–4238. doi: 10.1128/jvi.73.5.4230-4238.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piller SC, Jans P, Gage PW, Jans DA. Extracellular HIV-1 virus protein R causes a large inward current and cell death in cultured hippocampal neurons: implications for AIDS pathology. Proc Natl Acad Sci U S A. 1998;95:4595–4600. doi: 10.1073/pnas.95.8.4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rom I, Deshmane SL, Mukerjee R, Khalili K, Amini S, Sawaya BE. HIV-1 Vpr deregulates calcium secretion in neural cells. Brain Res. 2009;1275:81–86. doi: 10.1016/j.brainres.2009.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbah EN, Druillennec S, Morellet N, Bouaziz S, Kroemer G, Roques BP. Interaction between the HIV-1 protein Vpr and the adenine nucleotide translocator. Chem Biol Drug Des. 2006;67:145–154. doi: 10.1111/j.1747-0285.2006.00340.x. [DOI] [PubMed] [Google Scholar]
- Sabbah EN, Roques BP. Critical implication of the (70–96) domain of human immunodeficiency virus type 1 Vpr protein in apoptosis of primary rat cortical and striatal neurons. J Neurovirol. 2005;11:489–502. doi: 10.1080/13550280500384941. [DOI] [PubMed] [Google Scholar]
- Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem. 1999;274:11455–11458. doi: 10.1074/jbc.274.17.11455. [DOI] [PubMed] [Google Scholar]
- Sherman MP, Schubert U, Williams SA, de Noronha CM, Kreisberg JF, Henklein P, Greene WC. HIV-1 Vpr displays natural protein-transducing properties: implications for viral pathogenesis. Virology. 2002;302:95–105. doi: 10.1006/viro.2002.1576. [DOI] [PubMed] [Google Scholar]
- Snyder A, Alsauskas Z, Gong P, Rosenstiel PE, Klotman ME, Klotman PE, Ross MJ. FAT10: a novel mediator of Vpr-induced apoptosis in human immunodeficiency virus-associated nephropathy. J Virol. 2009;83:11983–11988. doi: 10.1128/JVI.00034-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornatore C, Meyers K, Atwood W, Conant K, Major E. Temporal patterns of human immunodeficiency virus type 1 transcripts in human fetal astrocytes. J Virol. 1994;68:93–102. doi: 10.1128/jvi.68.1.93-102.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornatore C, Nath A, Amemiya K, Major EO. Persistent human immunodeficiency virus type 1 infection in human fetal glial cells reactivated by T-cell factor(s) or by the cytokines tumor necrosis factor alpha and interleukin-1 beta. J Virol. 1991;65:6094–6100. doi: 10.1128/jvi.65.11.6094-6100.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verity MA, Torres M, Sarafian T. Paradoxical potentiation by low extracellular Ca2+ of acute chemical anoxic neuronal injury in cerebellar granule cell culture. Mol Chem Neuropathol. 1991;15:217–233. doi: 10.1007/BF03161061. [DOI] [PubMed] [Google Scholar]
- Vieira HL, Haouzi D, El Hamel C, Jacotot E, Belzacq AS, Brenner C, Kroemer G. Permeabilization of the mitochondrial inner membrane during apoptosis: impact of the adenine nucleotide translocator. Cell Death Differ. 2000;7:1146–1154. doi: 10.1038/sj.cdd.4400778. [DOI] [PubMed] [Google Scholar]
- Williams K, Burdo TH. Monocyte Mobilization, Activation Markers, and Unique Macrophage Populations in the Brain: Observations from SIV Infected Monkeys Are Informative with Regard to Pathogenic Mechanisms of HIV Infection in Humans. Journal of neuroimmune pharmacology: the official journal of the Society on Neuro Immune Pharmacology. 2011 doi: 10.1007/s11481-011-9330-3. [DOI] [PubMed] [Google Scholar]
- Xiao Y, Chen G, Richard J, Rougeau N, Li H, Seidah NG, Cohen EA. Cell-surface processing of extracellular human immunodeficiency virus type 1 Vpr by proprotein convertases. Virology. 2008;372:384–397. doi: 10.1016/j.virol.2007.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]