

Abstract
The “latent period” between brain injury and clinical epilepsy is widely regarded to be a seizure-free, pre-epileptic state during which a time-consuming cascade of molecular events and structural changes gradually mediates the process of “epileptogenesis.” The concept of the “latent period” as the duration of “epileptogenesis” implies that epilepsy is not an immediate result of brain injury, and that anti-epileptogenic strategies need to target delayed secondary mechanisms that develop sometime after an initial injury. However, depth recordings made directly from the dentate granule cell layers in awake rats after convulsive status epilepticus-induced injury have now shown that whenever perforant pathway stimulation-induced status epilepticus produces extensive hilar neuron loss and entorhinal cortical injury, hyperexcitable granule cells immediately generate spontaneous epileptiform discharges and focal or generalized behavioral seizures. This indicates that hippocampal injury caused by convulsive status epilepticus is immediately epileptogenic and that hippocampal epileptogenesis requires no delayed secondary mechanism. When latent periods do exist after injury, we hypothesize that less extensive cell loss causes an extended period during which initially subclinical focal seizures gradually increase in duration to produce the first clinical seizure. Thus, the “latent period” is suggested to be a state of “epileptic maturation,” rather than a prolonged period of “epileptogenesis,” and therefore the antiepileptogenic therapeutic window may only remain open during the first week after injury, when some delayed cell death may still be preventable. Following the perhaps unavoidable development of the first focal seizures (“epileptogenesis”), the most fruitful therapeutic strategy may be to interrupt the process of “epileptic maturation,” thereby keeping focal seizures focal.
Keywords: epileptogenesis, epilepsy, hippocampal formation, hippocampus, temporal lobe epilepsy, traumatic brain injury, status epilepticus, dentate gyrus
What is “epileptogenesis” exactly, and how can it be prevented?
Experimental and clinical studies have used antiepileptic drugs to abort post-injury epileptogenesis, but with minimal success (Dichter, 2009; Temkin, 2009; Löscher and Brandt, 2010; Pitkänen, 2010; Eastman et al., 2011; Langer et al., 2011). Although the right drugs may have been tested at the wrong doses, for the wrong duration, or at the wrong time after brain injury, it is also possible that antiepileptic drugs do not influence the injury-induced “epileptogenic” process. Because acquired epilepsy has not been prevented by any post-injury treatment, it is unknown whether acquired epileptogenesis can be stopped pharmacologically (Sloviter, 2011). But assuming that epileptogenesis can be aborted, it is unknown exactly what needs to be blocked, when the “therapeutic window” opens, and when it closes. Thus, the central challenges of antiepileptogenesis research are to: 1) develop animal models that reliably exhibit spontaneous seizures arising from a single identified brain region; and 2) determine exactly when and why spontaneous seizures develop in that region specifically. The need for an animal model with one primary epileptogenic site is a significant practical consideration because if an animal model exhibits widespread brain damage, multiple sites of seizure onset, and frequent seizures, a therapy capable of completely preventing epileptogenesis in one location might have no detectable effect on the overall seizure frequency.
In addition to practical model-related obstacles to progress in therapeutics, significant confusion surrounds the meaning of the term “epileptogenesis,” and therefore there is uncertainty about what it is exactly that we are trying to stop. Definitions are important because the way we think about the word “epileptogenesis” profoundly influences the way we conceptualize the disease process and how we devise strategies to interrupt it. In the case of acquired epilepsy, the term “epileptogenesis” describes an injury-initiated change that causes surviving neuron populations to generate abnormal, synchronous, and recurring epileptiform discharges that produce focal or generalized behavioral seizures. Historically, the most influential view of the acquired epileptogenic process envisaged a scenario in which an injury triggers a secondary process that takes time-- months, years, or even decades-- to mature and produce clinical seizures (Earle et al., 1953; French et al., 1993). As most frequently used in the experimental literature, the term “epileptogenesis” describes a process that begins with an initial insult, is presumed to involve a cascade of secondary epileptogenic events (Chang and Lowenstein, 2003; Rakhade and Jensen, 2009; Giblin and Blumenfeld, 2010), and ends when the first generalized behavioral seizure is observed (Bragin et al., 2000; Walker et al., 2002). More recently, however, the term “epileptogenesis” has been expanded to include the possibly never-ending process of clinical progression that involves changes in seizure frequency and the development of a refractory state (Pitkänen, 2010; Dudek and Staley, 2011; Pitkänen et al., 2011). Thus, there is a need to define “epileptogenesis” precisely, induce it reliably in experimental animals, identify its beginning and its end, and find a way to stop it. Does the first spontaneous focal seizure signal completion of epileptogenesis, or does epileptogenesis end only when the first of many clinically obvious seizures occur? Or, is epileptogenesis an all-inclusive, continuously evolving, and never fully completed process that underlies all stages of the disorder, including the possible progression from a pharmacosensitive- to a pharmacoresistant state?
Defining “epileptogenesis” and “epileptic maturation”
Perhaps the only indisputable thing that can be said about the term “epileptogenesis” is that the word literally and unambiguously means the “birth of epilepsy” or the “beginning of epilepsy.” The “genesis” phase in the life of an individual ends at birth, not in reaching maturity. The growth and maturation of the individual is a secondary process clearly distinct from “genesis.” Therefore, it seems logical to define the completion of “epileptogenesis” as the moment when the machinery and mechanisms necessary for spontaneous epileptiform behaviors first exist. A seizure focus may increase in size over time, it may transition from a focal to a secondarily generalized process, seizure frequency may change, and seizures may become pharmacoresistant, but these events would seem to be part of a “growth and maturation” process, not the “birth of epilepsy” phase.
Car manufacture provides an analogy for our argument that “epileptogenesis” is fundamentally a “birth” process. Every car is assigned a “build date” marking completion of the manufacturing process that allows a car to function for the first time as designed. A fully built car that has not yet been driven is still a car, and giving it a “build date” does not preclude making additional modifications that change the car’s attributes and capabilities. By analogy, we regard the end of “epileptogenesis” to be the brain’s unfortunate “build date” when all cellular and network changes needed for the spontaneous generation of epileptiform discharges are first present. Therefore, we define “epileptogenesis” to be the finite process that leads to the first of a series of spontaneous and recurring epileptiform events that disrupt behavior or ideation in any way, whether clinically obvious or not. We propose and define the term “epileptic maturation” to describe and encompass all processes that follow “epileptogenesis” and influence the secondary evolutionary changes in the clinical phenotype of the disorder. Therefore, “epileptogenesis” and “epileptic maturation” are viewed as two distinct processes and two separate targets for “disease prevention” and “disease modification,” respectively.
These definitions have important practical implications because there is a difference between coining a name for an amorphous conception of a process, and knowing something about its essential nature, i.e., being able to define it (Sloviter, 2002). If the name “epileptogenesis” describes an immediate change in network excitability and behavior caused by neuron loss, as we have proposed (Sloviter, 1994; Bumanglag and Sloviter, 2008), then an antiepileptogenesis strategy should incorporate the possibility that the brain may become immediately “epileptic” even if the disorder is not immediately obvious clinically. If so, a fruitful therapeutic strategy might focus on minimizing delayed cell death in the immediate post-injury period (Sloviter et al., 1996; Poirier et al., 2000; Brandt et al., 2003b; Langer et al., 2011; Sloviter, 2011). Conversely, if “epileptogenesis” necessarily involves the maturation of a slowly developing secondary process triggered by brain injury, then neuron loss may not be directly epileptogenic, and antiepileptogenesis strategies might logically focus on preventing one or more of the secondary processes that injuries initiate (Chang and Lowenstein, 2003; Rakhade and Jensen, 2009). Thus, the first questions that need to be answered are: when exactly after injury does epileptogenesis develop, i.e. when is epilepsy “born,” and what is the role of neuron loss? Also, which secondary effects triggered by injuries affect initial “epileptogenesis,” and which affect subsequent “epileptic maturation?” To answer these fundamental questions, it is necessary to have animal models that involve a restricted injury, minimal variability, and spontaneous seizures that develop in a known location that can be directly monitored starting immediately after brain injury.
Animal models of acquired temporal lobe epilepsy with hippocampal sclerosis
If chemoconvulsant-induced status epilepticus causes widespread brain damage and seizures of unknown origin, why has “hippocampal epileptogenesis” been so widely assumed?
Although epilepsy can arise from a variety of brain regions, most experimental studies have focused on modeling acquired temporal lobe epilepsy (TLE) with hippocampal sclerosis. Acquired TLE is of particular interest experimentally for several reasons. First and foremost, the pattern of selective hippocampal formation pathology associated with TLE in humans (Margerison and Corsellis, 1966; Du et al., 1993; Blümcke et al., 2007), as well as a chronic epileptic state, can be produced in animals by the same insults that cause epilepsy in humans (Sloviter, 1987; Lowenstein et al., 1992; Gorter et al., 2001; Bumanglag and Sloviter, 2008; Norwood et al., 2010). Second, the laminar structure of the “hippocampal formation” (an anatomical term that includes the entorhinal cortex, dentate gyrus, hippocampus “proper,” subiculum, parasubiculum, and presubiculum) facilitates the analysis and interpretation of anatomical, pathological, and electrophysiological data.
However, despite abundant evidence that the hippocampus is a frequent source of seizures in TLE patients (Spencer, 1998; 2002), the development of animal models that reliably exhibit both the human pattern of temporal lobe pathology (selective hippocampal neuron loss and limited extrahippocampal pathology) and hippocampal-onset seizures (Bumanglag and Sloviter, 2008; Kienzler et al., 2009; Norwood et al., 2010), has been a much more difficult task than originally envisaged by the early studies that promoted kainate- and pilocarpine-treated animals as models of TLE (Nadler, 1981; Ben-Ari, 1985; Leite et al., 1990). Although the first detailed study of the pathology caused by kainate-induced status epilepticus clearly showed that the hippocampus was less affected by convulsive status epilepticus than other more severely affected brain regions (Schwob et al., 1980), and despite the fact that the hippocampus was never shown to generate epileptiform discharges that preceded behavioral seizures, it was nonetheless asserted that kainate-induced status epilepticus reproduces the defining features of TLE with hippocampal sclerosis (Nadler, 1981; Ben-Ari, 1985).
However, it is now clear that prolonged convulsive status epilepticus produces variable hippocampal damage within a context of widespread extrahippocampal brain damage (Brandt et al., 2003a; Chen and Buckmaster, 2005; Harvey and Sloviter, 2005). In addition, hippocampal injury after chemoconvulsant-induced status epilepticus includes vascular/ischemic injury (Fig. 1; Sloviter, 2005; Biagini et al., 2008; Cardoso et al., 2011) that is unrelated to coincident seizure-induced excitotoxic hippocampal pathology (Sloviter et al., 1996), and this pattern of hippocampal injury does not resemble seizure-induced hippocampal sclerosis (Sloviter, 2005; Sloviter et al., 2007; Bumanglag and Sloviter, 2008; Norwood et al., 2010). The variable and relatively minor hippocampal injury that accompanies often extensive extrahippocampal injury (Schwob et al., 1980; Turski et al., 1983; Sloviter et al., 2003; Zappone and Sloviter, 2004; Bumanglag and Sloviter, 2008) indicates that the granule cells of chemoconvulsant-treated animals cannot be assumed to be “epileptic” in every animal simply because extensively brain-damaged animals are having spontaneous behavioral seizures of unknown origin (Harvey and Sloviter, 2005). This is a crucial issue because a multitude of studies of abnormalities in the dentate gyrus in particular, initiated by Nadler’s pioneering work on synaptic reorganization in kainate-treated rats (Nadler et al., 1980; Tauck and Nadler, 1985), have implicitly assumed that if an animal becomes epileptic, the granule cells of that animal must have undergone an “epileptogenic conversion.” However, to our knowledge, no studies that have recorded directly from the granule cell layers of awake kainate- or pilocarpine-treated rats during spontaneous seizures (Sloviter et al., 2003; Harvey and Sloviter, 2005; Bower and Buckmaster, 2008; Queiroz et al., 2009; Williams et al., 2009) have provided evidence of “hippocampal epileptogenesis” or made the case that dentate granule cells are a likely source of the spontaneous seizures that develop in chemoconvulsant-treated animals.
Figure 1.
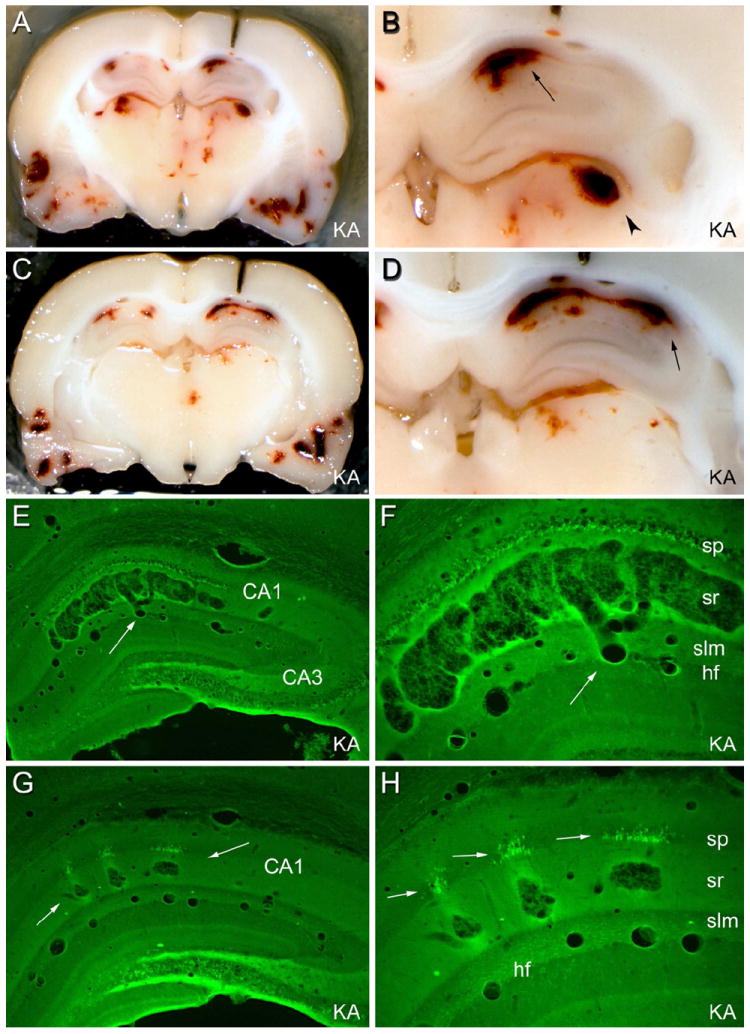
Brain structure 3 days after status epilepticus induced by systemic injection of kainic acid (KA; 12 mg/kg sc). In some animals given kainate or pilocarpine systemically, prolonged status epilepticus caused apparent hemorrhages in a multitude of brain structures, which were not cleared by vascular perfusion. (A) and (C): two coronal views of the same brain during the sectioning process. Note apparent hemorrhagic foci in the hippocampi, thalamus, and temporal cortices. (B) and (D): foci preferentially involve the CA1 pyramidal cell layer (arrow) and the dorsolateral thalamus (arrowhead). (E) and (F): in a Fluoro Jade B-stained section from the brain shown in (A)-(D), degenerating neurons are fluorescent. Note that the area CA1 pathology consists of a vascular expansion that is continuous with a capillary in the hippocampal fissure (hf; arrow), not an extravascular hemorrhage. (G): in a different kainate-treated rat, smaller focal vascular expansions occurred within the stratum radiatum (arrows). (H) At higher magnification, degenerating CA1 pyramidal cell somata were evident only adjacent to the vascular pathology. These results suggest that, in some cases, CA1 pyramidal cell layer injury in rats subjected to prolonged status epilepticus may be ischemic in nature, rather than excitotoxic. Abbreviations: sp: stratum pyramidale; sr: stratum radiatum; slm: stratum lacunosum-moleculare; hf: hippocampal fissure. Magnifications: 5X (A and C); 13.5X (B and D); 22X (E and G); 55X (F and H). From Sloviter, 2005.
Spontaneous granule cell layer events in awake, pilocarpine-treated epileptic rats
We monitored granule cell layer activity continuously in awake pilocarpine-treated rats during more than 200 spontaneous epileptic seizures, and found that the granule cells appeared to be hyperinhibited and minimally involved (Harvey and Sloviter, 2005). In the first days after pilocarpine-induced status epilepticus, behavioral seizures of unknown origin were able to recruit epileptiform discharges from initially hyperexcitable granule cells, but these spontaneous behavioral seizures always started before the granule cell recruitment (Figure 2, C). As synaptic reorganization (mossy fiber sprouting) slowly developed, granule cells gradually became hyperinhibited (Sloviter, 1992; Harvey and Sloviter, 2005; Sloviter et al., 2006), and spontaneous behavioral seizures failed to recruit epileptiform granule cell discharges (Figure 2, D2 and D3). Thus, there would appear to be little justification for assuming that the dentate granule cells of chemoconvulsant-treated rats become reliably and spontaneously “epileptic” simply because the animals are “epileptic.”
Figure 2.
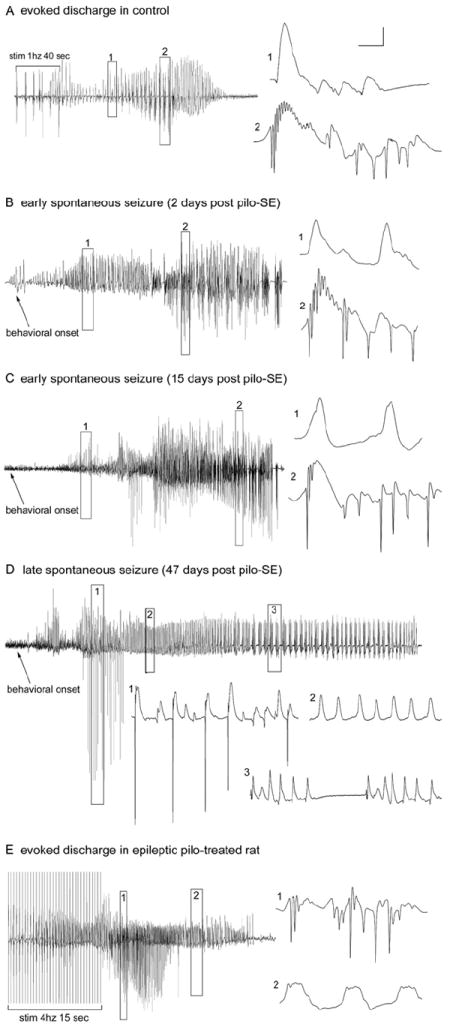
Evoked and spontaneous granule cell activity recorded from the granule cell layers of chronically epileptic, pilocarpine treated-rats. A: Granule cell layer seizure discharges evoked in a control animal (60 days post-saline treatment) by perforant path stimulation at 1 Hz (with paired-pulses 40 msec apart) for 40 sec. Note that evoked granule cell activity generated both low-frequency, positive-going field potentials (expanded box 1) and high-frequency, negative-going population spikes (expanded box 2). B: In an epileptic animal 2 days post-SE, spontaneous granule cell layer activity recorded during a spontaneous behavioral seizure exhibited both positive-going field potentials (expanded box 1) and negative-going epileptiform discharges (expanded box 2) that were qualitatively similar to those evoked in the control rat in A. C: Thirteen days later, spontaneous granule cell layer activity still exhibited high-frequency, negative-going epileptiform discharges. D: Forty-seven days after SE, the spontaneous granule cell layer activity recorded in the same animal during a spontaneous seizure exhibited single population spikes (expanded box 1) and positive-going field potentials (expanded boxes 2 and 3) but no seizure discharges (repetitive population spikes). Note that granule cells generated only single population spikes ~7 seconds after the onset of the observed behavioral seizure (expanded box 1) and then generated only field potentials for the duration of the behavioral seizure. E: Evoked granule cell seizure afterdischarges in the same chronically epileptic, pilocarpine-treated rat 45 days post-SE (tested 2 days prior to the activity shown in D), illustrating that, despite the lack of spontaneous seizure discharges in the chronic epileptic state, abnormally high-frequency (4 –10Hz) stimulation could evoke granule cell seizure discharges (expanded box 1), as well as positive-going field potentials (expanded box 2). The absence of seizure discharges recorded from the granule cell layers during spontaneous behavioral seizures was a consistent observation in epileptic animals subsequently shown to have dentate hilar cell loss and mossy fiber sprouting. Calibration bars: 3.5 mV, 30 msec in A; 5 mV, 2 sec for B; 4 mV, 30 msec for B1,2; 2.5 mV, 2 sec for C; 3 mV, 100 msec for C1–3; 3 mV, 2 sec for D; 4 mV, 25 msec for D1,2. From Harvey and Sloviter, 2005.
An important feature of the high acquisition-rate (10 kHz) recordings we made in epileptic pilocarpine-treated rats was the discovery that although granule cell layer electrographic activity during spontaneous seizures could be impressive high-amplitude events (Fig. 2), expansion and analysis of the traces revealed that the granule cell layer activity recorded during behavioral seizures mainly consisted of dendritic “field EPSPs” (Andersen et al., 1966) that lacked the high frequency, negative-going population spikes (Figures 2, D2 and 3) that define epileptiform discharges (Bragin et al., 1997). In addition, these largely spikeless field depolarizations followed, rather than preceded, the onset of the behavioral seizures, suggesting that extrahippocampal excitation can depolarize granule cells and produce high amplitude voltage deflections without producing granule cell epileptiform discharges (high frequency synchronous action potentials). Dentate granule cells were therefore not only not the source of the spontaneous behavioral seizures, they were ineffectively recruited by the generalized seizures that had apparently already started elsewhere (Harvey and Sloviter, 2005). Thus, these large-amplitude electrographic events, which might be reasonably assumed to be hippocampal seizures when analyzed in a compressed form, could be described more accurately as seizures of unknown origin. High amplitude EEG activity recorded from the granule cell layers can therefore be easily mistaken for a granule cell-derived epileptiform discharge, particularly if the activity is recorded at a low acquisition rate that misses all high-frequency events (Williams et al., 2009), or if a high acquisition-rate trace is not expanded and carefully analyzed for the presence of high-frequency population spikes.
Bower and Buckmaster (2008) also recorded directly from the granule cell layer in awake pilocarpine-treated rats, and focused on the discharge rates of individual granule cells before, during, and after spontaneous epileptic seizures. These authors reported that although some granule cells increased their basal firing rates to a limited extent before and during behavioral seizures of unknown origin, approximately half of the monitored cells either did not change their firing rates, or decreased their firing rates dramatically before and during seizures. Importantly, no granule cells exhibited discharge frequencies that approached the high frequencies that define epileptiform discharges (Figures 2 and 3). The finding by Bower and Buckmaster (2008) that granule cells failed to generate high-frequency discharges during severe EEG seizures recorded from the same electrodes suggests that their granule cell layer electrodes were recording distant, presumably extrahippocampal EEG seizures, and that these behavioral seizures had minor and variable effects on granule cell firing, with many cells being inhibited during seizures to 21% of their basal firing rate. We interpret these variable and limited changes in granule cell activity (Bower and Buckmaster, 2008) as being consistent with our finding that granule cells were hyperinhibited both interictally and ictally, and were relatively uninvolved during the chronic spontaneous behavioral seizures that occur in pilocarpine-treated rats (Harvey and Sloviter, 2005).
Figure 3.
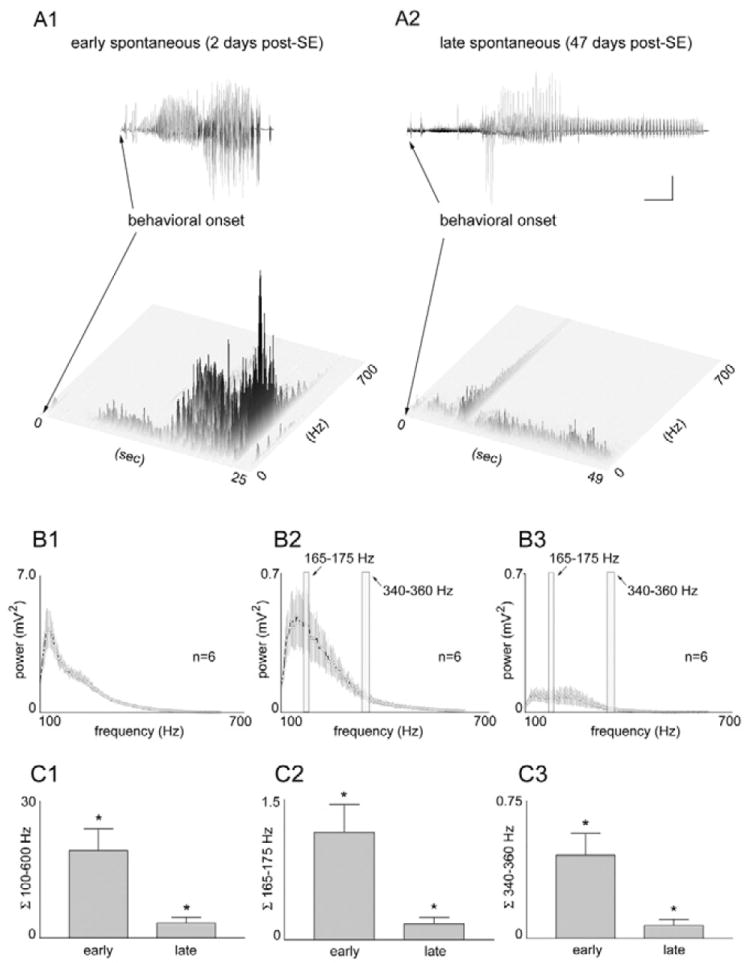
A–C: Quantitative frequency analysis of “early” and “late” spontaneous seizures recorded in the same chronically epileptic, pilocarpine-treated rats. A1,2: Joint time frequency analysis spectrograms of “early” (2 days post-SE) and “late” (47 days post-SE) seizures in the same awake, epileptic animal, indicating when granule cell layer activities of different frequencies occurred in relation to the behavioral seizure onsets. Note that all high-frequency components (100 – 600 Hz) were greater in magnitude (darkness and height of peaks) in the early vs. late post-status epilepticus (SE) states, indicating a loss of epileptiform discharges over time. Also note that these high-frequency components occurred after, rather than before, the spontaneous behavioral seizure onsets (onset of forepaw clonus and rearing). B1: Averaged power spectrum of six randomly selected, 1-minute-long traces of granule cell activity during SE. B2,3: The six first and last spontaneous seizures during the “early” and “late” spontaneous seizures, respectively, in a typical epileptic pilocarpine- treated rat. Note that all frequencies between 100 and 600 Hz were higher during “early” seizures (B2) than during “late” seizures (B3), indicating the loss of high-frequency epileptiform granule cell discharges in the chronic epileptic state. C1: The average integral taken from the averaged power spectrum (100 – 600 Hz) in all five chronically epileptic rats that were implanted before SE. Note the significant loss (P< 0.012) of all high-frequency components. C2,C3: The average integrals taken from the averaged power spectra of the two high-frequency bands (165–175 Hz and 340–360 Hz) in all five epileptic rats. Note the significant loss (P < 0.012 and 0.014, respectively) of both narrow frequency bands. Calibration bars: 5 mV, 4 sec. From Harvey and Sloviter, 2005.
Hippocampal c-Fos expression after spontaneous epileptic seizures
Consistent with the observed lack of hippocampal principal cell epileptiform discharges during spontaneous behavioral seizures in pilocarpine-treated rats (Harvey and Sloviter, 2005), the protein product of the immediate early gene c-Fos, which is expressed by cells after an epileptiform discharge (Dragunow and Robertson, 1987; Morgan et al., 1987), was not expressed by hippocampal granule cells and pyramidal neurons after each spontaneous epileptic seizure of unknown origin (Mello et al., 1996; Harvey and Sloviter, 2005). To the contrary, we unexpectedly found that consistent with the observed hippocampal hyperinhibition during the pilocarpine-induced chronic epileptic state, Fos protein expression after spontaneous seizures was primarily evident within hippocampal inhibitory interneurons (Harvey and Sloviter, 2005). However, in the rare instance when a behavioral seizure was able to recruit granule cells and CA1 pyramidal cells to discharge, both cell populations then expressed Fos immunoreactivity (Figure 4). This indicates that when hippocampal principal cells in a hyperinhibited hippocampus did not express Fos protein after a behavioral seizure, it was not due to an inability to do so, but was likely due to a lack of principal cell discharges during the spontaneous behavioral seizures.
Figure 4.
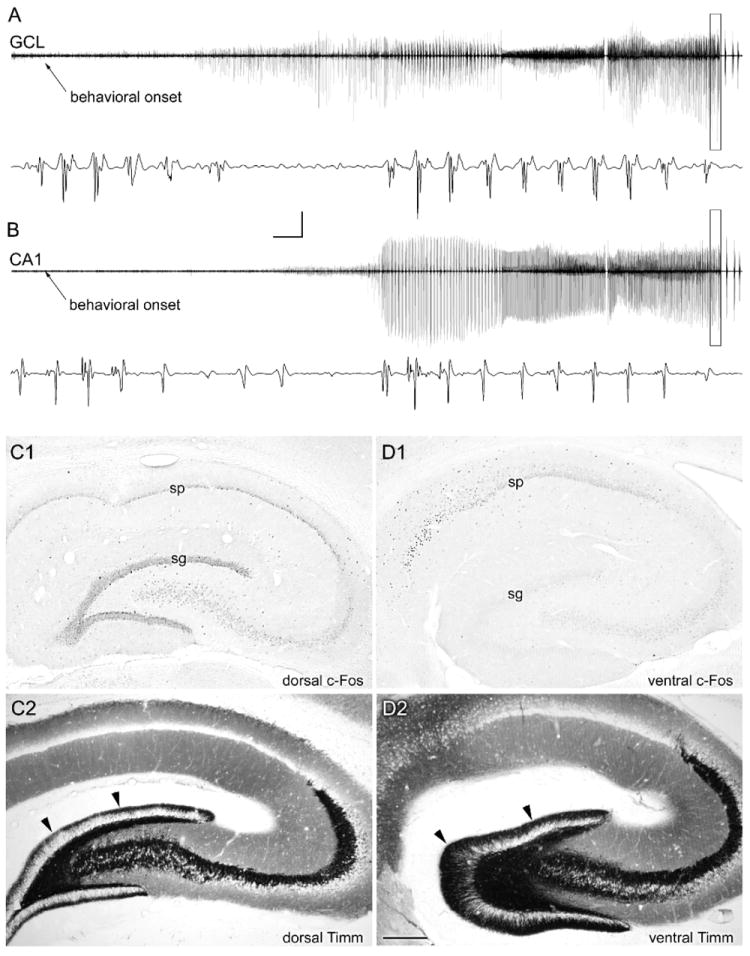
Atypical recruitment of granule cell and CA1 pyramidal cell discharges during a spontaneous seizure in a chronically epileptic rat. A,B: In this animal, perfusion fixed 56 days post-SE, seizure discharges were recorded from both dorsal hippocampal granule cell (GCL) and CA1 pyramidal cell layers beginning approximately 25 seconds after the behavioral seizure onset. C1: c-Fos immunostaining of the dorsal hippocampus in a coronal section revealed c-Fos expression in all hippocampal neurons. D1: c-Fos immunostaining of the ventral hippocampus in a horizontal section from the same rat revealed c-Fos expression in pyramidal layer neurons but not in the dentate gyrus (sg; stratum granulosum). C2,D2: Note that Timm staining revealed less extensive mossy fiber sprouting in the dorsal hippocampus (C2) than in the ventral hippocampus (D2), and a lack of c-Fos expression in the more heavily mossy fiber-sprouted ventral dentate gyrus. Calibration bars: 10 mV, 5 seconds (compressed areas); 6 mV, 75 msec (expanded boxes). Scale bar: 400 um in D (applies to C,D). From Harvey and Sloviter, 2005.
Addressing the same issue in spontaneously epileptic gerbils and in pilocarpine-treated epileptic mice, Mirzaeian and Ribak (2000) and Peng and Houser (2005) reported Fos expression in granule cells after generalized seizures, although neither study involved electrophysiological monitoring. Secondary recruitment of granule cells by seizures of extrahippocampal origin in these studies is strongly suggested by the finding of Mirzaeian and Ribak (2000) in epileptic gerbils that although all spontaneous seizures evoked Fos expression in extrahippocampal areas, including the piriform- and perirhinal cortex, only severe spontaneous seizures were associated with granule cell Fos expression. Thus, although severe seizures can apparently recruit dentate granule cells to discharge and express Fos protein (Fig. 4), a secondarily recruited dentate gyrus is not necessarily an “epileptogenic” dentate gyrus.
Spontaneous granule cell layer events in awake kainate-treated epileptic rats
Gorter and colleagues were the first to record directly from the granule cell layers in chronically epileptic kainate-treated rats (Queiroz et al., 2009), with results that were virtually identical to our findings of minimal granule cell involvement during spontaneous seizures in pilocarpine-treated rats (Harvey and Sloviter, 2005). Importantly, their experiments included the elegant use of two recording electrodes placed within the adjacent molecular- and granule cell layers, where the same local synaptic events exhibit opposite polarities (Andersen et al., 1966). By analyzing the recordings made simultaneously in two adjacent strata, Gorter and colleagues demonstrated that whereas evoked potentials originating from within the dentate gyrus exhibited different polarities, recordings from the same electrodes during spontaneous behavioral seizures exhibited identical polarities. This indicated that both electrodes were recording the same distant seizure events, rather than events of granule cell layer origin (Queiroz et al., 2009). Consistent with the conclusion that the granule cells were unlikely to be the generators of the spontaneous seizures in kainate-treated rats, their recordings rarely contained the high frequency, negative-going population discharges that define an epileptiform discharge, which we had also observed to be absent in pilocarpine-treated rats (Harvey and Sloviter, 2005).
The only other study of which we are aware in which granule cell layer activity was recorded during spontaneous seizures in kainate-treated rats was a study by Williams and colleagues (2009). This study showed that high-amplitude EEG events of unknown origin were recorded from granule cell layer electrodes during focal or generalized behavioral seizures, and that these events contained no high frequency, negative-going population discharges. However, Williams and colleagues (2009) used a “record-only” telemetry system with an acquisition rate (100 Hz) 100-fold lower than the rate (10 kHz) used in the other studies cited above. Because low acquisition-rate (100 Hz) recording cannot detect any high frequency information (Fig. 5), the data recorded by Williams and colleagues do not differentiate between: 1) granule cell field depolarizations (field “EPSPs”) that lacked granule cell epileptiform discharges, as recorded by Harvey and Sloviter (2005) and Queiroz et al (2009); 2) hippocampal discharges with high-frequency granule cell population spikes that were not detected by the low acquisition-rate recording method they used, or; 3) distant extrahippocampal EEG activity detected by granule cell layer electrodes, but containing no granule cell-derived information.
Figure 5.
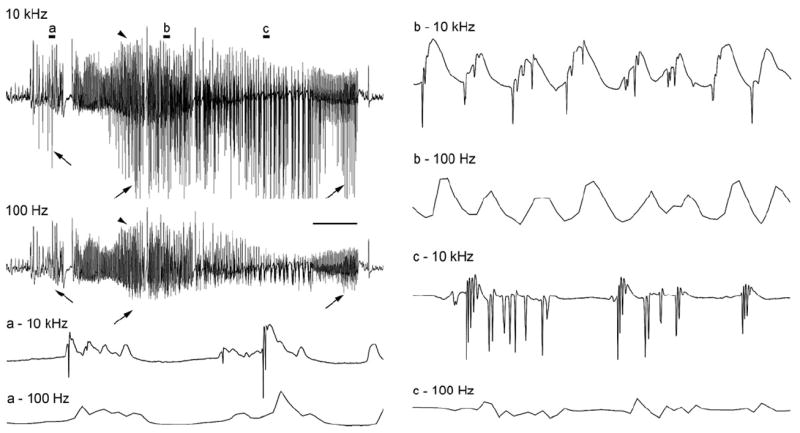
A spontaneous electrographic seizure recorded from the dentate granule cell layer of an awake rat 2 days after 3 hr of stimulation-induced status epilepticus. This first spontaneous granule cell-onset seizure was recorded at two acquisition rates (10 kHz and 100 Hz) simultaneously to illustrate the loss of high frequency data at the lower acquisition rate. Note that presentation of the recording at a compressed timescale (top left) makes it appear that both recordings are similar superficially. However, even at this compression, it can be seen that the slower 100 Hz recording lacks the negative-going population spikes (arrows). Conversely, the slower positive-going potentials are recorded by both methods (arrowheads). Expansion of the recordings clearly shows the selective loss of all high frequency data at the 100 Hz rate, making it impossible to know if population spikes (synchronous granule cell discharges) are present. Scale bar: 4 sec in the compressed traces and 60 msec in traces a-c.
Although the use of low acquisition-rate EEG methodology is clearly useful for documenting seizure occurrence somewhere within the brain, the inability of low acquisition-rate methodology to detect any high-frequency events (Williams et al., 2006; 2009) limits the utility of current telemetric methodology for analyzing spontaneous seizure recordings and determining whether they contain any high-frequency hippocampal neuronal discharges. An acquisition rate above 1 kHz, and preferably above 4 kHz, is necessary to record from identified cell populations and differentiate between: 1) high-amplitude “pseudo-seizure” events (presumed dendritic field depolarizations without epileptiform discharges) that only superficially resemble epileptiform discharges (Harvey and Sloviter, 2005), and; 2) high-frequency epileptiform discharges (population spikes) that involve synchronous action potentials that can propagate synaptically and cause seizures (Bumanglag and Sloviter, 2008). This is an important issue because if high-amplitude events are recorded from hippocampal electrodes and assumed to be hippocampal “seizures” on the basis of low acquisition-rate recordings (see Figure 2 of Williams et al., 2009), then a successful attempt to suppress these EEG events of undetermined nature and unknown location could be incorrectly interpreted as a successful attempt to inhibit “hippocampal epileptogenesis.” The issue of acquisition rate is also significant because spontaneous positive-going “field EPSPs” (“EEG spikes,” not to be confused with faster “population spikes;” Bragin et al., 2011) have been suggested to be biomarkers of future seizures (White et al., 2010), and it is important to know whether these transient events are of granule cell layer origin, and whether they do, or do not, contain high-frequency granule cell population spikes (Fig. 6C). This requires high acquisition-rate recording methods.
Figure 6.
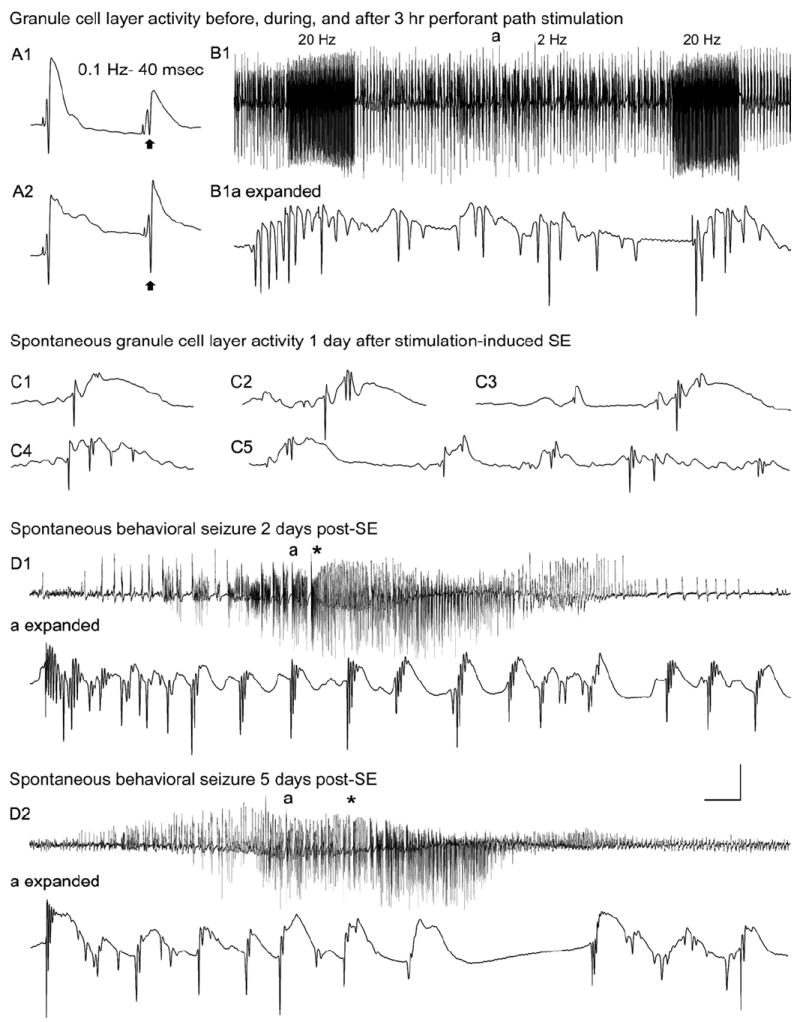
Dentate granule cell excitability and spontaneous activity before and 1–5 days after 3 hours of perforant pathway stimulation-induced convulsive status epilepticus (SE). A1: Before SE, paired-pulse perforant pathway stimulation at 0.1 Hz and an interstimulus interval of 40 msec evokes granule cell responses that exhibit partial suppression of the amplitude of the second population spike (arrow). A2: Three days after 3 hours of SE, the identical afferent stimulation failed to suppress the second population spike (arrow). B1: Granule cell layer activity during 3 hours of perforant pathway stimulation in the same awake rat. The stimulation paradigm involved continuous stimulation at 2 Hz with paired pulses delivered at a 40-msec inter- pulse interval plus 10-second-long 20-Hz trains delivered once per minute. Note the morphology of the granule cell epileptiform discharges during the 2 Hz intertrain interval (a) in the expanded trace (B1a expanded). C: On the first day after 3 hours of stimulation- induced SE, a granule cell layer electrode recorded spontaneous granule cell field “EPSPs” and population spikes that closely resemble the evoked responses in A. D: Granule cell layer activity during spontaneous behavioral seizures during the first week post-SE. D1: On day 2 post-SE, granule cell layer activity amplitude increased before the behavioral onset of the second behavioral seizure on that day (marked by asterisk). D1a (expanded): Expanded trace of the region above marked “a,” showing that the high-amplitude activity in D1 consisted of granule cell epileptiform discharges that preceded the behavioral seizure onset (asterisk). D2: Three days later, the fourth spontaneous behavioral seizure exhibited nearly identical features, including high-frequency granule cell epileptiform discharges (D2a expanded) that preceded the behavioral seizure-onset (asterisk). Calibration bars: 14 msec and 9mV in A; 7 seconds and 9 mV in B1; 46 msec and 9mV in B1 (expanded); 40 msec and 9 mV in C; 3.4 seconds and 9 mV in D1,D2; 53 msec and 9 mV in D1 (expanded); 60 msec and 9 mV in D2 (expanded). From Bumanglag and Sloviter, 2008.
From all of the data presented in the studies discussed above, we concluded that although most of the hypotheses about mechanisms of hippocampal epileptogenesis have come from studies of kainate- and pilocarpine-treated rats, hippocampal epileptogenesis does not appear to be a reliable feature of these models (Harvey and Sloviter, 2005; Queiroz et al., 2009). Therefore, the development of animal models with confirmed hippocampal-onset seizures became a necessity.
The “latent period” and the implication that epileptogenesis involves an obligatory secondary mechanism
In our studies of epileptogenesis in kainate and pilocarpine-treated rats, we noted unexpectedly that spontaneous behavioral seizures nearly always began within the first week following status epilepticus (Sloviter et al., 2003; Harvey and Sloviter, 2005), before any secondary mechanism could have had time to develop, and this has now been confirmed by other laboratories (Goffin et al., 2006; Raol et al., 2006; Jung et al., 2007). Although the lack of any detectable seizure-free “latent period” after chemoconvulsant-induced status epilepticus could mean that brain injury is immediately epileptogenic, rather than delayed, it is also possible that the earliest seizures could be caused by residual chemoconvulsant, and might therefore mask a real latent period during which an injury-initiated secondary epileptogenic mechanism slowly matures. The issue of residual excitotoxin, early seizures, and the latent period was crucial to resolve because the widely-accepted notion that convulsive status epilepticus is reliably followed by a seizure-free “latent period” lasting weeks or months (Bragin et al., 2002; Leite et al., 2002; Stables et al., 2003; El-Hassar et al., 2007) has been the main rationale for the idea that “epileptogenesis” cannot be caused by cell loss alone (Wasterlain et al., 1999). The lack of a latent period after chemoconvulsant-induced status epilepticus, and the lack of any evidence that the spontaneous seizures in chemoconvulsant-treated animals were of hippocampal origin (Harvey and Sloviter (2005), demonstrated the need to develop animal models that reliably exhibit the characteristics of human TLE with hippocampal sclerosis, including selective hippocampal pathology, hippocampal-onset seizures, and a confirmed latency to clinical epilepsy that might serve as a therapeutic window.
How soon after stimulation-induced hippocampal injury do spontaneous granule cell-onset behavioral seizures begin?
We developed an electrical stimulation-based status epilepticus model that was designed to reliably produce selective hippocampal neuron loss and confirmed granule cell-onset seizures in every animal, and to address the possibility that the earliest post-injury seizures in chemoconvulsant-treated rats might be due to residual chemoconvulsant. This model, involving bilateral perforant pathway stimulation-induced convulsive status epilepticus in awake rats, was designed to reliably evoke granule cell epileptiform discharges throughout the 3-hour duration of the experiment. This is an important feature of this model because by forcing the granule cells to discharge during status epilepticus, we avoided the variable hippocampal injury observed after kainate-induced status epilepticus (Sloviter, 1992; Sloviter et al., 2003; Zappone and Sloviter, 2004), or following stimulation-induced, but “self-sustained,” status epilepticus (Gorter et al., 2001). The use of recording electrodes implanted bilaterally within the granule cell somal layers, high acquisition-rate recording methods, and continuous (24/7) video monitoring enabled us to determine for the first time: 1) whether and when granule cells within a damaged dentate gyrus begin to generate spontaneous epileptiform discharges; 2) whether these granule cell discharges reliably initiate behavioral seizures, and; 3) how soon after a controlled injury spontaneous behavioral seizures begin when no residual chemoconvulsant is present.
The results from this stimulation-based model were as follows. First, after 3 hours of continuous bilateral granule cell epileptiform discharges and convulsive status epilepticus in awake rats, extensive hilar neuron loss was evident throughout the hippocampal longitudinal axis, and was coincident with immediate granule cell disinhibition and hyperexcitability in the awake state (Bumanglag and Sloviter, 2008). Second, continuous recording of spontaneous activity in the granule cell layers of awake rats revealed that starting immediately after injury, granule cells began to generate spontaneous high-amplitude “field EPSPs” and population spikes (synchronous granule cell population discharges) that were identical in polarity, amplitude, and waveform morphology to the potentials that were evoked by perforant pathway stimulation (Fig. 6). Third, spontaneous high-frequency granule cell layer epileptiform discharges (Fig. 6, D1a and D2a) were recorded on the second post-injury day, and each spontaneous epileptiform discharge immediately preceded, and apparently caused, either a focal- or a generalized behavioral seizure. Thus, early seizures were not related to the presence of residual chemoconvulsant, and there was no detectable latency to the first clinical seizure.
Fourth, spontaneous granule cell epileptiform discharges lasting ~45 seconds were reliably associated with focal behavioral seizures (staring without moving, facial automatisms, and “wet-dog” shakes), whereas spontaneous granule cell epileptiform discharges lasting ~2 minutes were reliably associated with generalized behavioral seizures (Bumanglag and Sloviter, 2008). Importantly, when spontaneous behavioral seizures were identified by first analyzing the video recordings, it was confirmed that all behavioral seizures were immediately preceded by granule cell epileptiform discharges. The reverse was similarly true; when granule cell epileptiform discharges were identified by first analyzing the electrographic recordings, all spontaneous granule cell epileptiform discharges longer than ~15 sec were immediately followed by either focal or generalized behavioral seizures. This finding is in contrast to the results we obtained in pilocarpine-treated rats, in which all spontaneous behavioral seizures of the chronic epileptic state appeared to minimally involve epileptiform granule cell discharges (Harvey and Sloviter, 2005).
The apparent temporal mismatch between initial neuron loss and the appearance of clinical epilepsy
The finding that confirmed granule cell epileptogenesis is an immediate consequence of prolonged status epilepticus resolves one fundamental issue regarding the nature of hippocampal epileptogenesis. We have hypothesized that the seizure-induced loss of dentate hilar mossy cells causes immediate granule cell disinhibition because mossy cells normally excite inhibitory basket cells and establish normal translamellar granule cell inhibition (the “dormant basket cell” hypothesis; Sloviter, 1987; 1991; 1994; Sloviter et al., 2003; Zappone and Sloviter, 2004). Our hypothesis that extensive mossy cell loss is immediately epileptogenic (Sloviter, 1994) was justifiably questioned by Wasterlain and colleagues who noted that, “this hypothesis does not tackle the problem of the ‘silent period’ between the initial injury and the development of spontaneous seizures. Because mossy cells are injured at the time of the original seizures and disappear within hours to days, it is hard to understand why spontaneous seizures are delayed by weeks to months, unless the loss of inhibition is permissive but not sufficient for epileptogenesis, and its role is only to permit further changes which, in turn, produce chronic epilepsy” (Wasterlain et al., 1999).
The notion that neuron loss and granule cell disinhibition cannot be epileptogenic because both occur immediately (Sloviter, 1987; Zappone and Sloviter, 2004), whereas clinical epilepsy is delayed for weeks or months (Wasterlain et al., 1999), received experimental support from a study in pilocarpine-treated rats in which occasional video monitoring resulted in a latent period estimate of 26 ± 3 days before the first behavioral seizure was observed (Kobayashi and Buckmaster, 2003). Despite using methods that involved no monitoring of animals for the majority (>75%) of the time after status epilepticus, these authors nonetheless concluded that, “reduced granule cell inhibition alone is insufficient to cause epilepsy, because it was present when rats were not experiencing spontaneous seizures. This finding suggests that epileptogenesis requires other changes” (Kobayashi and Buckmaster, 2003).
The use of continuous (24/7) monitoring by multiple laboratories has now demonstrated unequivocally that spontaneous behavioral seizures are not delayed, but begin within the first week after injury (Harvey and Sloviter, 2005; Goffin et al., 2006; Raol et al., 2006; Jung et al., 2007; Rattka et al., 2011), and often by day 2 post-injury (Bumanglag and Sloviter, 2008). Thus, there is no obligatory latent period after status epilepticus, and the prevalent perception to the contrary (Wasterlain et al., 1999; Leite et al., 2002; Walker et al., 2002; Kobayashi and Buckmaster, 2003; Stables et al., 2003; El-Hassar et al., 2007) is apparently a consequence of occasional behavioral observation that misses the earliest seizures, or continuous monitoring of minimally injured animals, which can have a very long latent period to clinical epilepsy (Navarro-Mora et al., 2009). Thus, we reiterate that granule cell hyperexcitability/ disinhibition closely associated with extensive loss of dentate hilar mossy cells and interneurons (Sloviter, 1987; 1991; Zappone and Sloviter, 2004; Jinde et al., 2008), and entorhinal cortex neurons (Du et al., 1995; Sloviter et al., 2012), could be a primary epileptogenic mechanism because pathology, granule cell disinhibition, and granule cell-onset epilepsy all occur concomitantly (Bumanglag and Sloviter, 2008), and because contralateral granule cell disinhibition caused by unilateral stimulation-induced injury was always and only associated with extensive contralateral hilar neuron loss (Sloviter, 1991). We suggest that neuron loss has a direct epileptogenic influence independent of any secondary processes that neuron loss or injuries may also trigger.
“Keeping focal seizures focal” as a primary therapeutic strategy
The concept of a virtually immediate “epileptogenic” change in network behavior, followed by a longer-lasting secondary process of “epileptic maturation,” suggests that “disease prevention” and “disease modification” are distinct therapeutic targets. Thus, antiepileptogenesis therapy might focus productively on neuroprotection, i.e., targeting potentially reversible neuronal injury during the immediate post-injury period (Langer et al., 2011; Jimenez-Mateos et al., 2011; Serrano et al., 2011). Once the first focal seizures begin, however, “disease modification” strategies might profitably include silencing (Noè et al., 2008; 2010; Gøtzsche et al., 2011; Shetty, 2011) or selectively ablating the seizure focus, targeting mechanisms of discharge elongation (“kindling”; Goddard et al., 1969; Giblin and Blumenfeld, 2010; Löscher and Brandt, 2010) and seizure spread, and other secondary changes that might alter the chronic clinical phenotype, including synaptic reorganization, inflammation, blood/brain barrier disruption, glial changes, changes in receptor and channel expression, and neurogenesis (Tauck and Nadler, 1985; Chang and Lowenstein, 2003; van Vliet et al., 2007; Parent and Murphy, 2008; Richichi et al., 2008; Ravizza et al., 2011). Therapies that target the immediate post-injury period, and are designed to “keep focal seizures focal,” might prevent initially subclinical seizures from becoming more life-disrupting clinical events, even if injury-induced epileptogenesis cannot be entirely prevented.
Caveats and clarifications
Several caveats and clarifications relate to our analyses and assertions. First, in terms of clarifications, we emphasize that acquired epilepsies involve a variety of insults that presumably produce clinically distinct epilepsies that originate in different locations and exhibit different latencies to clinical seizures. We do not suggest that there must be one common epileptogenic mechanism, and we recognize that future studies using chronic depth recording will need to determine whether other injuries, such as head trauma affecting the dentate gyrus (Lowenstein et al., 1992), produce granule cell-onset epilepsy similar to that caused by status epilepticus. In this paper, we are specifically addressing the assumptions underlying models that use convulsive status epilepticus to produce hippocampal epileptogenesis, and our assertions do not necessarily apply to other models, injuries, or disorders.
Second, we do not question the fact that latent periods exist after brain injury. In fact, most animals and people subjected to brain injuries exhibit a latent period, but we regard this to be a latency to the first clinical seizures, not a latency to epilepsy. Our working hypothesis (Sloviter et al, 2012) is that animals that exhibit a long latency to the first clinical seizures are simply less extensively injured (Sloviter, 1994; Gorter et al., 2001; Bumanglag and Sloviter, 2008; Navarro-Mora et al., 2009), although this relationship remains to be investigated in greater depth. The central issue is whether the latent period should be viewed as a seizure-free, pre-epileptic period of “epileptogenesis,” or a prolonged and variable period of “epileptic maturation” during which an initially subtle focal disorder gradually becomes more clinically obvious. We would argue that a lack of clinically obvious seizures shortly after brain injury does not permit the inference that epilepsy requires maturation of a slow secondary process, or that the latent period can be logically assumed to be the duration of “epileptogenesis.” If a patient develops clinical epilepsy 20 years or longer after prolonged febrile seizures in childhood, and assuming that the febrile seizures caused the subsequent afebrile epilepsy, can it be assumed that this patient’s brain was not “epileptic” a day, a year, or two decades before the first clinically obvious seizure? We don’t think so. Why clinical epilepsy sometimes takes decades to develop after an injury is a speculative topic beyond the focus of this paper, but may involve an age-related change in seizure threshold that eventually allows focal seizures to spread and cause the first clinically obvious seizure. In our view, these individuals do not have an incomplete epileptogenic process; they simply have an unrecognized, subclinical, focal seizure disorder.
Third, we refer repeatedly in this paper to “immediate” epileptogenesis after prolonged status epilepticus. By “immediate” we do not mean that all animals exhibit spontaneous generalized behavioral seizures within the first hour after the end of status epilepticus. Excitotoxic seizure activity produces both immediate and slightly delayed neuron death (Sloviter et al., 1996; Brandt et al., 2003b), and our main points are that: 1) there is no obligatory latent period of several weeks duration following status epilepticus, as widely believed and routinely restated (Wasterlain et al., 1999; Leite et al., 2002; Walker et al., 2002; Kobayashi and Buckmaster, 2003; Stables et al., 2003; El-Hassar et al., 2007), and 2) the time-course of epileptogenesis is coincident with initial cell death and all other early effects that are occurring throughout the first week post-injury. It is in this sense that we regard epileptogenesis to be “immediate.”
Fourth, in pointing out that kainate- and pilocarpine-induced status epilepticus produces widespread and often severe brain damage, and does not reproduce the human pattern of temporal lobe pathology (Schwob et al., 1980; Norwood et al., 2010), we are not suggesting that kainate- and pilocarpine-treated rats do not exhibit hippocampal injury; they do. The issue is whether convulsive status epilepticus, presumably initiated in multiple brain sites by a systemically circulating chemical, produces a pattern of pathology that resembles the limited brain pathology that defines most TLE patients (Van Paesschen et al., 1997), and whether such a severely damaged brain is a model of human TLE that can be used fruitfully to address the mechanisms of hippocampal epileptogenesis specifically. It is our contention that useful disease-relevant models should be shown to replicate the defining features of the human neurological condition, as we have sought to do in recent studies (Bumanglag and Sloviter, 2008; Kienzler et al., 2009; Norwood et al., 2010). Although kainate and pilocarpine-induced status epilepticus does not reliably cause classic hippocampal sclerosis in mice or rats under any circumstances (Kienzler et al., 2009; Norwood et al., 2010), convulsive status epilepticus often produces experimental “endfolium sclerosis” (Zappone and Sloviter, 2004; Harvey and Sloviter, 2005; Bumanglag and Sloviter, 2008), a pattern of cell loss characterized by the loss of hilar neurons and CA3c pyramidal cells (Margerison and Corsellis, 1966; Sloviter, 1994). However, when convulsive status epilepticus causes hippocampal damage, it does so, in our experience, within a context of widespread extrahippocampal injury that is the opposite of the pattern in human TLE with hippocampal sclerosis, i.e. extensive hippocampal neuron loss and relatively subtle extrahippocampal pathology (Sloviter et al., 2007; Sloviter, 2008; Kienzler et al., 2009; Norwood et al., 2010).
Fifth, it could be argued that the earliest post-injury seizures following electrical stimulation-induced status epilepticus are not individual seizures, but a recurrence of status epilepticus. We emphasize here that the spontaneous seizures that develop soon after injury in stimulated animals (Bumanglag and Sloviter, 2008) are brief (< 2 min) individual spontaneous seizures, which in all cases are initiated by granule cell epileptiform discharges that terminate spontaneously. Status epilepticus does not recur in these stimulation-based models when status epilepticus is effectively terminated. We can state this with some confidence because we monitor both electrographic and behavioral activity continuously.
As a final caveat, we acknowledge several problems of interpretation that are inherent in the studies from which all of our conclusions are generated. The mechanism by which granule cells become hyperexcitable and spontaneously epileptic is suggested to be a loss of inhibition caused primarily by neuron loss (Sloviter, 1987; 1994; Sloviter et al., 2012), but this is, and will remain, a correlational relationship until neuron subpopulations can be eliminated selectively (Jinde et al., 2008). Granule cell epileptogenesis could also be the result of another mechanism with a similarly rapid time course. Other issues of interpretation include the fact that the true source of seizures is difficult to determine, even with multiple electrode recording, that recording from implanted electrodes could involve issues related to the presence of electrodes (which is, in itself, a brain injury), and that the linear extrapolation of data from an animal model in which identical insults are forced upon genetically similar rodents, to a human neurological disorder in which variable insults occur in genetically dissimilar individuals, involves a leap of faith that can be reasonably questioned.
Highlights.
“Epileptogenesis” and “epileptic maturation” defined
Animal models of acquired temporal lobe epilepsy with hippocampal sclerosis
Spontaneous granule cell layer events in epileptic rats
Hippocampal c-Fos expression after spontaneous epileptic seizures
“Keeping focal seizures focal” as a primary therapeutic strategy
Acknowledgments
We thank Drs. Wolfgang Löscher (University of Veterinary Medicine Hannover), Daniel H. Lowenstein (University of California, San Francisco), Robert Schwarcz (University of Maryland), H. Steve White (University of Utah), and Hitten Zaveri (Yale University) for useful discussions and constructive criticism of the manuscript. Grant sponsor: National Institute of Neurological Disorders and Stroke, NIH; Grant NS18201.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andersen P, Holmqvist B, Voorhoeve PE. Entorhinal activation of dentate granule cells. Acta Physiol Scand. 1966;66:448–460. doi: 10.1111/j.1748-1716.1966.tb03223.x. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y. Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience. 1985;14:375–403. doi: 10.1016/0306-4522(85)90299-4. [DOI] [PubMed] [Google Scholar]
- Biagini G, Baldelli E, Longo D, Contri MB, Guerrini U, Sironi L, Gelosa P, Zini I, Ragsdale DS, Avoli M. Proepileptic influence of a focal vascular lesion affecting entorhinal cortex-CA3 connections after status epilepticus. J Neuropathol Exp Neurol. 2008;67:687–701. doi: 10.1097/NEN.0b013e318181b8ae. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blümcke I, Pauli E, Clusmann H, Schramm J, Becker A, Elger C, Merschhemke M, Meencke HJ, Lehmann T, von Deimling A, Scheiwe C, Zentner J, Volk B, Romstöck J, Stefan H, Hildebrandt M. A new clinico-pathological classification system for mesial temporal sclerosis. Acta Neuropathol. 2007;113:235–244. doi: 10.1007/s00401-006-0187-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bower MR, Buckmaster PS. Changes in granule cell firing rates precede locally recorded spontaneous seizures by minutes in an animal model of temporal lobe epilepsy. J Neurophysiol. 2008;99:2431–2442. doi: 10.1152/jn.01369.2007. [DOI] [PubMed] [Google Scholar]
- Bragin A, Csicsvári J, Penttonen M, Buzsáki G. Epileptic afterdischarge in the hippocampal-entorhinal system: current source density and unit studies. Neuroscience. 1997;76:1187–1203. doi: 10.1016/s0306-4522(96)00446-0. [DOI] [PubMed] [Google Scholar]
- Bragin A, Wilson CL, Engel J., Jr Chronic epileptogenesis requires development of a network of pathologically interconnected neuron clusters: a hypothesis. Epilepsia. 2000;41(Suppl. 6):S144–S152. doi: 10.1111/j.1528-1157.2000.tb01573.x. [DOI] [PubMed] [Google Scholar]
- Bragin A, Benassi SK, Kheiri F, Engel J., Jr Further evidence that pathologic high-frequency oscillations are bursts of population spikes derived from recordings of identified cells in dentate gyrus. Epilepsia. 2011;52:45–52. doi: 10.1111/j.1528-1167.2010.02896.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt C, Glien M, Potschka H, Volk H, Loscher W. Epileptogenesis and neuropathology after different types of status epilepticus induced by prolonged electrical stimulation of the basolateral amygdala in rats. Epilepsy Res. 2003a;55:83–103. doi: 10.1016/s0920-1211(03)00114-1. [DOI] [PubMed] [Google Scholar]
- Brandt C, Potschka H, Löscher W, Ebert U. N-methyl-D-aspartate receptor blockade after status epilepticus protects against limbic brain damage but not against epilepsy in the kainate model of temporal lobe epilepsy. Neuroscience. 2003b;118:727–740. doi: 10.1016/s0306-4522(03)00027-7. [DOI] [PubMed] [Google Scholar]
- Bumanglag AV, Sloviter RS. Minimal latency to hippocampal epileptogenesis and clinical epilepsy after perforant pathway stimulation-induced status epilepticus in awake rats. J Comp Neurol. 2008;510:561–580. doi: 10.1002/cne.21801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso A, Lukoyanova EA, Madeira MD, Lukoyanov NV. Seizure-induced structural and functional changes in the rat hippocampal formation: comparison between brief seizures and status epilepticus. Behav Brain Res. 2011;225:538–546. doi: 10.1016/j.bbr.2011.07.057. [DOI] [PubMed] [Google Scholar]
- Chang BS, Lowenstein DH. Mechanisms of disease; epilepsy. N Engl J Med. 2003;349:1257–1266. doi: 10.1056/NEJMra022308. [DOI] [PubMed] [Google Scholar]
- Chen S, Buckmaster PS. Stereological analysis of fore- brain regions in kainate-treated epileptic rats. Brain Res. 2005;1057:141–152. doi: 10.1016/j.brainres.2005.07.058. [DOI] [PubMed] [Google Scholar]
- Dichter MA. Emerging concepts in the pathogenesis of epilepsy and epileptogenesis. Arch Neurol. 2009;66:443–447. doi: 10.1001/archneurol.2009.10. [DOI] [PubMed] [Google Scholar]
- Dragunow M, Robertson HA. Kindling stimulation induces c-fos protein(s) in granule cells of the rat dentate gyrus. Nature. 1987;329:441–442. doi: 10.1038/329441a0. [DOI] [PubMed] [Google Scholar]
- Du F, Eid T, Lothman EW, Köhler C, Schwarcz R. Preferential neuronal loss in layer III of the medial entorhinal cortex in rat models of temporal lobe epilepsy. J Neurosci. 1995;15:6301–6313. doi: 10.1523/JNEUROSCI.15-10-06301.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du F, Whetsell WO, Jr, Abou-Khalil B, Blumenkopf B, Lothman EW, Schwarcz R. Preferential neuronal loss in layer III of the entorhinal cortex in patients with temporal lobe epilepsy. Epilepsy Res. 1993;16:223–233. doi: 10.1016/0920-1211(93)90083-j. [DOI] [PubMed] [Google Scholar]
- Dudek FE, Staley KJ. The time course of acquired epilepsy: Implications for therapeutic intervention to suppress epileptogenesis. Neurosci Lett. 2011;497:240–246. doi: 10.1016/j.neulet.2011.03.071. [DOI] [PubMed] [Google Scholar]
- Earle KM, Baldwin M, Penfield W. Incisural sclerosis and temporal lobe seizures produced by hippocampal herniation at birth. AMA Arch Neurol Psychiatry. 1953;69:27–42. doi: 10.1001/archneurpsyc.1953.02320250033003. [DOI] [PubMed] [Google Scholar]
- Eastman CL, Verley DR, Fender JS, Stewart TH, Nov E, Curia G, D’Ambrosio R. Antiepileptic and antiepileptogenic performance of carisbamate after head injury in the rat: blind and randomized studies. J Pharmacol Exp Ther. 2011;336:779–790. doi: 10.1124/jpet.110.175133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hassar L, Esclapez M, Bernard C. Hyperexcitability of the CA1 hippocampal region during epileptogenesis. Epilepsia. 2007;48(Suppl. 5):131–139. doi: 10.1111/j.1528-1167.2007.01301.x. [DOI] [PubMed] [Google Scholar]
- French JA, Williamson PD, Thadani VM, Darcey TM, Mattson RH, Spencer SS, Spencer DD. Characteristics of medial temporal lobe epilepsy: I. Results of history and physical examination. Ann Neurol. 1993;34:774–780. doi: 10.1002/ana.410340604. [DOI] [PubMed] [Google Scholar]
- Giblin KA, Blumenfeld H. Is epilepsy a preventable disorder? New evidence from animal models. Neuroscientist. 2010;16:253–275. doi: 10.1177/1073858409354385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard GV, McIntyre DC, Leech CK. A permanent change in brain function resulting from daily electrical stimulation. Exp Neurol. 1969;25:295–330. doi: 10.1016/0014-4886(69)90128-9. [DOI] [PubMed] [Google Scholar]
- Goffin K, Nissinen J, Van Laere K, Pitkänen A. Cyclicity of spontaneous recurrent seizures in pilocarpine model of temporal lobe epilepsy in rat. Exp Neurol. 2007;205:501–505. doi: 10.1016/j.expneurol.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Gorter JA, van Vliet EA, Aronica E, Lopes da Silva FH. Progression of spontaneous seizures after status epilepticus is associated with mossy fibre sprouting and extensive bilateral loss of hilar parvalbumin and somatostatin-immunoreactive neurons. Eur J Neurosci. 2001;13:657–669. doi: 10.1046/j.1460-9568.2001.01428.x. [DOI] [PubMed] [Google Scholar]
- Gøtzsche CR, Nikitidou L, Sørensen AT, Olesen MV, Sørensen G, Christiansen SH, Angehagen M, Woldbye DP, Kokaia M. Combined gene overexpression of neuropeptide Y and its receptor Y5 in the hippocampus suppresses seizures. Neurobiol Dis. 2011;45:288–296. doi: 10.1016/j.nbd.2011.08.012. [DOI] [PubMed] [Google Scholar]
- Harvey BD, Sloviter RS. Hippocampal granule cell activity and c-Fos expression during spontaneous seizures in awake, chronically epileptic, pilocarpine-treated rats; implications for hippocampal epileptogenesis. J Comp Neurol. 2005;488:441–462. doi: 10.1002/cne.20594. [DOI] [PubMed] [Google Scholar]
- Jimenez-Mateos EM, Bray I, Sanz-Rodriguez A, Engel T, McKiernan RC, Mouri G, Tanaka K, Sano T, Saugstad JA, Simon RP, Stallings RL, Henshall DC. miRNA expression profile after status epilepticus and hippocampal neuroprotection by targeting miR-132. Am J Pathol. 2011;179:2519–2532. doi: 10.1016/j.ajpath.2011.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinde S, Zsiros V, Kohno K, Nakazawa K. Neuroscience Meeting Planner. Chicago, IL: Society for Neuroscience; 2008. Generation and characterization of inducible-dentate mossy cell abla- tion mice. Program number 645.11. The URL is: http://www.abstractsonline.com/Plan/ViewAbstract.aspx?sKey=c9f36363-a10a-479-b-b930-57f8fdfc81f0&cKey=e4cbc77e-83bf-4-ca5-b871-df97324d0fbc&mKey={AFEA068D-D012-4520-8E42-10E4D1AF7944} [Google Scholar]
- Jung S, Jones TD, Lugo JN, Sheerin JH, Miller JW, D’Ambrosio R, Anderson AE, Poolos NP. Progressive dendritic HCN channelopathy during epileptogenesis in the rat pilocarpine model of epilepsy. J Neurosci. 2007;27:13012–13021. doi: 10.1523/JNEUROSCI.3605-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kienzler F, Norwood BA, Sloviter RS. Hippocampal injury, atrophy, synaptic reorganization, and epileptogenesis after perforant pathway stimulation-induced status epilepticus in the mouse. J Comp Neurol. 2009;515:181–196. doi: 10.1002/cne.22059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Buckmaster PS. Reduced inhibition of dentate granule cells in a model of temporal lobe epilepsy. J Neurosci. 2003;23:2440–2452. doi: 10.1523/JNEUROSCI.23-06-02440.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer M, Brandt C, Zellinger C, Löscher W. Therapeutic window of opportunity for the neuroprotective effect of valproate versus the competitive AMPA receptor antagonist NS1209 following status epilepticus in rats. Neuropharmacology. 2011;61:1033–1047. doi: 10.1016/j.neuropharm.2011.06.015. [DOI] [PubMed] [Google Scholar]
- Leite JP, Bortolotto ZA, Cavalheiro EA. Spontaneous recurrent seizures in rats: an experimental model of partial epilepsy. Neurosci Biobehav Rev. 1990;14:511–517. doi: 10.1016/s0149-7634(05)80076-4. [DOI] [PubMed] [Google Scholar]
- Leite JP, Garcia-Cairasco N, Cavalheiro EA. New insights from the use of pilocarpine and kainate models. Epilepsy Res. 2002;50:93–103. doi: 10.1016/s0920-1211(02)00072-4. [DOI] [PubMed] [Google Scholar]
- Löscher W, Brandt C. Prevention or modification of epileptogenesis after brain insults: experimental approaches and translational research. Pharmacol Rev. 2010;62:668–700. doi: 10.1124/pr.110.003046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenstein DH, Thomas MJ, Smith DH, McIntosh TK. Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J Neurosci. 1992;12:4846–4853. doi: 10.1523/JNEUROSCI.12-12-04846.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margerison JH, Corsellis JA. Epilepsy and the temporal lobes. A clinical, electroencephalographic and neuropathological study of the brain in epilepsy, with particular reference to the temporal lobes. Brain. 1966;89:499–530. doi: 10.1093/brain/89.3.499. [DOI] [PubMed] [Google Scholar]
- Mello LE, Kohman CM, Tan AM, Cavalheiro EA, Finch DM. Lack of Fos-like immunoreactivity after spontaneous seizures or reinduction of status epilepticus by pilocarpine in rats. Neurosci Lett. 1996;208:133–137. doi: 10.1016/0304-3940(96)12562-3. [DOI] [PubMed] [Google Scholar]
- Mirzaeian L, Ribak CE. Immunocytochemical mapping of Fos protein following seizures in gerbils indicates the activation of hippocampal neurons. Hippocampus. 2000;10:31–36. doi: 10.1002/(SICI)1098-1063(2000)10:1<31::AID-HIPO3>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Morgan JI, Cohen DR, Hempstead JL, Curran T. Mapping patterns of c-fos expression in the central nervous system after seizure. Science. 1987;237:192–197. doi: 10.1126/science.3037702. [DOI] [PubMed] [Google Scholar]
- Nadler JV. Minireview. Kainic acid as a tool for the study of temporal lobe epilepsy. Life Sci. 1981;29:2031–2042. doi: 10.1016/0024-3205(81)90659-7. [DOI] [PubMed] [Google Scholar]
- Nadler JV, Perry BW, Cotman CW. Selective reinnervation of hippocampal area CA1 and the fascia dentata after destruction of CA3– CA4 afferents with kainic acid. Brain Res. 1980;182:1–9. doi: 10.1016/0006-8993(80)90825-2. [DOI] [PubMed] [Google Scholar]
- Navarro-Mora G, Bramanti P, Osculati F, Chakir A, Nicolato E, Marzola P, Sbarbati A, Fabene PF. Does pilocarpine-induced epilepsy in adult rats require status epilepticus? PLoS One. 2009;4:e5759. doi: 10.1371/journal.pone.0005759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noè F, Pool AH, Nissinen J, Gobbi M, Bland R, Rizzi M, Balducci C, Ferraguti F, Sperk G, During MJ, Pitkänen A, Vezzani A. Neuropeptide Y gene therapy decreases chronic spontaneous seizures in a rat model of temporal lobe epilepsy. Brain. 2008;131:1506–1515. doi: 10.1093/brain/awn079. [DOI] [PubMed] [Google Scholar]
- Noè F, Vaghi V, Balducci C, Fitzsimons H, Bland R, Zardoni D, Sperk G, Carli M, During MJ, Vezzani A. Anticonvulsant effects and behavioural outcomes of rAAV serotype 1 vector-mediated neuropeptide Y overexpression in rat hippocampus. Gene Ther. 2010;17:643–652. doi: 10.1038/gt.2010.23. [DOI] [PubMed] [Google Scholar]
- Norwood BA, Bumanglag AV, Osculati F, Sbarbati A, Marzola P, Nicolato E, Fabene PF, Sloviter RS. Classic hippocampal sclerosis and hippocampal-onset epilepsy produced by a single “cryptic” episode of focal hippocampal excitation in awake rats. J Comp Neurol. 2010;518:3381–3407. doi: 10.1002/cne.22406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM, Murphy GG. Mechanisms and functional significance of aberrant seizureinduced hippocampal neurogenesis. Epilepsia. 2008;49(Suppl. 5):19–25. doi: 10.1111/j.1528-1167.2008.01634.x. [DOI] [PubMed] [Google Scholar]
- Peng Z, Houser CR. Temporal patterns of fos expression in the dentate gyrus after spontaneous seizures in a mouse model of temporal lobe epilepsy. J Neurosci. 2005;25:7210–7220. doi: 10.1523/JNEUROSCI.0838-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkänen A. Therapeutic approaches to epileptogenesis--hope on the horizon. Epilepsia. 2010;51(Suppl. 3):2–17. doi: 10.1111/j.1528-1167.2010.02602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkänen A, Bolkvadze T, Immonen R. Anti-epileptogenesis in rodent post-traumatic epilepsy models. Neurosci Lett. 2011;497:163–171. doi: 10.1016/j.neulet.2011.02.033. [DOI] [PubMed] [Google Scholar]
- Poirier JL, Capek R, De Koninck Y. Differential progression of Dark Neuron and Fluoro-Jade labelling in the rat hippocampus following pilocarpine-induced status epilepticus. Neuroscience. 2000;97:59–68. doi: 10.1016/s0306-4522(00)00026-9. [DOI] [PubMed] [Google Scholar]
- Queiroz CM, Gorter JA, Lopes da Silva FH, Wadman WJ. Dynamics of evoked local field potentials in the hippocampus of epileptic rats with spontaneous seizures. J Neurophysiol. 2009;101:1588–1597. doi: 10.1152/jn.90770.2008. [DOI] [PubMed] [Google Scholar]
- Rakhade SN, Jensen FE. Epileptogenesis in the immature brain: emerging mechanisms. Nat Rev Neurol. 2009;5:380–391. doi: 10.1038/nrneurol.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raol YH, Lund IV, Bandyopadhyay S, Zhang G, Roberts DS, Wolfe JH, Russek SJ, Brooks-Kayal AR. Enhancing GABA(A) receptor alpha 1 subunit levels in hippocampal dentate gyrus inhibits epilepsy development in an animal model of temporal lobe epilepsy. J Neurosci. 2006;26:11342–11346. doi: 10.1523/JNEUROSCI.3329-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattka M, Brandt C, Bankstahl M, Bröer S, Löscher W. Enhanced susceptibility to the GABA antagonist pentylenetetrazole during the latent period following a pilocarpine-induced status epilepticus in rats. Neuropharmacology. 2011;60:505–512. doi: 10.1016/j.neuropharm.2010.11.005. [DOI] [PubMed] [Google Scholar]
- Ravizza T, Balosso S, Vezzani A. Inflammation and prevention of epileptogenesis. Neurosci Lett. 2011;497:223–230. doi: 10.1016/j.neulet.2011.02.040. [DOI] [PubMed] [Google Scholar]
- Richichi C, Brewster AL, Bender RA, Simeone TA, Zha Q, Yin HZ, Weiss JH, Baram TZ. Mechanisms of seizure-induced ‘transcriptional channelopathy’ of hyperpolarization-activated cyclic nucleotide gated (HCN) channels. Neurobiol Dis. 2008;29:297–305. doi: 10.1016/j.nbd.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwob JE, Fuller T, Price JL, Olney JW. Widespread patterns of neuronal damage following systemic or intracerebral injections of kainic acid: a histological study. Neuroscience. 1980;5:991–1014. doi: 10.1016/0306-4522(80)90181-5. [DOI] [PubMed] [Google Scholar]
- Serrano GE, Lelutiu N, Rojas A, Cochi S, Shaw R, Makinson CD, Wang D, FitzGerald GA, Dingledine R. Ablation of cyclooxygenase-2 in forebrain neurons is neuroprotective and dampens brain inflammation after status epilepticus. J Neurosci. 2011;31:14850–14860. doi: 10.1523/JNEUROSCI.3922-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty AK. Progress in cell grafting therapy for temporal lobe epilepsy. Neurotherapeutics. 2011;8:721–735. doi: 10.1007/s13311-011-0064-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloviter RS. Decreased hippocampal inhibition and a selective loss of interneurons in experimental epilepsy. Science. 1987;235:73–76. doi: 10.1126/science.2879352. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. Permanently altered hippocampal structure, excitability, and inhibition after experimental status epilepticus in the rat: the dormant basket cell hypothesis and its possible relevance to temporal lobe epilepsy. Hippocampus. 1991;1:41–66. doi: 10.1002/hipo.450010106. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. Possible functional consequences of synaptic reorganization in the dentate gyrus of kainate-treated rats. Neurosci Lett. 1992;137:91–96. doi: 10.1016/0304-3940(92)90306-r. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. The functional organization of the hippocampal dentate gyrus and its relevance to the pathogenesis of temporal lobe epilepsy. Ann Neurol. 1994;35:640–654. doi: 10.1002/ana.410350604. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. Apoptosis: a guide for the perplexed. Trends Pharmacol Sci. 2002;23:19–24. doi: 10.1016/s0165-6147(00)01867-8. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. The neurobiology of temporal lobe epilepsy: too much information, not enough knowledge. CR Biol. 2005;328:143–153. doi: 10.1016/j.crvi.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. Hippocampal epileptogenesis in animal models of mesial temporal lobe epilepsy with hippocampal sclerosis; the importance of the “latent period” and other concepts. Epilepsia. 2008;49(Suppl. 9):85–92. doi: 10.1111/j.1528-1167.2008.01931.x. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. Progress on the issue of excitotoxic injury modification vs. real neuroprotection; implications for post-traumatic epilepsy. Neuropharmacology. 2011;61:1048–1050. doi: 10.1016/j.neuropharm.2011.07.038. [DOI] [PubMed] [Google Scholar]
- Sloviter RS, Dean E, Sollas AL, Goodman JH. Apoptosis and necrosis induced in different hippocampal neuron populations by repetitive perforant path stimulation in the rat. J Comp Neurol. 1996;366:516–533. doi: 10.1002/(SICI)1096-9861(19960311)366:3<516::AID-CNE10>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Sloviter RS, Zappone CA, Harvey BD, Bumanglag AV, Bender RA, Frotscher M. “Dormant basket cell” hypothesis revisited; relative vulnerabilities of dentate gyrus mossy cells and inhibitory interneurons after hippocampal status epilepticus in the rat. J Comp Neurol. 2003;459:44–76. doi: 10.1002/cne.10630. [DOI] [PubMed] [Google Scholar]
- Sloviter RS, Zappone CA, Harvey BD, Frotscher M. Kainic acid-induced recurrent mossy fiber innervation of dentate gyrus inhibitory interneurons: possible anatomical substrate of granule cell hyperinhibition in chronically epileptic rats. J Comp Neurol. 2006;494:944–960. doi: 10.1002/cne.20850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloviter RS, Zappone CA, Bumanglag AV, Norwood BA, Kudrimoti H. On the relevance of prolonged convulsive status epilepticus in animals to the etiology and neurobiology of human temporal lobe epilepsy. Epilepsia. 2007;48(Suppl. 8):6–10. doi: 10.1111/j.1528-1167.2007.01335.x. [DOI] [PubMed] [Google Scholar]
- Sloviter RS, Bumanglag AV, Schwarcz R, Frotscher M. Abnormal dentate gyrus network circuitry in temporal lobe epilepsy. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s Basic Mechanisms of the Epilepsies. fourth ed. Oxford University Press; 2012. [PubMed] [Google Scholar]
- Spencer SS. Substrates of localization-related epilepsies: biological implications of localizing findings in humans. Epilepsia. 1998;39:114–123. doi: 10.1111/j.1528-1157.1998.tb01349.x. [DOI] [PubMed] [Google Scholar]
- Spencer SS. Neural networks in human epilepsy: evidence of and implications for treatment. Epilepsia. 2002;43:219–227. doi: 10.1046/j.1528-1157.2002.26901.x. [DOI] [PubMed] [Google Scholar]
- Stables JP, Bertram E, Dudek FE, Holmes G, Mathern G, Pitkänen A, White HS. Therapy discovery for pharmacoresistant epilepsy and for disease-modifying therapeutics: summary of the NIH/NINDS/AES models II workshop. Epilepsia. 2003;44:1472–1478. doi: 10.1111/j.0013-9580.2003.32803.x. [DOI] [PubMed] [Google Scholar]
- Tauck DL, Nadler JV. Evidence of functional mossy fiber sprouting in hippocampal formation of kainic acid treated rats. J Neurosci. 1985;5:1016–1022. doi: 10.1523/JNEUROSCI.05-04-01016.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temkin N. Preventing and treating posttraumatic seizures: the human experience. Epilepsia. 2009;50(Suppl. 2):10–13. doi: 10.1111/j.1528-1167.2008.02005.x. [DOI] [PubMed] [Google Scholar]
- Turski WA, Cavalheiro EA, Schwarz M, Czuczwar SJ, Kleinrok Z, Turski L. Limbic seizures produced by pilocarpine in rats: behavioural, electroencephalographic and neuropathological study. Behav Brain Res. 1983;9:315–335. doi: 10.1016/0166-4328(83)90136-5. [DOI] [PubMed] [Google Scholar]
- Van Paesschen W, Duncan JS, Stevens JM, Connelly A. Etiology and early prognosis of newly diagnosed partial seizures in adults: a quantitative hippocampal MRI study. Neurology. 1997;49:753–757. doi: 10.1212/wnl.49.3.753. [DOI] [PubMed] [Google Scholar]
- van Vliet EA, da Costa Araújo S, Redeker S, van Schaik R, Aronica E, Gorter JA. Blood-brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain. 2007;130:521–534. doi: 10.1093/brain/awl318. [DOI] [PubMed] [Google Scholar]
- Walker MC, White HS, Sander JW. Disease modification in partial epilepsy. Brain. 2002;125:1937–1950. doi: 10.1093/brain/awf203. [DOI] [PubMed] [Google Scholar]
- Wasterlain CG, Mazarati AM, Shirasaka Y, Thompson KW, Penix L, Liu H, Katsumori H. Seizure-induced hippocampal damage and chronic epilepsy: a Hebbian theory of epileptogenesis. Adv Neurol. 1999;79:829–843. [PubMed] [Google Scholar]
- White A, Williams PA, Hellier JL, Clark S, Dudek FE, Staley KJ. EEG spike activity precedes epilepsy after kainate-induced status epilepticus. Epilepsia. 2010;51:371–383. doi: 10.1111/j.1528-1167.2009.02339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams P, White A, Ferraro D, Clark S, Staley K, Dudek FE. The use of radiotelemetry to evaluate electrographic seizures in rats with kainate-induced epilepsy. J Neurosci Methods. 2006;155:39–48. doi: 10.1016/j.jneumeth.2005.12.035. [DOI] [PubMed] [Google Scholar]
- Williams PA, White AM, Clark S, Ferraro DJ, Swiercz W, Staley KJ, Dudek FE. Development of spontaneous recurrent seizures after kainate-induced status epilepticus. J Neurosci. 2009;29:2103–2112. doi: 10.1523/JNEUROSCI.0980-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappone CA, Sloviter RS. Translamellar disinhibition in the rat hippocampal dentate gyrus after seizure-induced degeneration of vulnerable hilar neurons. J Neurosci. 2004;24:853–864. doi: 10.1523/JNEUROSCI.1619-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]