

Abstract
The interaction between human papillomavirus (HPV) and host cells is not well understood. We investigate the early stage of HPV infections by global expression profiling in a cell model, in which HaCaT cells were transfected with HPV11, HPV16 or HPV45 genomes. We report the differential expression of genes not previously implicated in HPV biology, such as the PSG family and ANKRD1, and of genes implicated in the biology of other viruses, e.g. MX1, IFI44 and DDX60. Carcinogenesis-related genes, e.g. ABL2, MGLL and CYR61, were upregulated by high-risk HPV16 and -45. The integrative analysis revealed the suppression of DNA repair by HPV11 and -16, and downregulation of cytoskeleton genes by all HPV types. Various signalling pathways were affected by the HPVs: IL-2 by HPV11; JAK-STAT by HPV16; and TGF-β, NOTCH and tyrosine kinase signalling by HPV45. This study uncovered novel strategies employed by HPV to establish infection and promote uncontrolled growth.
The early stages of human papillomavirus (HPV) infection and virus-host cell interaction are not well understood. The DNA genomes of HPVs encode six or seven early proteins E7, E6, E1, E2, E4 and E5 (HPV11 encodes two E5 genes) and two late structural genes L2 and L1 which assemble into the viral capsid1,2,3. The two major oncoproteins E6 and E7 have been extensively studied. They are multifunctional and are mainly involved in the deregulation of cell cycle control4. The E7 of malignant viruses inactivates the tumour-suppressor protein pRB and E6 degrades the tumour-suppressor protein p53 via interaction with E6AP. This leads to cell cycle progression from the G1 into the S-phase5. However, other aspects of HPV biology remain uncharacterised. For example, there is only fragmented knowledge concerning the function of the remaining early proteins. Additionally, since most research focuses on one protein at a time, little is known about how the viral proteins work together to establish and maintain viral infection.
Several studies have used global gene expression profiling to study the impact of HPV on the cellular transcriptome. Some of these studies used biopsy specimens infected with low- or high-risk HPVs6,7,8, while others used cell culture models bearing HPV genomes, e.g. W12 cell line with the HPV16 genome from natural infection9, keratinocytes transfected with HPV3110 or HPV11 episomes11 and keratinocytes with integrated or episomal HPV18 genomes12. In yet another approach, cell cultures have been transfected with HPV DNA encoding viral oncogenes. Gene expression analysis has been reported in cervical keratinocytes infected with retroviruses encoding HPV16 E6 and E713, HPV16 E7-expressing keratinocytes14 and keratinocytes infected with retroviruses encoding HPV18 E6 and E7 genes15.
In the present study, we transfected a human keratinocyte cell line, HaCaT, with the genomes of three HPV types: HPV11, HPV16 and HPV45. HPV11, which causes benign papillomas, is a prototype for non-malignant, low-risk virus. HPV16 is the most common high-risk virus, which is responsible for the majority of cervical and some head and neck cancers. HPV45 is the fifth most prevalent malignant virus type found in cervical lesions16 and is absent from the prophylactic vaccine implemented in Western countries. We conducted global expression profiling of the transfected cells and the control, followed by bioinformatics analysis integrating available knowledge of cellular pathways, protein-protein interactions and transcription factor binding sites. It is, to the best of our knowledge, the first direct comparison of global gene expression profiles of HPV11, -16 and -45 positive cells. Our study addresses the unexplored interactions between HPV and the host cell at early stages of infection. Secondly, we explore the contribution of the virus to uncontrolled growth and carcinogenesis. The expression profiles of 20,000 genes within our model are freely available online and constitute a valuable resourse for the HPV research community (ArrayExpress accession number: E-MTAB-1160).
Results
Cell culture growth is slowed by viral infection
The cell cultures were transfected with circular DNA genomes of HPV11, -16 or -45 and the control cultures were transfected with the plasmid encoding only the neomycin resistance. All three cell cultures transfected with HPV genomes grew more slowly than control-transfected cells (Figure 1). The reduction in absorbance within the first 24 hours in HPV positive cells indicates death of part of the cell population. This can be a result of virus killing the cells or cells switching on the apoptosis programme in response to viral infection. HPV11 slowed the growth of the cell culture to the greatest extent, while the HPV16 and -45 positive cultures grew at a similar rate. The presence of viral mRNA transcription was verified by qRT-PCR. The presence of the HPV genomes was confirmed by DNA PCR analysis ( Supplement 1).
Figure 1. HaCaT cells were transfected with full genomic DNA and grown under G418 selection for two weeks.
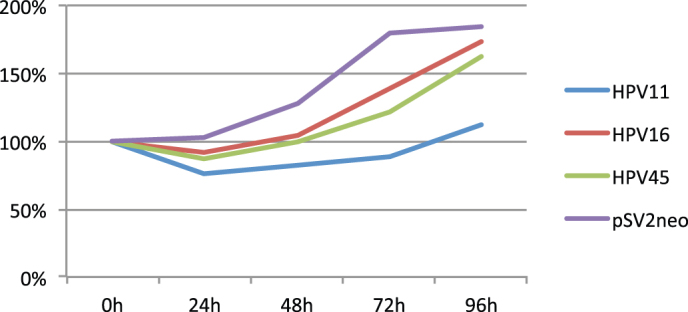
Cells were seeded for measurement of proliferation using the MTT assay,which was conducted without G418 selection. Cultures were harvested for proliferation measurement every 24 hours for a total of 96 hours. The cell cultures transfected with HPV genomes grew more slowly than the control (pSV2neo), and some cells died following the transfection.
Differential expression analysis
We profiled the global gene expression in the transfected cells and performed the analysis of differential expression between the cells transfected with each virus versus control. Throughout the article, we refer to the mRNA transcripts by their HUGO gene symbols. HPV11, -16 and -45 differentially expressed 391, 338 and 75 transcripts, respectively. The lower number of differentially expressed transcripts in HPV45 positive cells was due to failure of one array sample, which led to some loss of statistical power. Due to generally modest fold changes observed in differential expression (ranging from –4.1 to 4.5), we used a relatively stringent significance threshold (adjusted p-value <0.01). Tables of all the differentially expressed genes in cells transfected with HPV11, -16 or -45 genomes are available in Supplement 2. In order to improve the robustness of our results, we first focused on the genes that were differentially expressed by at least two HPV types and, in a further analysis, we integrated the prior knowledge of protein-protein interactions. This approach reduces the chances of false positives because the probability of observing a set of differentially expressed genes that interact at the protein level by chance only is considerably smaller.
We analysed the genes differentially expressed by more than one virus (Figure 2). Six members of the human pregnancy-specific glycoprotein (PSG) family (PSG3, 4, 5, 7, 8 and 9) were upregulated by all three HPV types. PSGs are known to be produced by placental syncytiotrophoblasts during pregnancy and have been shown to have an immune modulatory function and possibly proangiogenic effect17. ANKRD1 and IFIT2 were also found to be upregulated by all the studied HPVs. ANKRD1 is induced by IL-1 and TNF-α18, while IFIT2 is interferon-induced19. This suggests that these genes were upregulated by the host cell in response to the infection.
Figure 2.

Right panel: Venn diagram of differentially expressed genes (numbers of upregulated genes are shown in green and downregulated in red). Left panel: The heatmap of the expression of genes that were differentially expressed by at least two virus subtypes. The PSG family, ANKRD1 and IFIT2 were upregulated by all three HPV subtypes. ABL2, MGLL and CYR61, which have angiogenic and oncogenic potential, were upregulated by HPV16 and -45. IFI44 and DDX60 were upregulated by HPV11 and downregulated by HPV16 and -45. There were 16 genes that were significantly downregulated by at least two HPVs. These gene showed tendency to be downregulated across all 3 type, among them we find ANKRD11, AOAH, FOXN1 and the oncogene BCL11A. Lists of all the differentially expressed genes are available in Supplement 2. The upregulated genes are depicted in red, downregulated in blue, and not differentially expressed in white.
The group of genes upregulated upon HPV16 and -45 infection included ABL2 and MGLL, which can promote cancer cell migration, invasion and tumour growth by regulating the levels of fatty acids that serve as signalling molecules20. Other upregulated genes were: BNIP3, involved in protecting cells from virally-induced cell death; CTGF, a pro-inflammatory cytokine also upregulated by hepatitis C virus E2 protein21; and CYR61 (cysteine-rich, angiogenic inducer 61), a promoter of cell proliferation, chemotaxis and angiogenesis. This upregulation of proliferation, angiogenic and oncogenic genes by the high-risk HPV types suggests that these viruses may already exhibit their oncogenic potential during the early stages of infection.
Notably, IFI44 (interferon-induced protein 44) and DDX60 were upregulated by HPV11 and downregulated by HPV16 and -45 infections. IFI44 has previously been associated with hepatitis C virus infection22 and exhibits anti-proliferative activity23. DDX60 is a helicase which has recently been shown to have anti-viral function24,25.
The group of genes that was downregulated by at least 2 HPV types included: ANKRD11, which interacts with and enhances the activity of p53; AOAH, an immune response gene upregulated in response to swine fever virus26; and FOXN1, whose mutation in mice and rats causes hairlessness and a severely compromised immune system, and regulates keratin gene expression, which is consistent with the downregulation of a group of keratins in the HPV45 network (see the result section below on HPV45). Interestingly, the oncogene BCL11A was also downregulated by all the HPV types.
Integrative functional analysis
In order to ascertain the biological relevance of the differentially expressed mRNA transcripts, we integrated protein-protein interaction data to identify networks of differentially expressed genes that interact at the protein level. Throughout the article, we use the term ‘gene’, without referring specifically to mRNA or protein unless the distinction is necessary for clarification.
Assuming that functionally related genes will share an expression pattern within our model, we clustered all the genes that were differentially expressed by at least one virus type into six clusters. We then performed an enrichment analysis to ascertain the biological function of the genes within the clusters. We visualised the expression of differentially expressed genes across all three HPV types (Figure 3). Lists of genes for each of the six clusters are available in Supplement 3.
Figure 3. Heatmap showing the expression of differentially expressed genes upon transfection with HPV11, -16 or -45 genomes.

The genes were grouped into six clusters. Clusters 2, 5 and 6 were enriched with cell cycle and DNA repair genes from BRCA1 (clusters 2 and 5) and BRCA2 (cluster 6) networks27. Cluster 4 was enriched with JUN transcription factor target genes and cluster 3 was enriched with interferon response genes. The genes of each of the six clusters are listed in Supplement 3. The upregulated genes are depicted in red, downregulated in blue, and not differentially expressed in white.
HPV11
The protein-protein interaction (PPI) network of genes differentially expressed upon HPV11 infection (Figure 4A; later referred to as the HPV11 network) was dominated by downregulated genes involved in the (mitotic) cell cycle (26 genes), mostly centred around CENPA, PIN1, PLK1 and MCM2, -5, -6, -7 proteins. We also observed that the genes of clusters 2 and 5 (Figure 3), which were downregulated by HPV11, were enriched in cell cycle genes. This result is consistent with the observed growth curves demonstrating the slowest growth of HPV11 positive cells.
Figure 4. The networks represent the differentially expressed regions in the human protein-protein interaction (PPI) network upon infection with A) HPV11, B) HPV16 and C) HPV45.

The fold changes of differential expression are represented by colour of the nodes: upregulated – red, downregulated – green and not changed – white. The subcellular localisation is illustrated by shape of the nodes: ellipse – cytoplasm, triangle – extracellular, hexagon – nucleus, rectangle – plasma membrane and parallelogram – unknown.
The group of downregulated genes surrounding TP53 is involved in both the cell cycle and DNA damage repair. This group also connected the cell cycle group of genes to a group of genes involved in double-strand DNA repair, centred around BRCA1, RAD51 and FA family genes, FANCA, FANCE and FANCG, all of which were downregulated. In concordance with this result, clusters 2 and 5 (downregulated by HPV11) were enriched with 21 and 52 genes, respectively, from the BRCA1 signature. The signature of genes correlated with BRCA1 has been reported by others27. Additionally, cluster 2 contained five genes induced by BRCA1 (WELCSH_BRCA1_TARGETS_1_UP). Interestingly, PARP1, which plays a role in the repair of single-stranded DNA, was downregulated and was connected to CENPA in a cell cycle sub-network. The lowered expression of BRCA1 and genes interacting with it or correlated with its expression suggests a reduction of the activity of DNA damage mediated by BRCA1 and related proteins. BRCA1 regulates transcription of POLD1 and CHAF1A, present in the network. POLD1, CHAF1A and other genes surrounding PCNA were all downregulated and their biological functions include DNA replication (several DNA polymerases), but also DNA repair.
Some of the upregulated genes present in the network encode proteins involved in the cellular response to HPV infection, including MX1 (an interferon-induced anti-viral protein), TLR4 (an activator of innate immunity) and pro-inflammatory PTGS2. Interestingly, MX1 interacts with FA family proteins and IFI44 (STRING 9.0). Other upregulated groups included extracellular proteins IGFBP3, CP and TFPI2, which interacted with plasminogen (PLG). Their function in HPV infection is elusive.
HPV16
The PPI network of genes affected by HPV16 infection was enriched with metabolic genes, most of them being downregulated (Figure 4B). Forty-six genes of the network are involved in biopolymer metabolic processes, 25 of which are involved in transcription/RNA metabolism. Within the network, the RNA metabolism genes were placed around RB1, SP1 and NCOR1, but also around JUN and SMAD2. SMAD2 was also connected to cell cycle genes STAG1 and STAG2. There was also a group of downregulated cell cycle genes surrounding YWHAG. Supporting results come from the cluster analysis, where cluster 6, downregulated by HPV16 (Figure 3), was enriched with genes involved in the mitotic cell cycle. This downregulation of metabolic and cell cycle genes suggests the slowing down of growth and proliferation processes. Thus, this result is consistent with the growth curves (Figure 1). The role of JUN in the network is unclear, being surrounded by nine genes involved in RNA metabolism, three of which were upregulated. JUN itself was upregulated, and we observed the upregulation of its targets in cluster 4.
Secondly, we observed a group of 11 genes associated with signalling pathways for cytokines and growth factors. The genes were centred around PIK3R1, RASA1 and JAK2. Seven of these genes are involved in JAK-STAT signalling. The downregulation of these genes may represent the reduction in growth signalling, which is consistent with the observed downregulation of metabolic and cell cycle genes. However, the presence of IL7 and IL7R, which also stimulate the B- and T-cell response, may also have implications for the immune response. Only one gene in the sub-network, GRAP, was upregulated.
Genes grouped around APC, NIN, VIM, KIAA1377 and ATRX are related to the cytoskeleton and vimentin (VIM) was connected to the actin regulators ROCK1 and ROCK2, which are involved in the formation of stress fibres28. Cluster 6 was enriched with genes involved in cytoskeleton structures.
Additionally, we observed a group of downregulated genes responsible for DNA repair. The genes were centred around NBN, ATR, BRCA1, BRCA2 and RAD50. Further, cluster 6 (downregulated by HPV16) included 21 genes correlated with CHEK2 and 14 genes correlated with BRCA2.
Notably, several members of the PSG family, PSG3, 9 and 5, interacted with genes throughout the network.
HPV45
In the PPI network of genes differentially expressed upon HPV45 infection (Figure 4C), GRB2, which plays a role in signalling, was linked to two groups of surface proteins and to downstream signalling pathways. The first group consisted of membrane proteins EPHB2, EFNB1, SDCBP and MAGI3, and extracellular TGFA. EPHB2 was also connected to the upregulated oncogene ABL2 (tyrosine kinase). The second group included genes interacting with PLG, i.e. matrix remodelling proteins TFPI2 and MMP3 and cell surface glycoprotein F3, and genes interacting with PRNP, i.e. ADAM23, PVRL1 and PVRL4. Of note, the same interaction NEU3-GRB2-PRNP-PLG-TFPI2 was also observed in the HPV11 network. The genes interacting with PLG were upregulated, while the genes interacting with PRNP were downregulated by both viruses.
GRB2 was linked to a group of downregulated genes centred around members of the keratin family (KRT5, KRT6A and KRT14). Seven out of the nine proteins in this group are part of the cytoskeleton and four of these cytoskeletal genes are specifically involved in epidermis development. Additionally, four out of the nine genes (TRADD, TNFRSF1A (TNF receptor), PKP1 and DSP) are involved in apoptosis. GRB2, SHC3, AKT3, ADCY7 and PRKCD are part of the TRKA signalling from the plasma membrane.
The second part of the network was enriched with genes from the TGF-β pathway, which were placed around the INHBA-FST-SMAD9-SMAD3-CREBBP signalling chain. SMAD9 was linked to a group of eight extracellular/secreted proteins, seven of which were upregulated. SMAD9 interacted with SMAD3 which in turn was connected to a group of downregulated genes centred around CREBBP. Additionally, SMAD3 was linked to four downregulated members of the NOTCH pathway and three upregulated genes: JUN, SQSTM1 and IL1F7. JUN has binding sites on the promoters of four upregulated genes in the network: DDIT3 and HDAC9 in the SMAD3/CREBBP sub-network and F3 and TFPI2 in the PLG sub-network. Additionally, cluster 4, which was upregulated by HPV45, was enriched with genes having JUN binding sites in their promoter regions. The interaction chain SQSTM1-SMAD2/3-JUN-DDIT3 was shared between the high-risk types HPV16 and HPV45.
PRKCD, linked between CREBBP and PTK2B-GRB2, was also directly linked to plasma membrane proteins and the upregulated oncogene AKT3.
Discussion
The experimental model used here allows us to study and directly compare the differential expression of mRNAs mediated by different HPV types on the same cellular background. The model represents the early stages of viral infection and fills the gap between studies based on virus-induced cancers in human specimens and studies focused on single viral oncogenes like E6 and E7. Since only less than one percent of high-risk HPV infections leads to cancer, studies based on persistently infected tissues and infected cancers are biased towards the situations and mechanisms when the virus is not cleared by the immune system. We believe that a better understanding of the early stages of HPV infection can aid the development of treatment to clear out the infection in cases where it is not cleared spontaneously before it develops into malignancy. In contrast to studies which focus on single viral proteins like E6 or E7, our model represents the combined effect of all the early proteins working together in the infected cell, thus providing the possibility to observe the full effect of HPV genomes on host cells. It is especially useful for viral activities that rely on more than one viral protein. However, it should be kept in mind that the HaCaT cells used in the model are spontaneously immortalized skin derived cells, which harbour a mutated P53 and P16 inactivation. This may have an impact on the gene expression changes introduced by the HPV genome. We did not determine if the HPV genome remained in the episomal state after the transfection or if it was integrated in host's chromosomes during selection. In our opinion, it is unlikely that the HPV genomes would integrate into the host genome after only 3 weeks of growth as also demonstrated for HPV31 in a similar transfection protocol29.We have previously used this experimental model to study the impact of HPV11, -16 and -45 on cellular microRNA expression30,31.
Contrary to our expectations, transfection of host cells with the three studied HPV genomes has a negative impact on their growth. In concordance with this finding, the cluster analysis and PPI network show the downregulation of cell cycle and metabolic genes by HPV11 and -16, achieved by different sets of genes, e.g. genes of RNA metabolism are downregulated by HPV16 and genes of DNA metabolism are downregulated by HPV11. The downregulation of cell cycle genes is most profound in response to HPV11, which also induces the slowest cell culture growth. Notably, direct downregulation of cell cycle genes by HPV45 is not observed. We conclude that early HPV infection disrupts normal cellular processes, which hinders growth.
BRCA1, BRCA2 and CHEK2 are responsible for DNA repair, cell cycle arrest and apoptosis. The downregulation of BRCA1, genes correlated to BRCA1 expression as well as genes interacting with BRCA1 at the protein level, such as FANCA and RAD51, is mediated by HPV11. Transfection with HPV16 results in the downregulation of genes correlated to the expression of CHEK2 and BRCA2. Additionally, BRCA2, BRCA1, RAD50 and surrounding genes are also downregulated in the HPV16 PPI network. Different DNA repair genes are affected by HPV11 and -16. This suggests that HPVs can target different genes to achieve the same goal of lowering the capability of the host cell to repair its DNA.
The lowered expression of genes involved in DNA repair suggests a reduction of the activity of DNA damage repair and consequently a susceptibility to accumulation of mutations during repeated mitosis/DNA replication cycles. We propose that the increased activity of the DNA repair system in late stages of HPV infection and during HPV-driven carcinogenesis reported in previous studies6 is a secondary effect of substantial DNA damage present in the host cells and not induced by HPV. Contrarily, we suggest that HPV impairs the DNA damage detection and repair system early in infection, which allows the accumulation of mutations that can be beneficial for the initiation of a persistent viral infection and carcinogenesis process. The HPV16 E6 protein has also been found to interfere with single-strand repair by binding to XRCC1, leading the authors to conclude that the virus contributes to genomic instability32
It takes several months for the immune system to clear an HPV infection. Apart from avoiding immune surveillance by infecting only the basal layer of the epithelium and low expression of viral proteins, HPVs actively suppress the immune response (reviewed by33). It is not clear which factors lead to persistent infection that is considered a prerequisite for progression into cancer.
Our results from the PPI networks show the host cell response to infection both as an upregulation of anti-viral genes and a downregulation of genes involved in the immune response, which we deem to be part of the HPV strategy to avoid destruction by the immune system. In HPV11 positive cells, the pro-inflammatory PTGS2 and MX1 (which shows activity against influenza virus and VSV rhabdovirus and Hepatitis B virus34) are upregulated. However, DAXX, a protein which interacts with MX1, and TP53 which interacts with PTGS2 are downregulated. A similar pattern is seen for TLR4 (toll-like receptor 4). TLR4 is upregulated, yet SYK, GRB2 and other signalling genes functioning downstream in the signalling cascade are downregulated. We suggest that HPV may counteract the cellular response by downregulation of genes interacting with activated anti-viral proteins. Both MX1 and TLR4 have recently been shown to be involved in HPV infection35,36. However, MX1 was reported to be downregulated by high-risk HPVs35; MX1 expression is not changed by HPV16 and -45 in our experimental model. Notably, in HPV16 positive cells, a group of genes from the JAK-STAT signalling pathway is downregulated, including IL7, IL7R and JAK2. This finding is supported by a previous report showing that JAK2 is impaired by the E6 oncoprotein of HPV1837. Our results point to downregulation of JAK-STAT signalling pathway genes as the viral action to hinder the interferon-driven immune response. Another upregulated gene likely to be involved in the anti-viral response is SQSTM1 (EBI3-associated protein of 60 kDa where EBI3 represents Epstein-Barr virus induced 3)38. In the HPV45 network, IL1F7, which suppresses the immune response, is upregulated. This is likely caused by the virus. IL1F7 requires SMAD3 for its function, to which it is connected in the network and SMAD3 is further connected to SQSTM1 and JUN.
Interestingly, the PSG family (PSG3, 4, 5, 7, 8 and 9) is upregulated by all three HPV types. We suggest that HPV uses the immune modulatory function of the PSG family to suppress the activity of the immune system. However, PSGs have also been suggested to be possible receptors for mouse hepatitis virus39,40 and HIV41. The exact function of the PSGs in HPV infection remains unknown. Our study identified two further genes upregulated by all three HPV types, namely ANKRD1 and IFIT2. These are likely to be activated by the host cell in response to infection. In high-risk HPV16 and -45, BNIP3 and CTGF, which are known for their anti-viral activity21, are also upregulated.
IFI44 and DDX60 are very interesting because they are upregulated by low-risk HPV11 and downregulated by high-risk HPV16 and -45 infections. Both genes have anti-viral activity reported in other viruses22,24,25. IFI44 is induced by IFN-α and leads to microtubule aggregates in hepatitis C virus infected cells and overexpression inhibits cell proliferation22,23. DDX60 is a helicase and an integral part of the exosome. It is important for RNA stability25. The mechanism of action of these genes in HPV infection is currently unknown, but if elucidated, could lead to a better understanding of the differences between low- and high-risk viruses.
In epithelial cells, the cytoskeleton fibres maintain the cell structure. During viral infections, the actin fibres and microtubules are involved in both the uptake and release of virus particles42, whereas the function of the cytokeratins is less well understood. However, cytokeratins are important for the differentiation of epithelial cells and the fibres are composed of pairs of keratins defining the grade of differentiation. Keratins 5 and 14 are characteristic of the fibres in the proliferative basal layer, where they play a role in proliferation43. Interestingly, these keratins together with keratin 6A are downregulated in HPV45 positive cells, indicating a transition of the HPV positive cells into a more simple epithelial cell44. In HPV16 positive cells, there is no differential regulation of cytokeratins; however, a cluster of genes linking to vimentin (VIM) includes the actin regulators ROCK1 and ROCK2 involved in the formation of stress fibres28. There are no differentially regulated cytoskeleton genes in the HPV11 PPI network. However, the pseudogene ROCK1P1 is the most downregulated gene in HPV11 positive cells. The downregulation of cytokeratins 6A, 10 and 13 (all from the stratified epithelium) has also been described in high-grade cervical lesions (HSIL)45, thus validating the cell model system. Interestingly, FOXN1 induces keratin gene expression46 and is downregulated by all three HPV types.
Our results imply that HPVs affect cellular signalling pathways by changing the expression of genes involved in signalling. Several signalling pathways are represented in the HPV45 PPI network. First, NOTCH pathway genes are downregulated. The downregulation of NOTCH1 expression has been reported in cervical carcinoma cells and is thought to be important in the late stages of HPV-induced carcinogenesis47. We propose that the NOTCH pathway may already be modulated in the early stages of HPV infection.
Secondly, in the HPV45 network, TGF-α is upregulated and several genes of the TGF-β/SMAD signalling pathway are present. Interestingly, the genes of the extracellular part of TGF-β are upregulated, whereas the nuclear genes are downregulated. The TGF-β/SMAD signalling pathway is involved in many cellular processes; however, the molecular changes in the TGF-β/SMAD pathway in HPV infection are unclear. TGF-α is involved in mitogenesis and angiogenesis and is upregulated in several cancers48.
An interesting chain of interaction proteins, TFPI2-PLG-PRNP-GRB2-NEU3, is present in the PPI networks of both low-risk HPV11 and high-risk HPV45. The limited information on these genes and their involvement in HPV biology makes it difficult to deduce their function. The presence of upregulated extracellular PLG, TFPI2 and MMP3, which are involved in matrix remodelling, may lead to changes on the cell surface and of the extracellular matrix, and possibly more invasive growth. The presence of a GRB2-interacting sub-network, which is involved in TLR4 and tyrosine kinase receptor signalling, points to the possible implication of all five proteins in the immune response signalling. Interestingly, GRAP, a GRB2-related adaptor protein, is upregulated in the HPV16 PPI network and interacts with downregulated genes of the JAK-STAT and PIK3R1 signalling pathway.
The aim of the study was to look for the early changes in HPV genome-bearing cells and not at HPV-driven carcinogenesis. However, we have identified some expression changes which we believe imprint the cells for future progression to malignant growth. For example, in response to high-risk HPV16 and -45, we observe increased activity of known oncogenes: JUN, upregulation of oncogene ABL2, cancer-promoting MGLL, angiogenic CYR61 and histone deacetylase HDAC9 (HPV45 only). Additionally, the impact of HPV45 infection on signalling pathways such as NOTCH, TGF-α and TGF-β could be implicated in cancer development. Finally, the observed downregulation of DNA repair genes could lead to the accumulation of mutations and contribute to the acquisition of other hallmarks of cancer.
We believe that our experimental model provides an interesting and useful link between profiling studies based on the oncogenes E7 and/or E6 and tissue studies. Furthermore, the data obtained in the present study open up new avenues of research where genes not previously identified as involved in HPV infections, e.g. IFI44 and DDX60 can be studied in depth. We believe that an understanding of the early stages of HPV infection could lead to the development of treatment strategies that would aid the clearing of persistent HPV infections. For future studies, establishment of a similar model in primary kerationocytes would provide further validation of the results and could overcome any limitations introduced by using HaCaT cells in our model.
Methods
Transfection of cells and RNA extraction
HaCaT cells were cultured and transfected as previously described by Dreher et al.30. We conducted the transfection in three replicates for each virus type and control; however, one sample for HPV45 failed during microarray profiling.
The transfected cells were grown under G418 sulphate (Invitrogen) selection for three weeks. Total RNA was extracted in TriZOL reagent according to the protocol of the manufacturer (Invitrogen).
MTT growth assay
We performed the measurement of cell proliferation with the Cell Proliferation Kit 1 (MTT) (Roche A/S, Hvidovre, Denmark) according to the manufacturer's protocol. HaCaT cells were transfected with HPV11, -16 or -45 full genomes and after a two-week selection cells were plated in five 96-well plates (10,000 cells per well). The G418 selection was not used during the MTT assay. At time points 0 h, 24 h, 48 h, 72 h and 96 h, 10 µL MTT was added to each well of one plate and incubated at 37°C for four hours. Thereafter, 100µL solubilising buffer was added to each well and the plate incubated overnight. Absorbance was measured at 570 nm and 690 nm for reference using a Synergy HT Multi-Mode Microplate Reader (Bio-Tek, Winooski, USA).
qRT-PCR validation
RNA batches made from HPV11 transfected cells were validated for E7 transcription and RNA from HPV16 batches for E7and E6 transcripts by qRT-PCR. The primers were: 11-E7 fwd: 5′- nt.533 gctggaagacttgttaccc nt.551-3′ and 11-E7rev: 5′- nt.727 tcggacgttgctgtcacatcc nt.707-3′ 16-E7 fwd: 5′- nt.755 ttcggttgtgcgtacaaagc nt.774-3′ and 16-E7rev: 5′- nt.821 agtgtgcccattaacaggtcttc nt.799-3′ 16-E6 fwd: 5′- nt.215 ctgcgacgtgaggtatatgacttt nt.238-3′ and 16-E6 rev: 5′- nt.292 acatacagcatatggattcccatct nt.268-3′. The qRT-PCR reaction was performed according to standard procedures with the following hybridisation temperatures: for 11-E7 55°C, for 16-E6 and 16-E7 56°C. The PCR reaction was continued for 40 cycles.
RNA labelling and hybridisation to microarray
Total RNA (100 ng) was amplified and labelled using the Ambion WT Expression Kit (Applied Biosystems) according to the manufacturer's instructions. The labelled samples were hybridised to the Human Gene 1.0 ST GeneChip Array (Affymetrix, Santa Clara, CA, USA). Arrays were washed and stained with phycoerythrin-conjugated streptavidin (SAPE) using an Affymetrix Fluidics Station® 450 and subsequently scanned in an Affymetrix GeneArray® 2500 Scanner to generate fluorescent images according to the Affymetrix GeneChip® protocol. Cell intensity files (CEL files) were generated using Affymetrix GeneChip® Command Console® (AGCC) software.
Pre-processing of CEL files
The CEL files containing the array raw data were imported to the R environment and pre-processed using the OLIGO package from Bioconductor49. The expression data were normalised using the RMA (robust multi-array averaging) method and their probes were summarised by ‘core’ genes, resulting in log2 expression values for ~20,000 genes. A detailed description of all the analyses performed in R, as well as the source code to reproduce the results can be found in Supplement 4. The raw and pre-processed array are available online at ArrayExpress, accession number: E-MTAB-1160.
Differential expression
The analysis of differential expression was performed by means of the Limma package50, which uses moderated t-statistics. Samples transfected by each virus type were compared to controls transfected with empty vectors. The empirical Bayes approach employed in Limma ‘borrows’ information about variance across samples and results in stable inference when the number of arrays is small50. Therefore, we were able to perform the analysis of differential expression for HPV45 based on two samples. We did, however, observe some loss of statistical power, which resulted in a smaller number of differentially expressed genes for HPV45 compared to HPV11 and -16 (see Supplement 4 for the exact procedure and R code). The p-values were adjusted for multiple testing by the Benjamini-Hochberg correction method. The significance thresholds for differential expression were set to a) absolute log2 fold change above 0.6 (fold change >~1.5) and b) adjusted p-value <0.01.
Cluster analysis
All the genes that were differentially expressed in at least one virus type were clustered based on their expression profiles. d = 1 – r was used as the distance measure, where r is the Pearson correlation coefficient. The genes were clustered into six groups by PAM, a robust k-means-like clustering method. Each cluster was investigated for overlap with signatures from the Molecular Signatures Database v3.0, which includes the KEGG, GO, BioCarta and Reactome terms, as well as curated signatures of chemical and genetic perturbations. The significance of overlap/enrichment was calculated using the hypergeometric test implemented on the website (http://www.broadinstitute.org/gsea/msigdb/).
Integrated network analysis
The networks of differentially expressed networks of genes interacting at the protein-protein level were computed by means of BioNet51. This method identifies the differentially expressed functional module by integrating the p-values derived from the differential expression analysis and the human PPI network, as described in detail by52. The heuristic approach was used to calculate an approximation to the optimal scoring sub-network. The significance thresholds were HPV11, FDR = 0.001; HPV16, FDR = 0.002; HPV45, FDR = 0.02. The networks were exported to and visualised by Cytoscape53. The fold changes of differential expression were used to colour the nodes of the network; red was used for upregulated genes and green for downregulated genes. The function of the parts of the network was inferred using the enrichment/overlap analysis and literature search.
Author Contributions
BK, MR and BN designed the project. MR, DA, AD, MM and MV performed the wet-lab experiment. BK performed the statistical/bioinformatics analysis and has been instrumental in the biological interpretation of the data. BK prepared figures 2–4 and supplementary materials 2–4. BK, MR and BN wrote the manuscript with ideas and contributions from OW and FN. OW and FN critically revised the manuscript. BN supervised the project. All authors approved the final version of the manuscript.
Supplementary Material
Supplement 1 and 4
Supplement 2
Supplement 3
Acknowledgments
We wish to thank Susanne Smed for technical assistance, Tawny Abaniel for help with proofreading the manuscript and Nicolas Rapin for help with the preparation of the figures. Bogumil Kaczkowski was supported by the Novo Nordisk Foundation. Technician Melissa A. Visser was supported by a grant from the Læge Sophus Carl Emil Friis and hustru Olga Friis' Foundation. Ditte Andersen and Anita Dreher were supported by a student grant from the Danish Cancer Society. The work was funded by the following foundations: Beckett-Fonden, Fabrikant Einar Willumsens, Family Hede Nielsens, ML Jørgensen and Gunnar Hansen, and Lykfeldts.
References
- Hausen, zur H. Immortalization of human cells and their malignant conversion by high risk human papillomavirus genotypes. Semin. Cancer Biol. 9, 405–411 (1999). [DOI] [PubMed] [Google Scholar]
- Chow L. T., Broker T. R. & Steinberg B. M. The natural history of human papillomavirus infections of the mucosal epithelia. APMIS 118, 422–449 (2010). [DOI] [PubMed] [Google Scholar]
- Doorbar J. et al. Detection of novel splicing patterns in a HPV16-containing keratinocyte cell line. Virology 178, 254–262 (1990). [DOI] [PubMed] [Google Scholar]
- Antinore M. J., Birrer M. J., Patel D., Nader L. & McCance D. J. The human papillomavirus type 16 E7 gene product interacts with and trans-activates the AP1 family of transcription factors. EMBO J. 15, 1950–1960 (1996). [PMC free article] [PubMed] [Google Scholar]
- Ghittoni R. et al. The biological properties of E6 and E7 oncoproteins from human papillomaviruses. Virus Genes 40, 1–13 (2010). [DOI] [PubMed] [Google Scholar]
- Santegoets L. A. M. et al. Different DNA damage and cell cycle checkpoint control in low- and high-risk human papillomavirus infections of the vulva. Int. J. Cancer (2011).doi:10.1002/ijc.26345 [DOI] [PubMed] [Google Scholar]
- Santin A., Zhan F., Bignotti E., Siegel E. & Cané S. Gene expression profiles of primary HPV16- and HPV18-infected early stage cervical cancers and normal cervical epithelium: identification of novel candidate molecular markers for cervical cancer diagnosis and therapy. Virology (2005). [DOI] [PubMed] [Google Scholar]
- DeVoti J. A. et al. Immune dysregulation and tumor-associated gene changes in recurrent respiratory papillomatosis: a paired microarray analysis. Mol. Med. 14, 608–617 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alazawi W. et al. Changes in cervical keratinocyte gene expression associated with integration of human papillomavirus 16. Cancer Res. 62, 6959–6965 (2002). [PubMed] [Google Scholar]
- Chang Y. E. & Laimins L. A. Interferon-inducible genes are major targets of human papillomavirus type 31: insights from microarray analysis. Dis. Markers 17, 139–142 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas J. T., Oh S. T., Terhune S. S. & Laimins L. A. Cellular changes induced by low-risk human papillomavirus type 11 in keratinocytes that stably maintain viral episomes. Journal of Virology 75, 7564–7571 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karstensen B., Poppelreuther S., Bonin M. & Walter M. Gene expression profiles reveal an upregulation of E2F and downregulation of interferon targets by HPV18 but no changes between keratinocytes with integrated or …. Virology (2006). [DOI] [PubMed] [Google Scholar]
- Nees M. et al. Papillomavirus type 16 oncogenes downregulate expression of interferon-responsive genes and upregulate proliferation-associated and NF-kappaB-responsive genes in cervical keratinocytes. Journal of Virology 75, 4283–4296 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccardo E. et al. Expression of human papillomavirus type 16 E7 oncoprotein alters keratinocytes expression profile in response to tumor necrosis factor. Carcinogenesis 31, 521–531 (2010). [DOI] [PubMed] [Google Scholar]
- Garner-Hamrick P. A. et al. Global effects of human papillomavirus type 18 E6/E7 in an organotypic keratinocyte culture system. Journal of Virology 78, 9041–9050 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan P., Jones R. H. & Li N. Human papillomavirus (HPV) types in 115,789 HPV-positive women: A meta-analysis from cervical infection to cancer - Guan - International Journal of Cancer - Wiley Online Library. … Journal of Cancer (2012). [DOI] [PubMed] [Google Scholar]
- Lisboa F. A. et al. Pregnancy-specific glycoprotein 1 induces endothelial tubulogenesis through interaction with cell surface proteoglycans. J. Biol. Chem. 286, 7577–7586 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu W., Burns D. K., Swerlick R. A. & Presky D. H. Identification and characterization of a novel cytokine-inducible nuclear protein from human endothelial cells. J. Biol. Chem. 270, 10236–10245 (1995). [DOI] [PubMed] [Google Scholar]
- Wathelet M. G., Clauss I. M., Content J. & Huez G. A. Regulation of two interferon-inducible human genes by interferon, poly(rI).poly(rC) and viruses. Eur. J. Biochem. 174, 323–329 (1988). [DOI] [PubMed] [Google Scholar]
- Nomura D. K. et al. Monoacylglycerol lipase exerts dual control over endocannabinoid and fatty acid pathways to support prostate cancer. Chem. Biol. 18, 846–856 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming-Ju H., Yih-Shou H., Tzy-Yen C. & Hui-Ling C. Hepatitis C virus E2 protein induce reactive oxygen species (ROS)-related fibrogenesis in the HSC-T6 hepatic stellate cell line. J. Cell. Biochem. 112, 233–243 (2011). [DOI] [PubMed] [Google Scholar]
- Kitamura A., Takahashi K., Okajima A. & Kitamura N. Induction of the human gene for p44, a hepatitis-C-associated microtubular aggregate protein, by interferon-alpha/beta. Eur. J. Biochem. 224, 877–883 (1994). [DOI] [PubMed] [Google Scholar]
- Hallen L. C. et al. Antiproliferative activity of the human IFN-alpha-inducible protein IFI44. J. Interferon Cytokine Res. 27, 675–680 (2007). [DOI] [PubMed] [Google Scholar]
- Schoggins J. W. et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita M., Oshiumi H., Matsumoto M. & Seya T. DDX60, a DEXD/H box helicase, is a novel antiviral factor promoting RIG-I-like receptor-mediated signaling. Mol. Cell. Biol. 31, 3802–3819 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladue D. P. et al. Patterns of gene expression in swine macrophages infected with classical swine fever virus detected by microarray. Virus Res. 151, 10–18 (2010). [DOI] [PubMed] [Google Scholar]
- Pujana M. A. et al. Network modeling links breast cancer susceptibility and centrosome dysfunction. Nat. Genet. 39, 1338–1349 (2007). [DOI] [PubMed] [Google Scholar]
- Katoh K., Kano Y. & Noda Y. Rho-associated kinase-dependent contraction of stress fibres and the organization of focal adhesions. J R Soc Interface 8, 305–311 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Côté-Martin A. et al. Human papillomavirus E1 helicase interacts with the WD repeat protein p80 to promote maintenance of the viral genome in keratinocytes. Journal of Virology 82, 1271–1283 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreher A. et al. Differential expression of cellular microRNAs in HPV 11, -16, and -45 transfected cells. Biochemical and Biophysical Research Communications 412, 20–25 (2011). [DOI] [PubMed] [Google Scholar]
- Dreher A., Rossing M., Kaczkowski B., Nielsen F. C. & Norrild B. Differential expression of cellular microRNAs in HPV-11 transfected cells. An analysis by three different array platforms and qRT-PCR. Biochemical and Biophysical Research Communications 403, 357–362 (2010). [DOI] [PubMed] [Google Scholar]
- Iftner T. et al. Interference of papillomavirus E6 protein with single-strand break repair by interaction with XRCC1. EMBO J. 21, 4741–4748 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley M. A. Immune responses to human papilloma viruses. Indian J. Med. Res. 130, 266–276 (2009). [PubMed] [Google Scholar]
- Haller O., Staeheli P. & Kochs G. Interferon-induced Mx proteins in antiviral host defense. Biochimie 89, 812–818 (2007). [DOI] [PubMed] [Google Scholar]
- Reiser J. et al. High-risk human papillomaviruses repress constitutive kappa interferon transcription via E6 to prevent pathogen recognition receptor and antiviral-gene expression. Journal of Virology 85, 11372–11380 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daud I. I. et al. Association between toll-like receptor expression and human papillomavirus type 16 persistence. Int. J. Cancer 128, 879–886 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S. et al. The human papilloma virus (HPV)-18 E6 oncoprotein physically associates with Tyk2 and impairs Jak-STAT activation by interferon-alpha. Oncogene 18, 5727–5737 (1999). [DOI] [PubMed] [Google Scholar]
- Devergne O. et al. A novel interleukin-12 p40-related protein induced by latent Epstein-Barr virus infection in B lymphocytes. Journal of Virology 70, 1143–1153 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanga C., Cardellichio C. & Holmes K. Expression of the recombinant anchorless N-terminal domain of mouse hepatitis virus (MHV) receptor makes hamster of human cells susceptible to MHV infection. Journal of … (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D. S. et al. A pregnancy-specific glycoprotein is expressed in the brain and serves as a receptor for mouse hepatitis virus. Proc. Natl. Acad. Sci. U.S.A. 92, 12095–12099 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blinov V., Resenchuk S. & Chirikova G. Possible role of pregnancy-specific glycoprotein (PSG) in mother-child HIV infection transfer (… Conference On Aids: 1994). [Google Scholar]
- Taylor M. P., Koyuncu O. O. & Enquist L. W. Subversion of the actin cytoskeleton during viral infection. Nat. Rev. Microbiol. 9, 427–439 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam H. H., Sehgal L. L., Kundu S. T. S., Dalal S. N. S. & Vaidya M. M. M. Novel function of keratins 5 and 14 in proliferation and differentiation of stratified epithelial cells. CORD Conference Proceedings 22, 4068–4078 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper D. & Schermer A. Biology of disease. Laboratory investigation (1985). [Google Scholar]
- Arnouk H. et al. Characterization of Molecular Markers Indicative of Cervical Cancer Progression. Proteomics Clin Appl 3, 516–527 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes S. M., Ofstad T. A., Campbell D. H., Watt F. M. & Prowse D. M. Transient activation of FOXN1 in keratinocytes induces a transcriptional programme that promotes terminal differentiation: contrasting roles of FOXN1 and Akt. J. Cell. Sci. 117, 4157–4168 (2004). [DOI] [PubMed] [Google Scholar]
- Talora C., Sgroi D. & Crum C. Specific down-modulation of Notch1 signaling in cervical cancer cells is required for sustained HPV-E6/E7 expression and late steps of malignant transformation. Genes & development (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hose D. et al. Induction of angiogenesis by normal and malignant plasma cells. Blood 114, 128–143 (2009). [DOI] [PubMed] [Google Scholar]
- Carvalho B. S. & Irizarry R. A. A framework for oligonucleotide microarray preprocessing. Bioinformatics 26, 2363–2367 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth G. K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3, Article3 (2004). [DOI] [PubMed] [Google Scholar]
- Beisser D., Klau G. W., Dandekar T., Muller T. & Dittrich M. T. BioNet: an R-Package for the functional analysis of biological networks. Bioinformatics 26, 1129–1130 (2010). [DOI] [PubMed] [Google Scholar]
- Dittrich M. T., Klau G. W., Rosenwald A., Dandekar T. & Muller T. Identifying functional modules in protein-protein interaction networks: an integrated exact approach. Bioinformatics 24, i223–i231 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline M. S. et al. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc 2, 2366–2382 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement 1 and 4
Supplement 2
Supplement 3