

Abstract
M1 muscarinic acetylcholine receptors (mAChRs) represent a viable target for treatment of multiple disorders of the central nervous system (CNS) including Alzheimer's disease and schizophrenia. The recent discovery of highly selective allosteric agonists of M1 receptors has provided a major breakthrough in developing a viable approach for the discovery of novel therapeutic agents that target these receptors. Here we describe the characterization of two novel M1 allosteric agonists, VU0357017 and VU0364572, that display profound differences in their efficacy in activating M1 coupling to different signaling pathways including Ca2+ and β-arrestin responses. Interestingly, the ability of these agents to differentially activate coupling of M1 to specific signaling pathways leads to selective actions on some but not all M1-mediated responses in brain circuits. These novel M1 allosteric agonists induced robust electrophysiological effects in rat hippocampal slices, but showed lower efficacy in striatum and no measureable effects on M1-mediated responses in medial prefrontal cortical pyramidal cells in mice. Consistent with these actions, both M1 agonists enhanced acquisition of hippocampal-dependent cognitive function but did not reverse amphetamine-induced hyperlocomotion in rats. Together, these data reveal that M1 allosteric agonists can differentially regulate coupling of M1 to different signaling pathways, and this can dramatically alter the actions of these compounds on specific brain circuits important for learning and memory and psychosis.
Introduction
Selective activators of the M1 subtype of muscarinic acetylcholine receptor (mAChR) may provide an exciting new approach for treatment of schizophrenia and Alzheimer's disease (AD). M1 receptors play roles in multiple domains of cognitive function (Felder et al., 2000; Auld et al., 2002), and activation of M1 receptors has cognition-enhancing effects in a range of animal models (Shirey et al., 2009; Lebois et al., 2010). In addition, the M1/M4-preferring agonist xanomeline (Shannon et al., 2000; Perry et al., 2001; Jones et al., 2005) and the M1-selective agonist TBPB (Jones et al., 2008) have robust efficacy in rodent models used to predict antipsychotic efficacy. Consistent with this, clinical studies reveal that acetylcholinesterase (AChE) inhibitors or xanomeline can enhance cognitive function and have antipsychotic efficacy in patients (Bodick et al., 1997b; Shekhar et al., 2008; Massoud and Gauthier, 2010). These exciting findings provide a strong impetus for discovery of M1 receptor ligands as a novel treatment strategy for patients suffering from these CNS disorders (Lebois et al., 2010).
A major breakthrough in the development of selective activators of M1 came with the discovery of allosteric agonists. Multiple M1 allosteric or bitopic agonists have been discovered and are providing an exciting new approach for developing M1-selective compounds (Conn et al., 2009). We recently reported the discovery of two highly selective allosteric agonists VU0357017 and VU0364572 (Lebois et al., 2010, 2011). These compounds were optimized to achieve an excellent pharmacokinetic profile and VU0357017 had robust efficacy in improving hippocampal-dependent learning in rats (Lebois et al., 2010).
Interestingly, recent studies suggest that some M1 allosteric agonists may selectively activate M1 coupling to some but not all signaling pathways in cellular systems (Thomas et al., 2009). This could have critical implications for further development of M1 allosteric agonists as potential therapeutic agents. Multiple signaling pathways including induction of calcium release, β-arrestin recruitment, and extracellular regulated signal kinase (ERK) are activated following M1 stimulation to elicit a broad range of physiological effects, and each of these is likely to participate in the overall effects of M1 agonists in vivo. Thus, if novel M1 agonists differentially activate some but not all M1-mediated responses, this could dramatically alter the in vivo and potential therapeutic response to these agents.
We now report that VU0357017 and VU0364572 display profound differences in their efficacy in activating M1 coupling to calcium, β-arrestin, and ERK pathways. Using a cell line with inducible M1 expression, we found that the actions of these M1 allosteric agonists on calcium release and β-arrestin recruitment are dependent on receptor expression levels and suggest a role for differences in receptor reserve in regulating both of these responses. Interestingly, these differential effects on different responses to M1 activation lead to a dramatic effect on the ability to activate M1-mediated responses in different brain circuits. For example, M1 allosteric agonists have robust effects on M1-mediated electrophysiological responses in the hippocampus but have lower efficacy in activating M1 in the striatum of rats and are without measureable effects on established M1-mediated responses in mouse medial prefrontal cortex (mPFC) pyramidal cells. Consistent with these actions, both M1 agonists enhanced hippocampal-dependent cognitive function but did not reduce amphetamine-induced hyperlocomotion in rats. These data suggest that different M1-selective allosteric agonists can have fundamentally different effects on different M1-mediated responses in the CNS and this can fundamentally alter the in vivo and potentially therapeutic effects of different M1 agonists.
Materials and Methods
Compounds.
Muscarinic agonist carbachol (CCh) and acetylcholine (ACh) were purchased from Sigma-Aldrich. Chemical synthesis of VU0364572 and VU0357017 was performed at the Vanderbilt Center for Neuroscience Drug Discovery (Vanderbilt University, Nashville, TN).
Cell lines.
Initial agonist characterization was performed in stable CHO cell lines constitutively expressing human M1 receptors. To generate a tetracycline (tet)-inducible human M1 mAChR stable cell line, TREx Chinese hamster ovary (CHO) cells expressing a tet repressor (Invitrogen) were transfected with human M1-pcDNA5/TO expression plasmid, and subsequently went under hygromycin (450 μg/ml) selection in the presence of blasticidin (10 μg/ml) for maintaining tet repressors. The resulting hygromycin-resistant polyclones were further plated into a 96-well plate for monoclonal selection. Individual monoclones were screened for ACh-stimulated calcium mobilization across a range of tet concentrations. The TREx CHO cell line was cultured in medium containing 10% tet-tested fetal bovine serum (FBS) (Atlanta Biologicals) and 10 mm HEPES.
Calcium mobilization assays.
For all calcium assays, hM1-TREx CHO or hM1-CHO cells were seeded at a density of 50,000 cells per well in clear-bottomed, black-walled Costar 96-well plates (Corning) in media containing a range of tet concentrations. The following day, media was removed from the cells and replaced with 50 μl of calcium indicator dye, fluo-4 (2 μm), dissolved in HBSS (Invitrogen) containing 20 mm HEPES and 2.5 mm probenecid, pH 7.4. Cells were allowed to incubate in the fluo-4/HBSS solution for 45 min; solution was removed and replaced with 50 μl HBSS. Agonists were serial diluted into assay buffer for a 2× stock concentration in 1% dimethylsulfoxide (DMSO); stock compounds were added to assay for final concentration of 0.5% DMSO. Fifty microliters of agonist test solution was added to each well and fluorescent signals were measured at λ525 nm fluorescence emission after λ480 nm excitation at 1 s intervals for 60 s using either a Flexstation II or a Flexstation III (Molecular Devices). To generate concentration response curves (CRCs), baseline responses were subtracted from agonist-induced response and were normalized to the maximal response elicited by CCh.
ERK1/2 phosphorylation assays.
ERK1/2 phosphorylation was measured using SureFire Alpha Screen technology (PerkinElmer). Cells were plated at a density of 40,000 cells per well and were incubated in FBS-free media for 5 h before the assay. Cells were treated with agonist for 5 min and were lysed with 1× lysis buffer. phospho-ERK signals were acquired in 384-well plates (Corning #3705) using an Inspire plate reader (PerkinElmer).
β-arrestin recruitment assays.
β-arrestin recruitment was measured using PathHunter Express hM1 CHO cells (DiscoverX). Cells were plated in 96-well plates. The following day, cells were treated with drug and incubated at 37°C for 90 min. Substrate was added to each well and luminescence values were obtained using a BioTek Synergy2 luminometer.
Extracellular field potential recordings.
Young adult (4–6 week) male Sprague Dawley rats were obtained from Charles River. Rats were anesthetized with isoflurane, and the brains were removed and submerged in ice-cold cutting solution (Ayala et al., 2009; McCutchen et al., 2006) that was continuously bubbled with 95% O2/5% CO2. Transverse slices (400 μm) were made using a vibratome (Leica VT100S) and hippocampi were microdissected and transferred to a room temperature mixture containing equal volumes of cutting solution and artificial CSF (ACSF) for 30 min, followed by ACSF for 60 min (LTP; Ayala et al., 2009) or ACSF alone for 60 min (LTD; McCutchen et al., 2006). Recordings were performed using a submersion chamber with room temperature ACSF for LTD and 30−32°C ACSF for LTD. Stimulation was elicited using a bipolar-stimulating electrode placed in the stratum radiatum near the CA3–CA1 border. Recording electrodes were pulled with a Flaming/Brown micropipette puller (Sutter Instruments) and placed in the stratum radiatum of CA1. Field potential recordings were acquired using a MultiClamp 700B amplifier (Molecular Devices) and pClamp 9.2 software (Molecular Devices). Threshold and saturation LTP protocols were performed as described previously (Ayala et al., 2009) and LTD was performed as described previously (McCutchen et al., 2006). M1 compounds were diluted in ACSF and were bath applied for 10 min.
Whole-cell recordings.
In brief, 20- to 27-d-old C57BL/6Hsd mice (Harlan) were anesthetized with isoflurane. Brains were removed and placed in ice-cold modified oxygenated ACSF. Coronal brain slices (290–300 μm) containing the mPFC or striatum were cut using a vibratome, recovered at 32°C for 20 min, and transferred to a submersion chamber perfused with oxygenated ASCF at 30°C. Spontaneous EPSCs (sEPSCs) were recorded from layer V pyramidal neurons in voltage-clamp mode using a Warner 501A amplifier (Warner Instruments). Current-clamp recordings were performed on striatal medium spiny neurons (MSNs) using a MultiClamp 700B amplifier. Recording electrodes were prepared from borosilicate glass pipettes and had resistance of 2–4 MΩ. Electrical signals were low-pass-filtered at 2 kHz, digitized at 10 kHz, and acquired using a Clampex9.2/DigiData 1332 system.
Water maze testing.
Male Sprague Dawley rats (225–250 g) were tested on Morris water maze (MWM) assays to test spatial reference memory. A plastic tub (188 cm diameter) was filled with water (18°C) and made opaque using nontoxic paint. A hidden platform (10 cm wide) was positioned in the northeast quadrant. Rats were injected intraperitoneally with drug or vehicle 30 min before the first trial of the day. All treatment groups consisted of eight rats that were placed at a different starting point and given 60 s to locate the hidden platform. Rats were then placed into a heated cage until the next trial. The intertrial interval was ∼10 min. Swim path distance (centimeters) was used to assess performance via a video tracking system (Ethovision 3.1; Noldus Instruments). Spatial reference memory between testing days was determined by measuring swim distance from the last trial of day 4 and compared with the first trial on the following day (day 5). After all test trials on day 5, a 60 s probe trial (platform removed) was conducted to evaluate whether rats localized the platform to the spatial location.
Acquisition of contextual fear conditioning.
Contextual fear conditioning (CFC) studies were conducted in conditioning chambers housed in a sound-attenuating cubicle (Med Associates). Male Sprague Dawley rats (225–250 g) were pretreated with M1 agonist (0–1 mg/kg, i.p.) or vehicle 30 min before conditioning. Following a 2 min habituation period, one footshock (1 s, 0.5 mA) was delivered through the stainless steel grid floor, and after 45 s rats were returned to their homecage. After ∼24 h, animals were exposed to the same chambers for assessment of freezing behavior. All trials were video recorded (VID-CAM-MONO-2A; Med Associates) and scored blinded.
GPCRProfiler assay.
GPCRProfiler assay buffer was HBSS supplemented with 20 mm HEPES and 2.5 mm probenecid (Sigma P8761). Probenecid was prepared by placing 710 mg into a 15 ml conical vial. Five milliliters of 1 N NaOH was then added and the mixture was vortexed until dissolved. An additional 5 ml of HBSS/20 mm HEPES was then added. Ten milliliters of probenecid solution was then mixed into 1 L of HBSS 20 mm HEPES, pH 7.4, with NaOH. Cells were seeded (from cultures that were <90% confluent) at 12,500 cells per well of a 384-well plate. Plates were then incubated at room temperatures for time lengths that varied depending upon the specific cell line and then incubated at 37°C for 24 h before assay. Assays were performed using Fluo-8 No Wash Ca2+ dye. A 2× dye concentration was prepared in GPCRProfiler assay buffer. Cells were washed with GPCRProfiler assay buffer and 20 μl of buffer was retained in the plate. Twenty microliters per well of 2× dye was added and the plates were incubated at 30°C, 5% CO2 for 90 min. Compound plates were prepared to add 20 μl per well during the first addition and compounds were prepared at 3× the final concentration (in this case, 10 μl with a final volume after compound addition of 60 μl). To assess allosteric modulation, a second additional plate was prepared using a 4× stock of reference agonist which was diluted in fourfold dilutions into an eight point concentration series. Twenty microliters was then added to the plate for a final volume of 80 μl. Plates were read on a FLIPR kinetic plate reader. Sixty reads at 1 s per read were performed, followed by 60 reads at 2 s per read. Data were analyzed as follows: the response of the compound (VU0357017) alone was normalized to the maximal response of the relevant agonist to identify potential agonist activity of VU0357017. To assess modulator effects of VU0357017, complete agonist concentration responses were performed in the presence and absence of 10 μm ACh.
[3H]NMS saturation binding assays.
Saturation binding studies were performed to determine the Bmax values of membranes prepared from human M1-TREx CHO cells treated across a range of tet concentrations. Binding studies were performed by incubating membranes isolated from cells with a range of [3H]NMS (GE Heathcare) concentrations dissolved in radioligand binding buffer containing 100 mm NaCl, 20 mm HEPES, and 10 mm MgCl2, pH 7.4. Five micrograms of protein were used per well and serial dilutions of test compounds were added to 96-well deep-well plates using a final assay volume of 0.5 ml. Nonspecific binding was determined in the presence of 10 μm atropine. Binding reactions were performed at room temperature for 3 h and reactions were terminated by rapid filtration through GF/B filter plates. Following termination, plates were washed three times with ice-cold harvesting buffer using a 96-well Brandel harvester. Plates were allowed to dry overnight and radioactivity was determined using a TopCount NXT microplate scintillation and luminescence counter (PerkinElmer Life and Analytical Sciences).
Data analysis.
sEPSCs were quantified and analyzed using Minianalysis software (Synaptosoft). Statistical analysis between control and drug-induced effects was performed using student's paired or unpaired t test or one-way ANOVA followed post hoc by Tukey's multiple comparison or Dunnett's test. Prism (GraphPad Software) was used to generate bar graphs and CRCs. Cumulative probability plots were made using Origin (v6, OriginLab). LTP and LTD sampled data were analyzed using Clampfit 9.2 and analyzed using a one-way ANOVA and a post hoc Tukey's multiple comparison. For enhancement of the CF acquisition, percentage freezing data were analyzed using an AVOVA and a post hoc Dunnett's test was used to compare all dose groups to the vehicle treated group.
Results
VU0364572 and VU0357017 have robust effects on M1-activation of calcium mobilization and ERK1/2 phosphorylation but have little effect on β-arrestin recruitment
We previously reported that VU0364572 and VU0357017 are highly selective agonists of M1 relative to other mAChR subtypes (Lebois et al., 2010, 2011). To further assess selectivity of VU0357017, we took advantage of the GPCRProfiler service offered by Millipore to determine the effect of this compound on Ca2+ responses of 168 G-protein-coupled receptors (GPCRs), including 32 Family A nonpeptide GPCRs and 14 Family B and C GPCRs in a functional screening paradigm (Fig. 1a,b). VU0357017 showed clean ancillary pharmacology with very small responses at monoamine receptors including D4 dopamine receptors and β3-adrenergic receptors. These data are consistent with our previous findings suggesting that VU0364572 is also highly selective for M1 when it was profiled against 68 GPCRs, ion channels, and transporters. VU0364572 had little effect on D3 receptors and inhibited only 10% of binding and required a concentration of >10 μm to induce effects (Lebois et al., 2011).
Figure 1.

Selectivity profile of VU0357017 among families. A–C, GPCRs. a, Selectivity profile of VU0357017 when tested as an agonist against multiple Family A GPCRs using GPCRProfiler calcium assay; 10 μm VU0357017 was applied to cells expressing various GPCRs. VU0357017 induced a significant response in cells expressing M1 muscarinic receptors but had little activity at other receptor subtypes. A subsequent addition of a full agonist CRC (ACh) for each receptor allowed measurement of potential PAM or antagonist activity (data not shown). Interestingly, VU0357017 did show potentiation of responses to ACh at D4 and at β3 adrenergic receptors. b, Selectivity profile of VU0357017 when tested as an agonist against multiple Family B and C GPCRs. 5-HT, serotonin; M, muscarinic; A, adenosine; Alpha, α-adrenergic; Beta, β-adrenergic; CB, cannabinoid; D, dopamine; H, histamine; P2Y, purinergic; CGRP, calcitonin gene-related peptide; CCK, cholecystokinin; GIP, glucose-dependent insulinotropic peptide; GLP2, glucagon-like peptide receptor; PAC1, pituitary adenylate cyclase-activating polypeptide type I receptor; PTH, parathyroid hormone; VPAC, vasoactive intestinal peptide; CaS, calcium sensing; GABAB, γ amino butyric acid, mGlu, metabotropic glutamate receptor.
We next performed studies to evaluate the effects of VU0364572 and VU0357017 (Fig. 2a) on M1 coupling to multiple signaling pathways, including Ca2+ mobilization, ERK1/2 phosphorylation, and β-arrestin recruitment in CHO cells stably expressing M1. For comparison, we used CCh as a prototypical orthosteric agonist. CCh is a close analog of ACh and is a full agonist but is resistant to AChE activity; therefore, it is useful for both cell line and brain slice studies. Consistent with our previous reports, CCh and both allosteric agonists induced concentration-dependent increases in Ca2+ mobilization in hM1 CHO cells (Fig. 2b). The overall efficacies of VU0364572 (% CChmax 70.8 ± 6.43) and VU0357017 (% CChmax 41.7 ± 2.37) were comparable to values reported in previous studies (Lebois et al., 2011). We also performed saturation binding assays to determine receptor density values (Fig. 2e).
Figure 2.

VU0364572 and VU0357017 induce calcium release and ERK phosphorylation but are without effects on β-arrestin recruitment. a, Chemical structures of the two M1 agonists VU0357017 and VU0364572. b, CRCs of receptor-induced calcium release for CCh (filled circles), VU0364572 (open circles), and VU0357017 (crosses) in CHO-K1 cells stably expressing human M1 mAChRs. Data are normalized to the CCh maximum response. Data points represent mean ± SEM of four independent experiments performed in duplicate or triplicate. c, CRCs of agonist-induced ERK1/2 phosphorylation (pERK1/2) assessed using the SureFire ERK phosphorylation assay in hM1 CHO cells. Data are expressed as fold change over basal ERK levels and is normalized to the maximum response elicited by CCh. Data represent the mean ± SEM of 7–8 independent experiments performed in duplicate or triplicate. d, CRCs of agonist-induced β-arrestin recruitment in hM1 CHO cells using PathHunter detection kit. Data points represent mean ± SEM of three independent experiments performed in duplicate or triplicate and are normalized to % CCh max. e, Saturation isotherms of [3H]-NMS binding to membranes prepared from hM1 CHO cells. Receptor density values (1479 ± 129 fmol/mg protein) were obtained from three independent experiments. f, Representative saturation isotherms of [3H]-NMS binding to membranes prepared from hM1 CHO cells used in β-arrestin recruitment assays. Receptor density values (11701.600 ± 1411.21 fmol/mg protein) were obtained from five independent experiments.
We next determined the effects of each agonist on M1-mediated activation of ERK1/2 phosphorylation and β-arrestin recruitment, two well established responses of M1 activation. CCh and VU0364572 induced robust increases in ERK1/2 phosphorylation in the same cellular background that was used for studies of calcium mobilization (Fig. 2c). VU0357017 induced a modest ERK1/2 phosphorylation response and also exhibited reduced potency relative to its effect on calcium mobilization (Fig. 2c; for a complete list of EC50 values and EMAX values see Table 1).
Table 1.
CCh, VU0364572, and VU0357017 induce Ca release, ERK phosphorylation, and β-arrestin responses in hM1 CHO cells
| CCh |
VU0364572 |
VU0357017 |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| EC 50 (in μm) | % CCh max | n | EC 50 (in μm) | % CCh max | n | EC 50 (in μm) | % CCh max | n | |
| Ca release | 0.280 (0.016) | 99.6 (0.10) | 4 | 0.890 (0.070) | 70.8 (0.430) | 4 | 1.60 (0.180) | 41.7 (2.37) | 4 |
| ERK | 1.60 (0.90) | 99.9 (12.6) | 8 | 10.0 (7.10) | 90.5 (13.3) | 7 | 37.0 (20.0) | 37.9 (8.80) | 7 |
| β-arrestin | 62.0 (26.7) | 100 (0.0) | 3 | NA | NA | 3 | NA | NA | 3 |
EC50 values are represented in μm and Emax values are normalized to the percentage maximum CCh response. VU0364572 and VU0357017 induce responses in ERK and Ca assays but are without effects in arrestin recruitment assays. Values in parentheses are SEM.
CCh also induced robust increases in β-arrestin recruitment in hM1CHO PathHunter cells (DiscoverX). In contrast, VU0364572 and VU0357017 had little or no effect on β-arrestin recruitment (Fig. 2d, Table 1). Saturation binding experiments confirmed that the cell line used in this assay has robust expression of hM1 receptors (Fig. 2f). Together these data confirm previous reports that VU0364572 and VU0357017 act as M1 agonists in activating calcium mobilization. They also reveal that these agents have agonist activity when measuring M1-mediated increases in ERK1/2 phosphorylation but not for inducing β-arrestin recruitment.
The finding that VU0364572 and VU0357017 have little or no effect on β-arrestin recruitment may suggest that these allosteric agonists display stimulus bias for activation of M1 coupling to phospholipase C (PLC), calcium mobilization, and ERK relative to β-arrestin recruitment. In addition, the finding that VU0357017 has partial agonist activity in activating both calcium mobilization and ERK1/2 phosphorylation suggests that this compound, and possibly VU0364572, may be a partial agonist and may have differential effects on signaling in systems with lower receptor expression and less receptor reserve. If VU0364572 and VU0357017 are weak partial agonists in inducing calcium mobilization and ERK phosphorylation, responses to these allosteric agonists should be diminished in cells expressing lower levels of M1. To test this hypothesis, we created a cell line system using a tet-inducible (TREx) human M1 receptor expression system. This allowed us to systematically vary the levels of M1 expression and measure each response to M1 activation in a single cellular background with different levels of receptor reserve. As shown in Figure 3a,b, treatment of TREx hM1 cells with tetracycline-induced concentration-dependent increases in M1 expression and caused progressive leftward shifts in the CCh CRC for activation of calcium mobilization. The potency values of CCh with 0 ng/ml tet (EC50 = 2.4 ± 0.7 μm) or 25 ng/ml tet (EC50 = 1.9 ± 0.6 μm) are consistent with an estimated CCh affinity at M1 (which is in the low micromolar range) (Fisher and Snider, 1987), suggesting there is no receptor reserve in these conditions. In contrast, incubation with 50 ng/ml or 1 μg/ml tet induced leftward shifts in the CCh CRC, suggesting the presence of receptor reserve for the calcium mobilization response (EC50 = 0.06 ± 0.01 μm for 1 μg/ml tet).
Figure 3.
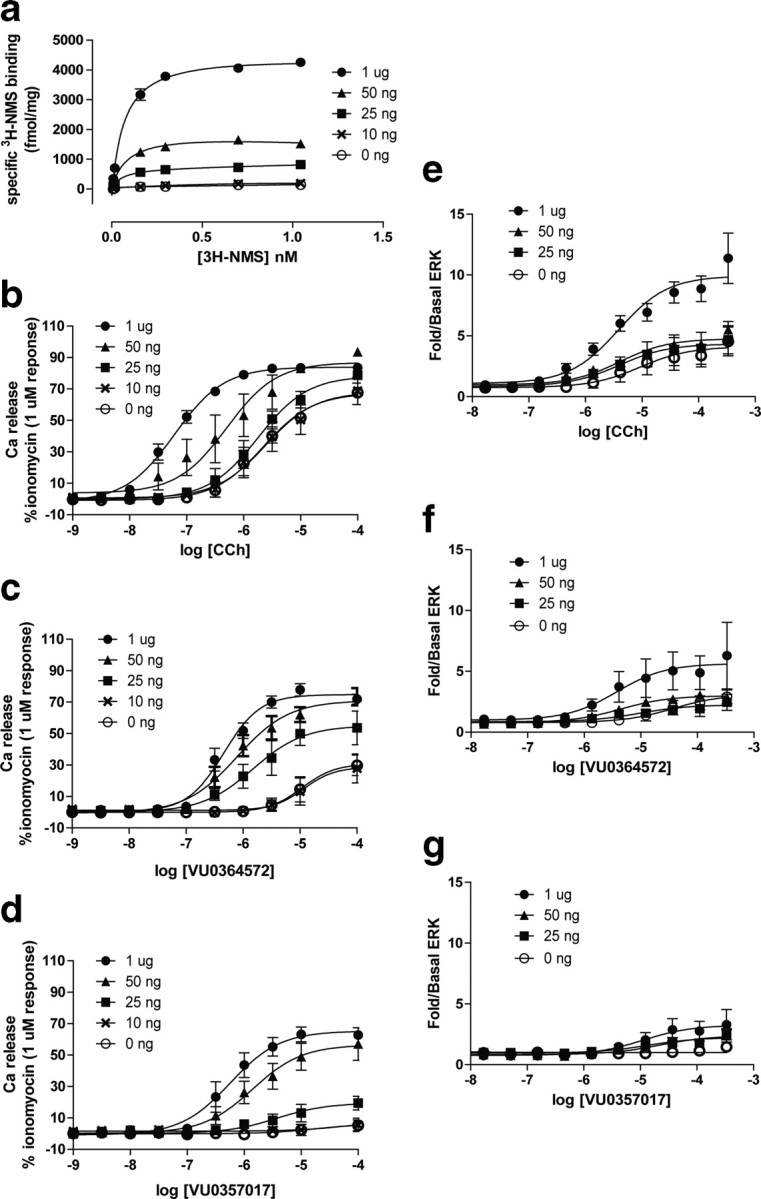
VU0364572 and VU0357017 induce responses in a cell line with variable receptor expression. a, Saturation isotherms of [3H] NMS binding to membranes prepared from hM1 TREx CHO cells treated with varying concentrations of tet. Membranes were prepared 24 h after treatment with tet and specific binding values increased following treatment indicating increased receptor density (in fmol/mg protein; 0 ng/ml tet = 268.5 ± 88.0, 10 ng/ml tet = 188.33 ± 37.0, 25 ng/ml tet = 666.9 ± 130, 50 ng/ml tet = 2277.2 ± 619.6, 1 μg/ml tet = 4435.6 ± 1431.1, n = 3). b–d, CRCs of calcium release for CCh, VU0364572, and VU0357017 in hM1 TREx CHO cells that were treated overnight with 1 μg/ml (closed circles), 50 ng/ml (closed triangles), 25 ng/ml (closed squares), 10 ng/ml (crosses), or 0 ng/ml tet (open circles). Data points represent mean ± SEM of three independent experiments performed in duplicate or triplicate. Data are normalized to % ionomycin (1 μm). e–g, M1-induced ERK phosphorylation measured in TREx CHO cells treated across a range of tet concentrations. An increase in CCh's maximal response was present in cells treated with 1 μg/ml tet. There was little effect on the EC50 values. Induction of M1 expression with 50 ng/ml or 1 μg/ml tet had a small effect on the maximal response to VU0364572, but had little effect on the maximal response to VU0357017. Data points represent the mean ± SEM of two or three independent experiments performed in duplicate or triplicate. Data are expressed as fold over basal ERK response.
As shown in Figure 3c and d, VU0364572 and VU0357017 both behaved as partial agonists in inducing calcium mobilization responses in the TREx hM1 cell line. Both allosteric agonists induced weak concentration-dependent increases in calcium mobilization in cells treated with 10 ng/ml tet. Further induction of M1 expression with 25 ng/ml or 1 μg/ml tet increased maximal responses to both allosteric agonists. Table 2 presents EC50 values and maximal responses seen under each condition. Based on this weak partial agonist activity, these agonists may induce robust physiological responses that depend on activation of PLC and calcium mobilization in CNS systems that express high levels of M1 but may have weaker effects on responses in neuronal populations that lack M1 receptor reserve for this signaling pathway.
Table 2.
VU0364572 and VU0357017 induce Ca responses in hM1 TREx CHO cells treated across a range of TET concentrations
| TET concentration | CCh |
VU0364572 |
VU0357017 |
|||
|---|---|---|---|---|---|---|
| EC 50 (in μm) | % CCh max | EC 50 (in μm) | % CCh max | EC 50 (in μm) | % CCh max | |
| 0 ng/ml (n =3) | 2.46 (0.72) | 100 (3.45) | 30.5 (12.5) | 44.2 (9.58) | NA | 8.24 (5.06) |
| 10 ng/ml (n =3) | 2.94 (0.82) | 100 (12.8) | 146 (120) | 39.9 (13.5) | 16 (11) | 7.21 (6.24) |
| 25 ng/ml (n =3) | 1.99 (0.69) | 100 (6.40) | 2.3 (1.7) | 68.1 (13.6) | 16 (14) | 24.5 (5.53) |
| 50 ng/ml (n =3) | 1.08 (0.84) | 100 (2.93) | 1.0 (0.4) | 78.1 (5.95) | 1.6 (0.57) | 60.4 (11.1) |
| 1 μg/ml (n =3) | 0.06 (0.01) | 100 (1.29) | 0.5 (0.1) | 86.2 (3.75) | 0.69 (0.24) | 75.3 (1.07) |
CCh induces a near maximum response in untreated cells. A decrease in the potency values was present in cells treated with 50 or 1 μg of TET suggesting the presence of receptor reserves. In cells treated 25, 50, or 1 μg TET, VU0364572 and VU0357017 induces an increase in Emax values. Both compounds were found to be more potent following treatment of cells across a range of TET concentrations. Values in parentheses are SEM.
CCh-induced increases in ERK 1/2 phosphorylation were also dependent on levels of M1 expression. CCh induced a relatively weak increase in ERK1/2 phosphorylation in M1 TREx cells treated with 25 ng/ml tet with a slight increase in the maximal response with 50 ng/ml tet and a larger increase in the maximal response after M1 induction with 1 μg/ml tet (Fig. 3e). However, the potency of CCh did not change with different levels of M1 expression, suggesting that there is little or no receptor reserve for the ERK1/2 response in this cell line even with strong induction of M1 expression. VU0357017 and VU0364572 were weak agonists in the TREx hM1 cell line treated with 25 ng/ml tet. Interestingly, induction of M1 expression with 50 ng/ml or 1 μg/ml tet had little effect on the maximal responses to VU0357017 and did not alter the EC50 values (Fig. 3f,g; for EC50 values see Table 3).
Table 3.
VU0364572 and VU0357017 induce ERK responses in hM1 TREx CHO cells treated across a range of TET concentrations
| TET concentration | CCh |
VU0364572 |
VU0357017 |
|||
|---|---|---|---|---|---|---|
| EC 50 (in μm) | % CCh max | EC 50 (in μm) | % CCh max | EC 50 (in μm) | % CCh max | |
| 0 ng/ml (n = 3) | 14.9 (7.39) | 96.3 (3.79) | 108 (63.3) | 56.4 (17.8) | NA | 27.4 (9.96) |
| 25 ng/ml (n = 3) | 1.73 (0.97) | 80.3 (8.87) | 9.88 (1.40) | 50.6 (10.3) | NA | 50.3 (2.29) |
| 50 ng/ml (n = 3) | 4.36 (2.16) | 91.8 (2.93) | 9.44 (4.93) | 52.6 (9.31) | 35.9 (0.00) | 45.9 (12.5) |
| 1 μg/ml (n = 3) | 4.12 (2.07) | 93.3 (2.88) | 3.30 (1.50) | 49.3 (8.04) | 3.17 (0.00) | 30.3 (13.7) |
CCh induced large responses in cells treated with 1 μg of TET but had little effect on potency values across all TET concentrations. VU0364572 induced an increase in Emax values in cells treated with higher concentrations of TET, but little effect on potency was detected. VU0357017 induced a small response in cells treated with 1 μg TET. Values in parentheses are SEM.
Unfortunately, it is not possible to use the PathHunter system for quantitative measures and CRCs of agonist-induced β-arrestin recruitment in the TREx-inducible cell line. Thus, we used confocal imaging analysis to qualitatively assess β-arrestin2-YFP recruitment in the hM1 TREx cells in which M1 was induced with 50 ng/ml tet. CCh induced a robust β-arrestin2 response in hM1 TREx CHO cells treated with 50 ng/ml tet (Fig. 4). In contrast, VU0357017 and VU0364572 had no effect on β-arrestin recruitment in these cells. VU0364572 did induce responses after induction of M1 expression with 1 μg/ml tet. Together, these data suggest that VU0357017 and VU0364572 are partial agonists with robust effects on some M1-mediated responses in systems where M1 expression is high and VU0364572 is the stronger of the two agonists across multiple assays. However, their actions depend on the signaling pathways engaged and on differences in receptor expression. Previous reports indicate that varying degrees of receptor reserve for multiple M1-mediated responses in the CNS (Conn et al., 2009). Thus, we next determined the effects of both compounds across multiple responses in brain slices and in behaving animals.
Figure 4.

CCh and M1 compound VU0364572 induce β-arrestin recruitment in TREx CHO cells. a, Preagonist and postagonist confocal scans of hM1 TREx CHO cells expressing β-arrestin2-YFP. Treatment of cells with CCh (100 μm) induces β-arrestin recruitment (black arrows, bottom) in cells that were exposed overnight to tet (50 ng/ml). Treatment of cells with VU0364572 (100 μm) induces β-arrestin2 recruitment (black arrows, bottom) in cells that were exposed overnight to tet (1 μg/ml). VU0357017 (100 μm) did not induce β-arrestin2 recruitment in cells treated overnight with 50 ng/ml or 1 μg/ml tet. b, Quantification of the effects of each agonist on the number of puncta. A one-way ANOVA revealed that puncta in CCh-treated cells (50 ng/ml tet) differed significantly when compared with VU0357017 (1 μg/ml tet) and VU0364572 (1 μg/ml tet)-treated cells (F(3,26) = 4.82, *p < 0.001). Neither CCh nor M1 agonist induced arrestin recruitment in cells that were not treated with tet (data not shown).
VU0364572 and VU0357017 enhance synaptic plasticity in the hippocampus
Activation of M1 has a broad range of physiological effects in multiple brain regions that are critical for the overall in vivo actions of M1 agonists. Given the above responses to VU0364572 and VU0357017 in cell lines, we postulated that these compounds may have differential effects in activating M1-mediated responses in different brain regions. To test this hypothesis we determined the effects of CCh, VU0364572, and VU0357017 on clearly established M1-mediated responses in three brain regions that are thought to be important for in vivo and therapeutic effects of M1 agonists. One of the most well established effects of M1 activation is in area CA1 of the hippocampus, a brain region where M1 has been shown to be the predominant receptor subtype expressed (∼60%) (Flynn et al., 1995). M1 activation potentiates NMDA receptor currents in this brain region (Marino et al., 1998), enhances LTP of excitatory synaptic transmission (Anagnostaras et al., 2003; Buchanan et al., 2010), and induces LTD, another form of hippocampal synaptic plasticity (Scheiderer et al., 2006; Volk et al., 2007). Each of these actions is thought to play a critical role in the cognitive enhancing effects of M1 activation (Anagnostaras et al., 2003; Ma et al., 2009). We previously reported that both VU0364572 and VU0357017 have similar effects to CCh in potentiating NMDA receptor currents in hippocampal pyramidal cells (Lebois et al., 2010, 2011). We now evaluated the effects of VU0364572 and VU0357017 on hippocampal LTP by determining the effects of each compound on potentiation of threshold theta-burst-stimulated (TBS) LTP at the Schaffer collateral-CA1 synapse in rat hippocampus (Ayala et al., 2009). This response is thought to be mediated exclusively by M1 receptors and is absent in mice where the M1 receptor is genetically deleted (Anagnostaras et al., 2003). Dendritic field potential recordings revealed that threshold TBS induced a slight potentiation of field EPSPs (fEPSPs) (Fig. 5a,c, 17.1 ± 5.2% over basal at 55 min postdrug). Saturated LTP was used as a positive control showing that robust LTP is induced after a 4× TBS (Fig. 5a,c, 45.3 ± 4.4% over basal). Interestingly, when slices were incubated with either VU0364572 or VU0357017, the threshold TBS protocol induced a robust LTP response that was significantly potentiated relative to vehicle control (Fig. 5b,c, one-way ANOVA; F(3,26) = 4.82, p < 0.01) and was similar to the saturated LTP response. Thus, both allosteric agonists potentiate threshold TBS LTP in a manner similar to what has been observed with other M1 agonists (Buchanan et al., 2010).
Figure 5.

M1 agonists VU0364572 and VU0357017 significantly enhance threshold theta-burst LTP and VU0364572 induces LTD at the Schaffer collateral-CA1 synapse of rodent hippocampal slices. Insets for each figure are representative fEPSP traces measured at baseline (black) or 50 (LTD) or 55 (LTP) minutes after compound washout (gray). Scale bars: x-axis, 2 ms; y-axis, 0.6 mV. a, The standard TBS protocol (TBS saturation) induces significant LTP (n = 9), whereas the threshold TBS protocol induces only a slight potentiation of fEPSP slope (n = 8) at the SC-CA1 synapse. b, Bath application of 500 nm VU0364572 (n = 5) or VU0357017 (n = 5 of 9 experiments) for 10 min before threshold TBS induced a significant potentiation of fEPSP slope. c, Significant differences (*p < 0.01) were observed in the mean percentage potentiation induced by threshold TBS compared with TBS saturation or TBS threshold plus compound. d, Addition of 50 μm (n = 5) CCh for 10 min induced LTD of fEPSP slope, whereas addition of 30 μm CCh (n = 4) had no effect. e, Bath application of 30 μm VU0364572 (n = 6) for 10 min induced LTD, whereas addition of 30 μm VU0357017 (n = 6) had no effect on LTD. f, Mean percentage maximal depression induced by each compound. VU0364572 induced a significant depression in fEPSP slope compared with 30 μm CCh (*p < 0.01).
We next evaluated the ability of VU0364572 and VU0357017 to induce muscarinic LTD (mLTD) in rat brain slices. Previous studies reveal that CCh induces mLTD when added at higher concentrations than those used for potentiation of LTP (Scheiderer et al., 2006). This effect is mediated by M1 receptors and is blocked by the M1-toxin MTx7 (Scheiderer et al., 2006). We replicated these studies and also found that the addition of 50 μm CCh induced robust LTD, whereas 30 μm CCh was without effect on this response (Fig. 5d). VU0364572 (30 μm) also induced robust LTD (Fig. 5e,f, closed circles, one-way ANOVA F(3,20) = 5.3, p < 0.01). Interestingly, VU0357017 (30 μm) did not induce a significant LTD response (Fig. 5e, open circles). Thus, while the two allosteric M1 agonists have similar effects on modulation of NMDA receptor currents (Lebois et al., 2010, 2011) and induction of hippocampal LTP, they differ in their abilities to induce hippocampal LTD.
VU0357017 and VU0364572 exhibit weak efficacy in inducing excitatory effects in striatal MSNs
M1 also plays a major role in regulating the function of the striatum where activation of this receptor induces excitation and increased firing of MSNs, the primary striatal projection neurons (Pisani et al., 2007; Xiang et al., 2011). Activation of M1 in striatal MSNs is thought to be responsible for the effects of M1 agonists on locomotor activity, including M1-mediated reductions of amphetamine-induced hyperlocomotion (Gerber et al., 2001; Jones et al., 2008). Also, actions of M1 in the ventral striatum have been postulated to be important for the antipsychotic-like effects of M1 agonists, and mAChR-mediated reductions in amphetamine-induced hyperlocomotion have been used as an animal model to assess potential antipsychotic effects of these compounds. Studies measuring [3H]QNB binding in mice where M1 has been genetically deleted have confirmed that M1 receptors are the predominate mAChR subtype expressed in this brain region (Miyakawa et al., 2001). To determine whether VU0364572 and VU0357017 induce responses in striatum isolated from rats, we measured modulation of spike discharge frequency by performing whole-cell current-clamp recordings in MSNs. Striatal MSNs were identified based on their electrophysiological characteristics, including hyperpolarized resting membrane potentials, inward rectification, and delayed action potential discharges in response to a depolarization current injection (Shen et al., 2007). Excitability of MSNs was assessed by monitoring the changes of membrane potential and the number of spike discharges in response to depolarization current pulse (1.5 s in duration). The amplitude of the depolarization pulse was adjusted such that only 1–3 spikes per second were elicited before the application of agonists. As previously reported, CCh (10 μm) induced a robust excitatory effect in MSNs that included an increase in the number of spikes in response to the depolarization current pulse (Fig. 6a,d). VU0357017 (30 μm) had very weak efficacy in inducing excitatory effects in MSNs and caused only a slight increase in spiking frequency (Fig. 6c,d). VU0364572 (30 μm) also had a relatively weak excitatory effect in MSNs, though the effect of VU036572 was more pronounced than that of VU0357017 (Fig. 6b,d). The effects of VU0364572 and VU0357017 were significantly different from CCh suggesting that both compounds act as weak partial agonists for inducing changes in spike frequency in MSNs (F(2,14) = 16.1, p < 0.0002, post hoc analysis using Tukey's test; VU0364572, p < 0.01 and VU0357017, p ≤ 0.001). Comparison of VU0364572 and VU0357017 with post hoc Tukey's test suggests these two responses were not statistically different (p ≥ 0.05). Interestingly, the VU0364572 and VU0357017 responses mirror the effects of each compound in heterologous expression systems in that VU0357017 is consistently shown to be a weaker partial agonist when compared with VU0364572.
Figure 6.

M1-selective agonists VU0364572 and VU0357017 induce small changes in action potential spiking frequency in MSNs. a, The membrane potential response to a current step before and after application of CCh (10 μm) in MSNs of rats. CCh (n = 7) induces a robust increase in evoked action potential firing. b, Response to current injection following application of VU0364572 (n = 5). c, Response to current injection following application of VU0357017 (n = 5). d, Bar graph showing the change in number of spikes/pulse following addition of test compounds. *p < 0.0002 for VU0364572; *p ≤ 0.001 for VU0357017. For comparison of VU0364572 and VU0357017, *p ≥ 0.05. See Results for details.
VU0357017 and VU0364572 are devoid of activity in inducing M1-mediated responses in the mPFC
To further evaluate the effects of VU0364572 and VU0357017 in brain circuits thought to be critical for the potential therapeutic effects of M1 agonists, we determined the effects of these compounds on mPFC pyramidal cells where M1 activation induces depolarization and increases excitatory drive (Shirey et al., 2009). These effects are thought to be important for efficacy of mAChR agonists and M1-selective PAMs in enhancing mPFC-dependent forms of cognitive function, including working memory and reversal learning (Shirey et al., 2009). Furthermore, M1 receptors are thought to represent up to 40% of all muscarinic receptor subtypes in cortex (Levey et al., 1995). Whole-cell voltage-clamp recordings were performed on layer V pyramidal neurons of mice to monitor agonist-induced changes in membrane potential and sEPSC frequency following treatment with CCh, VU0364572, or VU0357017. CCh was used as a positive control to confirm agonist-induced generation of sEPSCs. The response to CCh is clearly mediated by activation of M1 as it is absent in M1 knock-out mice, is blocked by an M1-selective antagonist, and is potentiated by the M1-selective positive allosteric modulator, BQCA (Shirey et al., 2009). Consistent with our previous report, 100 μm CCh induced a robust increase in sEPSC frequency (Fig. 7a). Cumulative probability plots of the interevent interval (IEI) demonstrate a large shift in the frequency of events that was reversible upon washout (Fig. 7a). The decrease in IEI after addition of 100 μm CCh reveals a significant increase in sEPSC frequency (p < 0.0001, one-sample t test vs normalized baseline; Fig. 7d). In contrast to CCh, VU0357017 and VU0364572 (100 μm) had no significant effect on IEI (p > 0.05; Fig. 7). An unpaired t test revealed that IEI in CCh-treated slices was significantly different from responses to VU0357017 (p < 0.0001) or VU0364572 (p = 0.0002).Together, the above results suggest that VU0364572 and VU0357017 induce brain region-specific responses, in that they have robust activity in hippocampus, weaker efficacy in striatal MSNs, and are inactive in eliciting M1-mediated prefrontal cortical electrophysiological responses. It is possible that the lack of an effect in PFC is directly related to low receptor reserve inherent to this signaling response as this response requires >50 μm CCh for induction.
Figure 7.

VU0357017 and VU0364572 are devoid of agonist activity in mouse mPFC. a, Sample traces from single neurons and changes in probability plots of the IEI from representative cells following treatment with CCh showing that CCh induces increases in sEPSCs (n = 15). b, c Sample traces and probability plots of IEI in representative cells treated with VU0357017 (n = 7) or VU0364572 (n = 4). d, Bar graphs depicting mean changes in sEPSC frequency. All changes in frequency represent the mean ± SEM and are compared with baseline controls. #p < 0.0001, **p < 0.0001, for VU037017 and **p = 0.0002 for VU0364572. See Results for details.
Systemic dosing of VU0364572 and VU0357017 enhances two forms of hippocampal-dependent cognitive function
While it is impossible to definitively ascribe any given behavioral effect of M1 agonists to a specific electrophysiological response, the finding that M1 agonists can differentially activate distinct M1-mediated responses in cell lines and in brain slices raises the possibility that these compounds may have different in vivo effects, depending on their specific actions on M1 signaling in the CNS. For instance, based on the present findings, it is possible that VU0364572 and VU0357017 may have efficacy in animal models that reflect M1-mediated effects on hippocampal-dependent learning but may not have activity in models reflective of M1 activation in other brain regions. We tested the effects of both compounds in rodent animal models that reflect efficacy in improving hippocampal-dependent cognitive function. First, we evaluated spatial memory using the MWM test after intraperitoneal injection of either vehicle or one of the M1 agonists. Interestingly, VU0364572 enhanced performance in the MWM when injected before testing, suggesting that it enhanced spatial learning. As seen in Figure 8b, VU0364572 decreased swim distance at the 0.1 mg/kg and 10 mg/kg doses on day 5 (F(2,42) = 6.107, p < 0.001 vs vehicle-treated animals). This VU0364572-induced enhancement was also seen on day 4 for the 0.1 mg/kg dose (Fig. 8b; p < 0.05 vs vehicle-treated animals). These data also showed comparably enhanced overnight memory retention in the 0.1 mg/kg group from day 4 to day 5 (Fig. 8c; F(2,21) = 5.557, p ≤ 0.02 vs vehicle-treated animals for day 5). The 0.1 mg/kg dose of VU0364572 also increased platform crossings during the first 30 s of the probe trial (Fig. 8d; F(2,21) = 4.766, p < 0.05 vs vehicle-treated animals) suggesting improved spatial localization. In contrast to VU0364572, VU0357017-treated groups did not differ from controls in swim path distance from days 4–5 (Fig. 8f; p ≥ 0.445), overnight memory retention (Fig. 8g; p ≥ 0.755), or platform crossing on the probe trial (Fig. 8h; p ≥ 0.981). Interestingly, these effects mirror many of the in vitro and electrophysiology studies suggesting that VU0364572 has greater efficacy than VU0357017 in activating M1. However, it is also possible that the procognitive effects of VU0364572 are due to better brain exposure of this compound (Lebois et al., 2011).
Figure 8.

M1 agonists VU0357017 and VU0364572 enhance performance in MWM and CFC in rats. a, Swim distance across the 5 d of testing in the water maze. Data are collapsed across four daily trials and across groups of VU0364572 or vehicle-treated animals. b, Effects of treatment with VU0364572 on spatial memory on days 4 (*p < 0.05) and 5 (*p < 0.001) of testing in MWM assays. c, Effects of treatment with VU0364572 on memory retention between day 4 Trial 4 and day 5 Trial 1 (n = 8) +p < 0.05 versus day 4 for vehicle-treated animals only. *p ≤ 0.02 versus vehicle-treated animals for day 5 (n = 8). d, Effects of treatment with VU0364572 on platform crossings during the first 30 s of the probe trial. *p < 0.05. e, Swim distance across the 5 d of testing in the water maze. Data are collapsed across four daily trials and across groups of VU0357017 or vehicle-treated animals. f, Effects of treatment with VU0357017 on spatial memory on days 4 and 5 of testing (n = 8) (p > 0.445). g, Effects of treatment with VU357017 on spatial memory retention between day 4 Trial 4 and day 5 Trial 1 (n = 8) (p > 0.755). h, Effects of treatment with VU0357017 on platform crossings during the first 30 s of the probe trial (p > 0.981). i, Effects of treatment of VU0364572 on acquisition of contextual fear in rats (n = 4–6, each group) (*p < 0.05). j, Effects of VU0357017 treatment on acquisition of contextual fear in rats (n = 4–6, each group) (*p < 0.05).
As a second measure to evaluate the effects of these agents on hippocampal-dependent cognitive function, we determined the effects of both compounds on CFC, a behavioral model previously shown to be impaired by muscarinic antagonists (Sutherland et al., 1982; Anagnostaras et al., 1995). Interestingly, VU0364572 improved acquisition of contextual fear learning at the 0.056, 0.3, and 0.56 mg/kg doses (Fig. 8i, p < 0.05 vs vehicle-treated animals). VU0357017 induced robust improvements in acquisition of contextual fear at doses of 0.1, 0.3, 0.56, 1, and 3 mg/kg as measured by an increased duration of freezing (Fig. 8j, p < 0.05 vs vehicle-treated animals). Interestingly, these effects did not follow a typical dose–response pattern, which may have been a consequence of the extremely low doses used for both compounds (0.01 mg/kg, etc.). Together, these results support the idea that M1-selective partial agonists may provide a novel approach for enhancing hippocampal-dependent forms of cognitive function.
VU0364572 and VU0357017 fail to reverse amphetamine-induced hyperlocomotion suggesting that these agents have limited efficacy as antipsychotic agents
A number of previous studies suggests that activation of M1 receptors in the striatum induces behavioral effects that reflect increases in dopaminergic transmission (Gerber et al., 2001; Miyakawa et al., 2001; Ellis, 2002). This can be assessed by measuring the ability of M1 agonists to reduce behavioral responses to amphetamine and other psychostimulants (Jones et al., 2008; Vanover et al., 2008; Ma et al., 2009). The effects of xanomeline on reversal of amphetamine-induced hyperlocomotion have been postulated to be relevant for the antipsychotic efficacy of this compound in patients suffering from schizophrenia (Stanhope et al., 2001; Andersen et al., 2003). These findings also contribute to the basis for efforts to develop selective M1 agonists for treatment of this disorder (Jones et al., 2008; Conn et al., 2009). To determine the effects of our compounds on reversing amphetamine-induced hyperlocomotion, we first established a working dose of amphetamine by performing a dose–response curve. As can be seen in Figure 9a, a 1 mg/kg dose of amphetamine induces robust hyperlocomotion that is not accompanied by stereotypy, an effect consistent with previously published data (Jones et al., 2008; Rodriguez et al., 2010). A 3 mg/kg dose, on the other hand, induced a decrease in locomotor activity, an effect consist with the induction of stereotypy (Fig. 9a). We next evaluated the ability of both VU0364572 and VU0357017 to reverse amphetamine-induced hyperlocomotion in rats after intraperitoneal dosing. In contrast to previously reported effects of xanomeline and other M1-selective compounds (Stanhope et al., 2001; Andersen et al., 2003; Jones et al., 2008; Ma et al., 2009), neither VU0364572 nor VU0357017 (3–56.6 mg/kg) reduced amphetamine-induced hyperlocomotor activity (Fig. 9b,c). These results suggest that these functionally selective M1 agonists lack efficacy in eliciting an established behavioral response that is often used to predict potential antipsychotic effects of M1 agonists. Interestingly, the 56.6 mg/kg dose of VU0364572 reduced locomotor activity in rats before injection of amphetamine (Fig. 9c). However, this effect was not accompanied by a reduction in hyperlocomotion.
Figure 9.
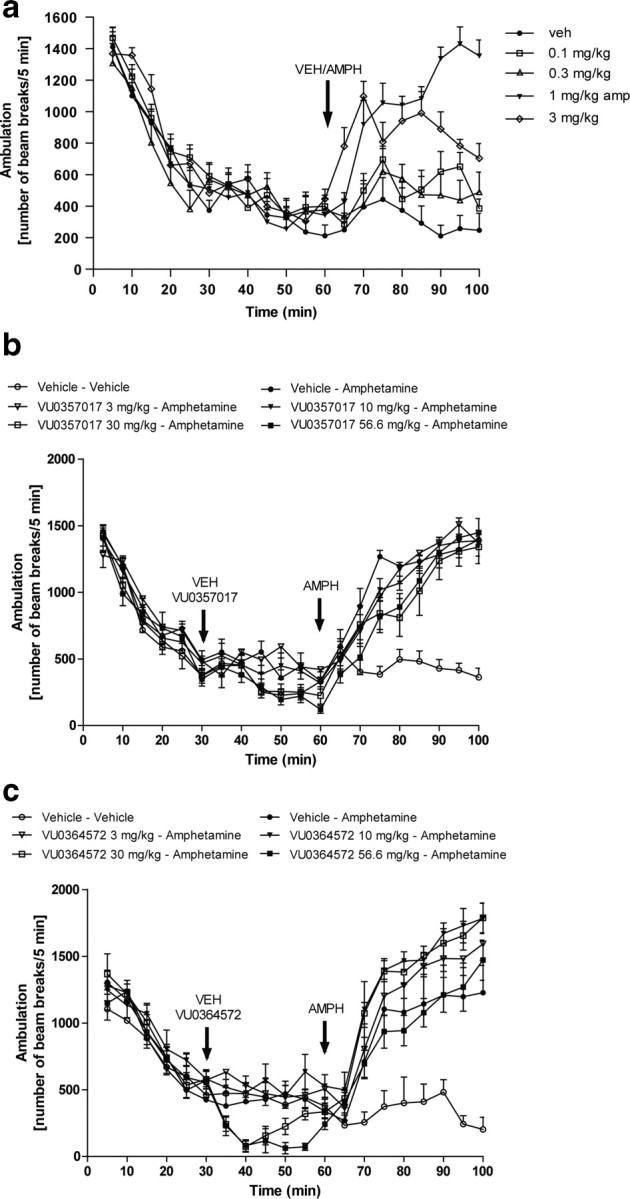
VU0357017 and VU0364572 do not reverse amphetamine-induced hyperlocomotion in rats. a, An amphetamine dose–response curve showing that 1 mg/kg amphetamine induces robust hyperlocomotion. b, VU0357017 fails to reverse hyperlocomotion induced by amphetamine treatment in rats suggesting that M1 agonism by this compound does not have an antipsychotic-like profile. VU0357017 or vehicle was injected intraperitoneally 30 min before amphetamine injection (4 mg/kg, s.c.). c, A broad dose range of VU0364572 also fails to reverse this response.
Discussion
In recent years, the M1 mAChR has emerged as an exciting new target for treatment of schizophrenia and other major brain disorders (Langmead et al., 2008; Conn et al., 2009). Recent studies suggest that mAChR agonists could provide a fundamental advance in providing efficacy in treatment of all major symptom clusters in schizophrenia patients, including positive symptoms, negative symptoms, and impaired cognitive function (Shekhar et al., 2008; Conn et al., 2009). Currently available antipsychotic drugs have efficacy in reducing positive symptoms (hallucinations, delusions, and thought disorder) but have little or no efficacy in the treatment of negative symptoms (social withdrawal, anhedonia, and apathy) or cognitive impairments (deficits in perception, attention, short- and long-term memory, and executive function) that are characteristic of this disease (Conn et al., 2009). However, cognitive deficits and negative symptoms are major components of the disabilities associated with schizophrenia and are considered to be especially important predictors of long-term disability and treatment outcome (Meltzer et al., 1999; Conn et al., 2008). Recent clinical studies have revealed that the M1/M4-preferring mAChR agonist, xanomeline, has robust efficacy in improving positive and negative symptoms and improving cognitive function in schizophrenic patients (Shekhar et al., 2008). In addition, xanomeline has efficacy in reducing hallucinations, delusions, and related behavioral disturbances, in addition to improving cognitive function, in patients suffering from AD and other neurodegenerative disorders (Bodick et al., 1997a,b).
Unfortunately, efforts to develop xanomeline and other traditional mAChR agonists have failed because of a lack of selectivity of these agents for individual mAChR subtypes and prominent adverse effects that are mediated by activation of M2 and M3 mAChRs (Conn et al., 2009). However, a major breakthrough was established with the discovery of allosteric agonists and positive allosteric modulators (PAMs) that are highly selective for individual mAChR subtypes. Interestingly, selective M1 PAMs and allosteric agonists have efficacy in multiple animal models used to predict antipsychotic activity, such as reversal of amphetamine-induced hyperlocomotor activity (Jones et al., 2008), and induce robust improvements in both hippocampal and prefrontal cortical-dependent domains of cognitive function in mouse and rodent models (Ma et al., 2009; Shirey et al., 2009). In addition, M1 has been clearly established to be the primary mAChR subtype involved in several electrophysiological responses that are thought to be critical for the efficacy of mAChR agonists in schizophrenia patients. These include excitatory effects on striatal MSNs (Shen et al., 2007; Xiang et al., 2011), increased excitatory drive and depolarization of mPFC neurons (Shirey et al., 2009), and induction of both LTP and LTD in the hippocampal formation (Anagnostaras et al., 2003; Shinoe et al., 2005; Scheiderer et al., 2006; Volk et al., 2007). The present finding that VU0357017 and VU0364572 selectively activate some but not all physiological responses that are associated with M1 activation in the CNS provides critical new insights that will be important in guiding any future efforts focused on optimization of M1 allosteric agonists as therapeutic agents or as research tools. To achieve maximal efficacy in treatment of the major symptom clusters observed in patients with schizophrenia, it will be important to develop M1 activators that mimic or potentiate the effects of ACh on each of the major systems that are thought to be critical for therapeutic efficacy. This is consistent with our finding that VU0357017 and VU0364572 have efficacy in improving hippocampal-dependent forms of cognitive function, but do not have efficacy in reversing amphetamine-induced hyperlocomotion in rodents, an established model of positive symptoms, where xanomeline (Shannon et al., 2000; Perry et al., 2001; Stanhope et al., 2001; Andersen et al., 2003; Jones et al., 2008) and previous M1-selective PAMs (Ma et al., 2009) and allosteric agonists (Bradley et al., 2009; Ma et al., 2009) are efficacious. In addition, the finding that VU0357017 and VU0364572 are inactive in eliciting known physiological effects of M1 activation in the mPFC raises the possibility that compounds with this profile may not have efficacy in improving cognitive function that relies of activation of the PFC in patients with schizophrenia. Interestingly, we recently reported that the M1-selective PAM, BQCA, has robust effects on M1-mediated responses in mPFC pyramidal cells, increases firing of mPFC neurons in vivo, and improves mPFC-dependent forms of cognitive function (Shirey et al., 2009). Clinical studies suggest that deficits in mPFC activation represent a key component of the pathophysiology in patients with schizophrenia and that these patients are especially impaired in cognitive tasks that require activation of the PFC (Barch et al., 2001; Arnsten, 2011). Also, as discussed above, M1 actions on striatal MSNs may be important for the potential antipsychotic efficacy of M1 agonists. Thus, it will be critical to advance M1 allosteric activators into clinical development that have robust actions on M1-mediated responses in mPFC neurons and in MSNs.
Importantly, it may also be possible to take advantage of the ability to develop M1 agonists that have limited actions of M1 in the CNS for other indications. For instance, patients suffering from Parkinson's disease (PD) also suffer from cognitive impairments and there is a need to develop strategies for improving cognitive function in these patients. Muscarinic agonists have not been viewed as a viable option in PD patients because of their actions in the striatum that could worsen parkinsonian motor symptoms (McCarthy et al., 2011). However, the present findings suggest that it may be possible to develop M1 agonists that do not alter motor function. As we develop a more complete understanding of the signaling pathways required for different actions of M1 agonists in the CNS, it may be possible to specifically target those that are required for optimal efficacy.
The finding that responses to VU0357017 and VU0364572 on different signaling pathways in cell lines can be altered by changes in levels of M1 expression is also important for a mechanistic understanding of actions of these agents in the CNS. In studies of calcium mobilization, VU0357017 and VU0364572 behaved as classical partial agonists and had robust efficacy in settings of high receptor expression (i.e., high receptor reserve) and relatively low efficacy in cell lines with low receptor expression. Also, effects of these agents on ERK1/2 phosphorylation were highly influenced by levels of M1 expression. Previous studies reveal that levels of receptor reserve for M1-mediated responses are highly variable in the CNS and other native systems (Conn et al., 2009). Thus, different levels of M1 expression are likely to contribute to the differential responses to VU0357017 and VU0364572 observed in these studies. However, it was interesting to find that VU0357017 never achieved full efficacy in activation of ERK1/2 phosphorylation, even in cell lines with strong induction of M1 expression to levels that induced high receptor reserve in the calcium mobilization assay. Also, VU0357017 did not induce robust β-arrestin responses in the original cell line or in the TREx hM1 cells. Thus, the differential effects of these M1 agonists on CNS responses may reflect a combination of partial agonist activity that is impacted by differences in receptor reserve and by an inherent stimulus bias at M1 so that these compounds are not capable of fully activating some responses, even in systems in which the receptor is highly expressed.
In addition to the importance of these findings for our understanding of regulation of M1 signaling and functional responses in the CNS, these findings provide critical new insights into issues for chemical lead optimization efforts focused on optimizing novel M1 allosteric agonists as potential therapeutic agents. Lead optimization efforts often focus on a single in vitro assay to drive chemical optimization as a way to streamline chemistry efforts. M1 agonist optimization commonly relies on measures of M1-mediated calcium mobilization as the primary assay and uses overexpressing cell lines with high levels of receptor reserve to maximize the signal. The present findings suggest that reliance on a streamlined strategy of optimizing with a single readout of M1 function could yield compounds that may not have the desired effects. At a practical level, our studies raise the importance of measuring effects of key compounds on multiple signaling pathways under conditions of relatively low receptor expression to drive lead optimization efforts. Also, measuring physiological effects of advanced compounds in multiple CNS systems is important to reduce the risk of inadvertently advancing drug candidates that have more restricted CNS actions.
Footnotes
This work was supported by the National Institutes of Health Grants U54 MH084659, R01 MH082867, U01 MH087956, RO1 MH073676, F32 MH087039, F32NS071746, T32 MH065215, and 1R01 NS065867. C.W.L. receives funding from the National Institutes of Health–National Institute of Mental Health and National Institute on Drug Abuse, Johnson & Johnson, Seaside Therapeutics, Vanderbilt University Medical Center (Department of Pharmacology), and the American Chemical Society (as Editor-in-Chief of ACS Chemical Neuroscience). In the recent past, C.W.L. has also received funding from the Michael J. Fox Foundation and the Alzheimer's Association and has consulted for Merck, Eisai, GlaxoSmithKline, Biogen, and Amgen.
P.J.C. has served as a consultant over the past three years for Merck and Co., Johnson & Johnson, Hoffman-La Roche, GlaxoSmithKline, Lundbeck, Epix Pharmaceuticals, Invitrogen Life Technologies, Evotech, Addex Pharmaceuticals, Michael J. Fox Foundation, Cephalon, LEK Consulting, The Frankel Group, Prestwick Chemical, IMS Health, Primary Insight, Otsuka Pharmaceuticals, AstraZeneca USA, NeurOP, Seaside Therapeutics, Millipore, Genentech, Abbott Laboratories, AMRI, Bristol-Myers Squibb, and PureTech. P.J.C. receives research support that includes salary support from Seaside Therapeutics and Johnson & Johnson.
References
- Anagnostaras SG, Maren S, Fanselow MS. Scopolamine selectively disrupts the acquisition of contextual fear conditioning in rats. Neurobiol Learn Mem. 1995;64:191–194. doi: 10.1006/nlme.1995.0001. [DOI] [PubMed] [Google Scholar]
- Anagnostaras SG, Murphy GG, Hamilton SE, Mitchell SL, Rahnama NP, Nathanson NM, Silva AJ. Selective cognitive dysfunction in acetylcholine M1 muscarinic receptor mutant mice. Nat Neurosci. 2003;6:51–58. doi: 10.1038/nn992. [DOI] [PubMed] [Google Scholar]
- Andersen MB, Fink-Jensen A, Peacock L, Gerlach J, Bymaster F, Lundbaek JA, Werge T. The muscarinic M1/M4 receptor agonist xanomeline exhibits antipsychotic-like activity in Cebus apella monkeys. Neuropsychopharmacology. 2003;28:1168–1175. doi: 10.1038/sj.npp.1300151. [DOI] [PubMed] [Google Scholar]
- Arnsten AF. Prefrontal cortical network connections: key site of vulnerability in stress and schizophrenia. Int J Dev Neurosci. 2011;29:215–223. doi: 10.1016/j.ijdevneu.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auld DS, Kornecook TJ, Bastianetto S, Quirion R. Alzheimer's disease and the basal forebrain cholinergic system: relations to beta-amyloid peptides, cognition, and treatment strategies. Prog Neurobiol. 2002;68:209–245. doi: 10.1016/s0301-0082(02)00079-5. [DOI] [PubMed] [Google Scholar]
- Ayala JE, Chen Y, Banko JL, Sheffler DJ, Williams R, Telk AN, Watson NL, Xiang Z, Zhang Y, Jones PJ, Lindsley CW, Olive MF, Conn PJ. mGluR5 positive allosteric modulators facilitate both hippocampal LTP and LTD and enhance spatial learning. Neuropsychopharmacology. 2009;34:2057–2071. doi: 10.1038/npp.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barch DM, Carter CS, Braver TS, Sabb FW, MacDonald A, 3rd, Noll DC, Cohen JD. Selective deficits in prefrontal cortex function in medication-naive patients with schizophrenia. Arch Gen Psychiatry. 2001;58:280–288. doi: 10.1001/archpsyc.58.3.280. [DOI] [PubMed] [Google Scholar]
- Bodick NC, Offen WW, Shannon HE, Satterwhite J, Lucas R, van Lier R, Paul SM. The selective muscarinic agonist xanomeline improves both the cognitive deficits and behavioral symptoms of Alzheimer disease. Alzheimer Dis Assoc Disord. 1997a;11(Suppl 4):S16–S22. [PubMed] [Google Scholar]
- Bodick NC, Offen WW, Levey AI, Cutler NR, Gauthier SG, Satlin A, Shannon HE, Tollefson GD, Rasmussen K, Bymaster FP, Hurley DJ, Potter WZ, Paul SM. Effects of xanomeline, a selective muscarinic receptor agonist, on cognitive function and behavioral symptoms in Alzheimer disease. Arch Neurol. 1997b;54:465–473. doi: 10.1001/archneur.1997.00550160091022. [DOI] [PubMed] [Google Scholar]
- Bradley SR, Lameh J, Ohrmund L, Son T, Bajpai A, Nguyen D, Friberg M, Burstein ES, Spalding TA, Ott TR, Schiffer HH, Tabatabaei A, McFarland K, Davis RE, Bonhaus DW. AC-260584, an orally bioavailable M(1) muscarinic receptor allosteric agonist, improves cognitive performance in an animal model. Neuropharmacology. 2010;58:365–373. doi: 10.1016/j.neuropharm.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Buchanan KA, Petrovic MM, Chamberlain SE, Marrion NV, Mellor JR. Facilitation of long-term potentiation by muscarinic M(1) receptors is mediated by inhibition of SK channels. Neuron. 2010;68:948–963. doi: 10.1016/j.neuron.2010.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PJ, Tamminga C, Schoepp DD, Lindsley C. Schizophrenia: moving beyond monoamine antagonists. Mol Interv. 2008;8:99–107. doi: 10.1124/mi.8.2.7. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Jones CK, Lindsley CW. Subtype-selective allosteric modulators of muscarinic receptors for the treatment of CNS disorders. Trends Pharmacol Sci. 2009;30:148–155. doi: 10.1016/j.tips.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis J. Muscarinic receptors. In: Pangalos MN, Davis CH, editors. Understanding g protein-coupled receptors and their role in the CNS. London: Oxford UP; 2002. [Google Scholar]
- Felder CC, Bymaster FP, Ward J, DeLapp N. Therapeutic opportunities for muscarinic receptors in the central nervous system. J Med Chem. 2000;43:4333–4353. doi: 10.1021/jm990607u. [DOI] [PubMed] [Google Scholar]
- Fisher SK, Snider RM. Differential receptor occupancy requirements for muscarinic cholinergic stimulation of inositol lipid hydrolysis in brain and in neuroblastomas. Mol Pharmacol. 1987;32:81–90. [PubMed] [Google Scholar]
- Flynn DD, Ferrari-DiLeo G, Mash DC, Levey AI. Differential regulation of molecular subtypes of muscarinic receptors in Alzheimer's disease. J Neurochem. 1995;64:1888–1891. doi: 10.1046/j.1471-4159.1995.64041888.x. [DOI] [PubMed] [Google Scholar]
- Gerber DJ, Sotnikova TD, Gainetdinov RR, Huang SY, Caron MG, Tonegawa S. Hyperactivity, elevated dopaminergic transmission, and response to amphetamine in M1 muscarinic acetylcholine receptor-deficient mice. Proc Natl Acad Sci U S A. 2001;98:15312–15317. doi: 10.1073/pnas.261583798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CK, Eberle EL, Shaw DB, McKinzie DL, Shannon HE. Pharmacologic interactions between the muscarinic cholinergic and dopaminergic systems in the modulation of prepulse inhibition in rats. J Pharmacol Exp Ther. 2005;312:1055–1063. doi: 10.1124/jpet.104.075887. [DOI] [PubMed] [Google Scholar]
- Jones CK, Brady AE, Davis AA, Xiang Z, Bubser M, Tantawy MN, Kane AS, Bridges TM, Kennedy JP, Bradley SR, Peterson TE, Ansari MS, Baldwin RM, Kessler RM, Deutch AY, Lah JJ, Levey AI, Lindsley CW, Conn PJ. Novel selective allosteric activator of the M1 muscarinic acetylcholine receptor regulates amyloid processing and produces antipsychotic-like activity in rats. J Neurosci. 2008;28:10422–10433. doi: 10.1523/JNEUROSCI.1850-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead CJ, Starr KR, Teriakidis A, Wood MD, et al. Characterization of a CNS penetrant, selective M1 muscarinic receptor agonist, 77-LH-28–1. Br J Pharmacol. 2008;154:1104–1115. doi: 10.1038/bjp.2008.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebois EP, Bridges TM, Lewis LM, Dawson ES, Kane AS, Xiang Z, Jadhav SB, Yin H, Kennedy JP, Meiler J, Niswender CM, Jones CK, Conn PJ, Weaver CD, Lindsley CW. Discovery and characterization of novel subtype-selective allosteric agonists for the investigation of M(1) receptor function in the central nervous system. ACS Chem Neurosci. 2010;1:104–121. doi: 10.1021/cn900003h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebois EP, Digby GJ, Sheffler DJ, Melancon BJ, Tarr JC, Cho HP, Miller NR, Morrison R, Bridges TM, Xiang Z, Scott Daniels J, Wood MR, Conn PJ, Lindsley CW. Development of a highly selective, orally bioavailable and CNS penetrant M1 agonist derived from the MLPCN probe ML071. Bioorg Med Chem Lett. 2011;21:6451–6455. doi: 10.1016/j.bmcl.2011.08.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levey AI, Edmunds SM, Koliatsos V, Wiley RG, Heilman CJ. Expression of m1–m4 muscarinic acetylcholine receptor proteins in rat hippocampus and regulation by cholinergic innervation. J Neurosci. 1995;15:4077–4092. doi: 10.1523/JNEUROSCI.15-05-04077.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, et al. Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc Natl Acad Sci U S A. 2009;106:15950–15955. doi: 10.1073/pnas.0900903106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino MJ, Rouse ST, Levey AI, Potter LT, Conn PJ. Activation of the genetically defined m1 muscarinic receptor potentiates N-methyl-D-aspartate (NMDA) receptor currents in hippocampal pyramidal cells. Proc Natl Acad Sci U S A. 1998;95:11465–11470. doi: 10.1073/pnas.95.19.11465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massoud F, Gauthier S. Update on the pharmacological treatment of Alzheimer's disease. Curr Neuropharmacol. 2010;8:69–80. doi: 10.2174/157015910790909520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MM, Moore-Kochlacs C, Gu X, Boyden ES, Han X, Kopell N. Striatal origin of the pathologic beta oscillations in Parkinson's disease. Proc Natl Acad Sci U S A. 2011;108:11620–11625. doi: 10.1073/pnas.1107748108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutchen E, Scheiderer CL, Dobrunz LE, McMahon LL. Coexistence of muscarinic long-term depression with electrically induced long-term potentiation and depression at CA3-CA1 synapses. J Neurophysiol. 2006;96:3114–3121. doi: 10.1152/jn.00144.2006. [DOI] [PubMed] [Google Scholar]
- Meltzer HY, Park S, Kessler R. Cognition, schizophrenia, and the atypical antipsychotic drugs. Proc Natl Acad Sci U S A. 1999;96:13591–13593. doi: 10.1073/pnas.96.24.13591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyakawa T, Yamada M, Duttaroy A, Wess J. Hyperactivity and intact hippocampus-dependent learning in mice lacking the M1 muscarinic acetylcholine receptor. J Neurosci. 2001;21:5239–5250. doi: 10.1523/JNEUROSCI.21-14-05239.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry KW, Nisenbaum LK, George CA, Shannon HE, Felder CC, Bymaster FP. The muscarinic agonist xanomeline increases monoamine release and immediate early gene expression in the rat prefrontal cortex. Biol Psychiatry. 2001;49:716–725. doi: 10.1016/s0006-3223(00)01017-9. [DOI] [PubMed] [Google Scholar]
- Pisani A, Bernardi G, Ding J, Surmeier DJ. Re-emergence of striatal cholinergic interneurons in movement disorders. Trends Neurosci. 2007;30:545–553. doi: 10.1016/j.tins.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Rodriguez AL, Grier MD, Jones CK, Herman EJ, Kane AS, Smith RL, Williams R, Zhou Y, Marlo JE, Days EL, Blatt TN, Jadhav S, Menon UN, Vinson PN, Rook JM, Stauffer SR, Niswender CM, Lindsley CW, Weaver CD, Conn PJ. Discovery of novel allosteric modulators of metabotropic glutamate receptor subtype 5 reveals chemical and functional diversity and in vivo activity in rat behavioral models of anxiolytic and antipsychotic activity. Mol Pharmacol. 2010;78:1105–1123. doi: 10.1124/mol.110.067207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiderer CL, McCutchen E, Thacker EE, Kolasa K, Ward MK, Parsons D, Harrell LE, Dobrunz LE, McMahon LL. Sympathetic sprouting drives hippocampal cholinergic reinnervation that prevents loss of a muscarinic receptor-dependent long-term depression at CA3-CA1 synapses. J Neurosci. 2006;26:3745–3756. doi: 10.1523/JNEUROSCI.5507-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon HE, Rasmussen K, Bymaster FP, Hart JC, Peters SC, Swedberg MD, Jeppesen L, Sheardown MJ, Sauerberg P, Fink-Jensen A. Xanomeline, an M(1)/M(4) preferring muscarinic cholinergic receptor agonist, produces antipsychotic-like activity in rats and mice. Schizophr Res. 2000;42:249–259. doi: 10.1016/s0920-9964(99)00138-3. [DOI] [PubMed] [Google Scholar]
- Shekhar A, Potter WZ, Lightfoot J, Lienemann J, Dubé S, Mallinckrodt C, Bymaster FP, McKinzie DL, Felder CC. Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am J Psychiatry. 2008;165:1033–1039. doi: 10.1176/appi.ajp.2008.06091591. [DOI] [PubMed] [Google Scholar]
- Shen W, Tian X, Day M, Ulrich S, Tkatch T, Nathanson NM, Surmeier DJ. Cholinergic modulation of Kir2 channels selectively elevates dendritic excitability in striatopallidal neurons. Nat Neurosci. 2007;10:1458–1466. doi: 10.1038/nn1972. [DOI] [PubMed] [Google Scholar]
- Shinoe T, Matsui M, Taketo MM, Manabe T. Modulation of synaptic plasticity by physiological activation of M1 muscarinic acetylcholine receptors in the mouse hippocampus. J Neurosci. 2005;25:11194–11200. doi: 10.1523/JNEUROSCI.2338-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirey JK, Brady AE, Jones PJ, Davis AA, Bridges TM, Kennedy JP, Jadhav SB, Menon UN, Xiang Z, Watson ML, Christian EP, Doherty JJ, Quirk MC, Snyder DH, Lah JJ, Levey AI, Nicolle MM, Lindsley CW, Conn PJ. A selective allosteric potentiator of the M1 muscarinic acetylcholine receptor increases activity of medial prefrontal cortical neurons and restores impairments in reversal learning. J Neurosci. 2009;29:14271–14286. doi: 10.1523/JNEUROSCI.3930-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanhope KJ, Mirza NR, Bickerdike MJ, Bright JL, Harrington NR, Hesselink MB, Kennett GA, Lightowler S, Sheardown MJ, Syed R, Upton RL, Wadsworth G, Weiss SM, Wyatt A. The muscarinic receptor agonist xanomeline has an antipsychotic-like profile in the rat. J Pharmacol Exp Ther. 2001;299:782–792. [PubMed] [Google Scholar]
- Sutherland RJ, Whishaw IQ, Regehr JC. Cholinergic receptor blockade impairs spatial localization by use of distal cues in the rat. J Comp Physiol Psychol. 1982;96:563–573. doi: 10.1037/h0077914. [DOI] [PubMed] [Google Scholar]
- Thomas RL, Mistry R, Langmead CJ, Wood MD, Challis RA. G protein coupling and signaling pathway activation by m1 muscarinic receptor orthosteric and allosteric agonists. J Pharmacol Exp Ther. 2008;327:365–374. doi: 10.1124/jpet.108.141788. [DOI] [PubMed] [Google Scholar]
- Vanover KE, Veinbergs I, Davis RE. Antipsychotic-like behavioral effects and cognitive enhancement by a potent and selective muscarinic M-sub-1 receptor agonist, AC-260584. Behav Neurosci. 2008;122:570–575. doi: 10.1037/0735-7044.122.3.570. [DOI] [PubMed] [Google Scholar]
- Volk LJ, Pfeiffer BE, Gibson JR, Huber KM. Multiple Gq-coupled receptors converge on a common protein synthesis-dependent long-term depression that is affected in fragile X syndrome mental retardation. J Neurosci. 2007;27:11624–11634. doi: 10.1523/JNEUROSCI.2266-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Z, Thompson AD, Jones CK, Lindsley CW, Conn PJ. Roles of M1 muscarinic acetylcholine receptor subtype in regulation of basal ganglia function and implications for treatment of Parkinson's disease. J Pharmacol Exp Ther. 2011 doi: 10.1124/jpet.111.187856. [DOI] [PMC free article] [PubMed] [Google Scholar]