

Abstract
Objective:
Sporadic, genetically complex essential tremor (ET) is one of the most common movement disorders and may lead to severe impairment of the quality of life. Despite high heritability, the genetic determinants of ET are largely unknown. We performed the second genome-wide association study (GWAS) for ET to elucidate genetic risk factors of ET.
Methods:
Using the Affymetrix Genome-Wide SNP Array 6.0 (1000K) we conducted a two-stage GWAS in a total of 990 subjects and 1,537 control subjects from Europe to identify genetic variants associated with ET.
Results:
We discovered association of an intronic variant of the main glial glutamate transporter (SLC1A2) gene with ET in the first-stage sample (rs3794087, p = 6.95 × 10−5, odds ratio [OR] = 1.46). We verified the association of rs3794087 with ET in a second-stage sample (p = 1.25 × 10−3, OR = 1.38). In the subgroup analysis of patients classified as definite ET, rs3794087 obtained genome-wide significance (p = 3.44 × 10−10, OR = 1.59) in the combined first- and second-stage sample. Genetic fine mapping using nonsynonymous single nucleotide polymorphisms (SNPs) and SNPs in high linkage disequilibrium with rs3794087 did not reveal any SNP with a stronger association with ET than rs3794087.
Conclusions:
We identified SLC1A2 encoding the major glial high-affinity glutamate reuptake transporter in the brain as a potential ET susceptibility gene. Acute and chronic glutamatergic overexcitation is implied in the pathogenesis of ET. SLC1A2 is therefore a good functional candidate gene for ET.
Sporadic, genetically complex essential tremor (ET) is one of the most frequent neurologic disorders with a prevalence between 0.9% and 4.6% in the population older than 65 years.1 The clinical hallmark of the disease is a postural or kinetic tremor of the upper extremities. Severe ET not only causes permanent disability but often also social stigmatization. In a large proportion of patients, ET can be acutely alleviated by moderate doses of ethanol.2,3 The diagnosis of ET is based on the clinical examination because no biomarker or specific diagnostic test exists. The most stringent research diagnostic criteria for ET are the consensus criteria proposed by the Tremor Investigation Group (TRIG).1 Family history studies and twin studies demonstrated high heritability with concordance rates of up to 95% in monozygotic vs 29% in dizygotic twins.4 The pathogenesis of ET is largely unknown. Two competing hypothesis based on neurophysiologic, pathologic, and animal data state that ET could either be a neurofunctional disorder caused by abnormal oscillations possibly generated in the inferior olivary nucleus or a neurodegenerative disorder of the cerebellum.5 However, it can also not be excluded that both hypotheses apply, e.g., that permanent longstanding abnormal oscillations transmitted to the cerebellum ultimately lead to neurodegeneration. The first genome-wide association study (GWAS) for ET identified an association between a variant in the LINGO1 gene and ET in a first-stage sample composed of Icelandic patients.6 This finding was confirmed by several but not all other groups attempting replication.7–9 Here, we present the second GWAS for ET.
METHODS
Standard protocol approvals, registrations, and patient consents.
The local ethics committees approved the study. Written informed consent was obtained from all participants.
Study samples.
ET was diagnosed according to the TRIG criteria.1 In the first stage we analyzed 436 patients and 928 control subjects of German origin. The second-stage independently collected verification sample consisted of 554 patients and 609 control subjects of European (Germany, Denmark, and Austria) origin. Control samples were of German, Austrian, and Danish origin. Recent publications only found minor genetic differences between Germans, Austrians, and Danes.10,11 Therefore, we combined the samples from these 3 different populations in stage 2. In this study we analyzed only patients with definite or probable ET but not possible ET, which is the weakest diagnostic category according to the TRIG criteria, to achieve maximal diagnostic certainty. All control subjects were screened negative for ET by a validated screening procedure.12 Control subjects were not matched to patients regarding the sex ratio, and no adjustment for gender was performed because no sex-specific differences in epidemiologic data, pathogenesis, or clinical presentation are known. The 847 German patients from Kiel used in stages 1 and 2 were recruited by movement disorder specialists at the Department of Neurology of the University Hospital of Schleswig-Holstein, campus Kiel, and also in cooperation with the PopGen Biobank.13 The 928 German control subjects for the genome-wide scan were obtained from the PopGen Biobank.13 For the second-stage analyses, 75 healthy German spouses were recruited as control subjects at the University Hospital of Schleswig-Holstein, Kiel campus, and 478 German control subjects were recruited from the Kiel aging project within PopGen.14 The recruitment of the Danish patient and control sample (stage 2) has been described elsewhere.4 German patients with ET from Tübingen (stage 2) were recruited from the movement disorder clinic at the Center of Neurology, University of Tübingen. Patients were systematically examined, and ET was diagnosed by movement disorder specialists. After the initial diagnosis, patients were followed for at least 5 years to exclude diagnoses other than definite ET. Patients showing clinical signs or imaging abnormalities not compatible with a diagnosis of ET, such as progressive cerebellar dysfunction, parkinsonism, dystonia, or MRI abnormalities, were excluded. A total of 64 Austrian patients were recruited by movement disorder specialists from the movement disorder clinic at the Department of Neurology, Innsbruck Medical University.
Genotyping and quality control.
Samples with >10% missing genotypes in the single nucleotide polymorphism (SNP) sets analyzed were excluded in all analyses in the first and second stage and in the LINGO1 analysis. The first-stage genotyping of this study was performed using the Affymetrix Genome-Wide SNP Array 6.0 (1000K) (Affymetrix, Santa Clara, CA). Affymetrix processed more than 99% of the samples. Genotypes were called using the Affymetrix Birdseed v2 algorithm. We used visual inspection of cluster plots to exclude false-positive associations among the highest ranking SNPs typed in the first stage. As part of the comprehensive quality control (QC) of the first stage, only markers with a call rate ≥95% in patients and control subjects, with a minor allele frequency (MAF) ≥0.05 in control subjects, and with p > 0.0001 for Hardy-Weinberg equilibrium were considered for association analysis. We investigated the first-stage sample for population stratification as well as for identical or related individuals based on pairwise identity-by-state distance and calculated the genomic inflation factor with the use of PLINK (version 1.04; http://pngu.mgh.harvard.edu/∼purcell/plink/) and of R (version 2.10.0; http://www.r-project.org/).15,16 Quantile-quantile plots were created using the qq.chisq function of the R package snpMatrix (version 1.0.33, http://www-gene.cimr.cam.ac.uk/clayton/software/).17 Genotyping of samples for the second verification step and subsequent fine mapping was performed with SNPlex (Applied Biosystems, Foster City, CA), MassARRAY iPLEX platform (Sequenom Inc, San Diego, CA) and TaqMan technology (Applied Biosystems) following standard protocols. QC criteria for second-stage genotyping were minimal genotype call rate >95% for each SNP, MAF ≥0.05, and p > 0.01 for Hardy-Weinberg equilibrium. Multiple testing correction for the second-stage SNPs was performed using the mperm 1000 permutation command of PLINK. SNPs for subsequent fine mapping were selected from the European HapMap CEU reference data (http://hapmap.org/).
Statistical analysis.
All association analyses were performed using R (version 2.10.0)16 and PLINK (version 1.04).15 Genotype-phenotype associations were assessed for statistical significance using a χ2 test. In addition to the comparison of the allelic frequencies of patients and control subjects, association was also calculated using genotype frequencies as well as recessive and dominant genetic models. The different models did not differ significantly from the allelic model, and therefore these models were disregarded.
RESULTS
To identify additional ET susceptibility loci, we performed a two-stage GWAS. The demographical data for all samples are found in table 1. The first-stage sample had 80% power to detect a disease-associated variant with an odds ratio (OR) of ≥1.6 at the 5% significance level, assuming a MAF of ≥0.1 in healthy control subjects (figure e-1 on the Neurology® Web site at www.neurology.org). After extensive QC, 620,077 SNPs were tested in the exploratory first stage. The estimated genomic inflation factor of λ = 1.077 indicated very low population substructure in the sample, obviating the need for correction. In the first stage, no marker reached genome-wide significance (p ≤ 8 × 10−8). An overview of the GWAS p values in form of a Manhattan plot is given in figure e-2. The quantile-quantile plot (figure e-3) showed only a slight excess of signal. Visual inspection of the cluster plots of all 535 SNPs with p < 10−4 led to removal of 462 SNPs with bad genotyping quality. The first-stage association results for all SNPs p < 10−4,which passed the visual inspection of the cluster plots, are shown in table e-1. All remaining SNPs with p < 10−5 (n = 17) as well as SNPs with p < 10−4 (n = 9), which showed support of at least 1 additional correlated SNP with p < 10−4 and 4 SNPs located in functional candidate genes (SLC1A2, GARB1, KCNU1, and SMOC2) with p < 10−4 were selected for second-stage genotyping in an independent verification sample of 554 patients and 609 control subjects of European origin. Four SNPs were excluded from the analysis because of insufficient genotyping quality, leaving 26 SNPs for the second-stage analysis (table e-2). Two SNPs showed nominally significant p values. Only one of these variants (rs3794087), located in intron 4 of the SLC1A2 gene (OMIM 600300) on chromosome 11p13, passed the correction for multiple testing and reached a corrected allelic p = 0.037 in the second-stage sample (table e-2). SNP rs3794087 yielded a nominal p = 1.16 × 10−7 in the combined first- and second-stage sample (table 2). In the subgroup analysis of the most stringent phenotype of definite ET (n = 658), the SNP rs3794087 achieved a genome-wide significant p = 3.44 × 10−10 in the combined sample (table 2). The sample sizes are too small for a meaningful further subgroup analyses (e.g., patients with a positive family history or patients with a positive alcohol response).
Table 1.
Demographic data for the samples used in the first and second stage of the GWAS

Abbreviations: ET = essential tremor; FH = family history; GWAS = genome wide association study; NA = not applicable; TÜ a Tübingen.
All demographic data refer to study samples before genotyping quality control was applied.
Table 2.
Association results for rs3794087
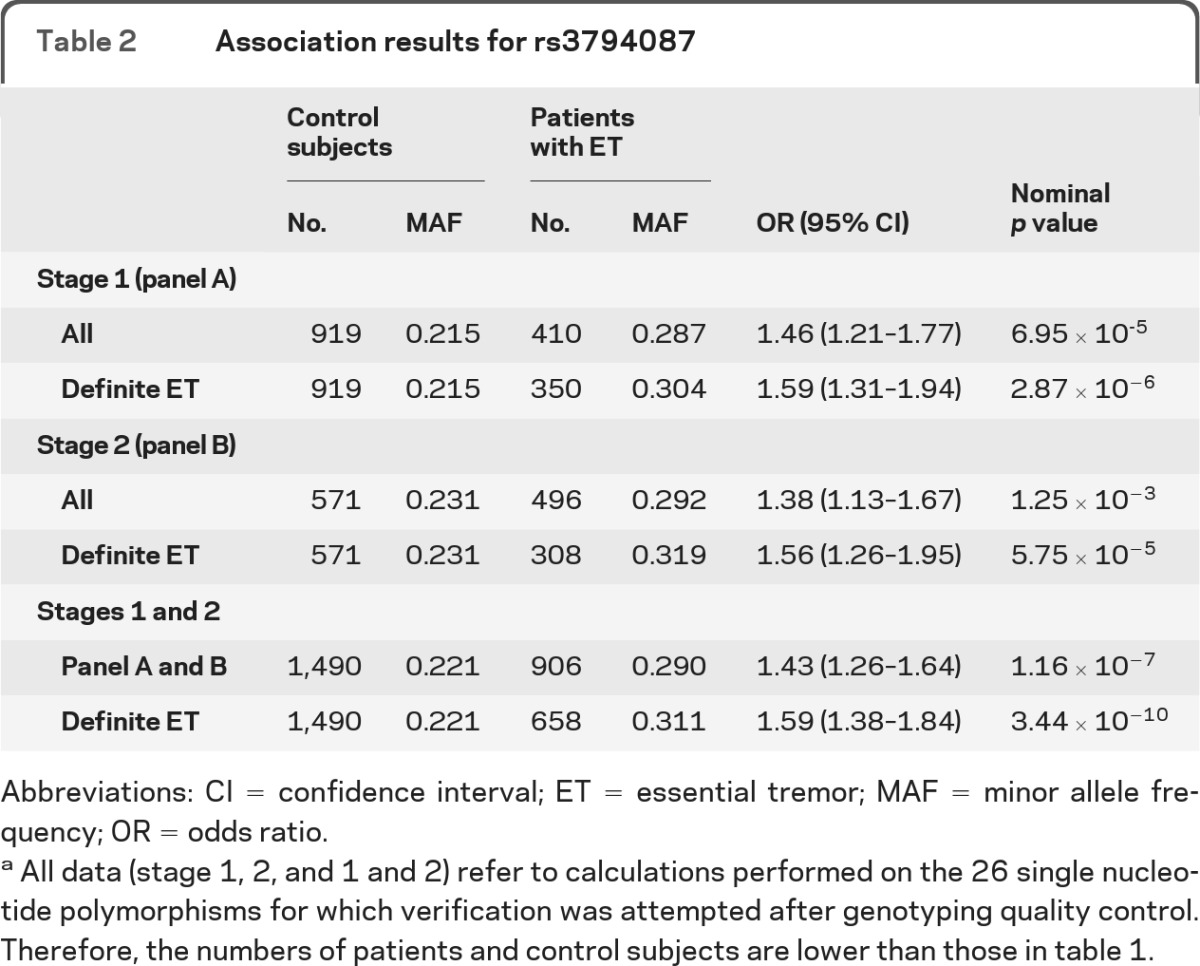
Abbreviations: CI = confidence interval; ET = essential tremor; MAF = minor allele frequency; OR = odds ratio.
All data (stage 1, 2, and 1 and 2) refer to calculations performed on the 26 single nucleotide polymorphisms for which verification was attempted after genotyping quality control. Therefore, the numbers of patients and control subjects are lower than those in table 1.
For fine mapping, we tested 7 SNPs in high linkage disequilibrium with rs3794087 (r2 > 0.8) and in addition 8 nonsynonymous coding SNPs located in the SLC2A1 gene (figure 1). Seven of these 15 SNPs tested for fine mapping were monomorphic in the study population. None of the remaining 8 SNPs showed a stronger association with ET than rs3794087. The genetic region of the SLC1A2 gene is characterized by a weak linkage disequilibrium structure (figure 1).
Figure 1. Disease associations and linkage disequilibrium (LD) structure of the genomic region containing SLC1A2.

(A) The nominal allele-based p values (−log10 P) of all genotyped single nucleotide polymorphisms (SNPs) were plotted against their physical position (in megabases) on chromosome 11. Red rhombi, first-stage (genome-wide scan) SNPs; green squares, finemapping SNPs; triangles, results for SNP rs3794087; black triangle, first stage; blue triangle, second stage; lilac triangle, combined first and second stage; yellow triangle, combined first and second stage in the subsample with definite essential tremor (ET). (B) Schematic representation of SLC1A2 gene structure. (C) LD plot of the locus based on the measure r2 in CEU HapMap (http://hapmap.org/).24
We also investigated 3 published LINGO1 SNPs.6,8 These SNPs were not included on the Affymetrix SNP Array 6.0, and no other SNP in the region of LINGO1 present on the array showed a significant association in the first GWAS stage. We were just able to replicate the association of the LINGO1 SNP rs9652490 with ET in the combined sample of the first and second stage with p = 0.0135 and OR = 1.20. The other 2 SNPs (rs8030859 and rs11856808) showed no significant association (data not shown). Recently, we replicated the association of variants in the LINGO1 gene in a German sample of ET patients, which partly shared subjects with the first and second samples used in this GWAS.8
DISCUSSION
The present GWAS revealed an association between the SNP rs3794087 in intron 4 of the SLC1A2 gene and ET. The estimated OR of ∼1.5 is in the typical range of ORs found in GWAS. However, it should be kept in mind that it does not allow any risk prediction on an individual patient basis. The function of this noncoding SNP is unknown. Independent replication studies are needed, especially because rs3794087 did not achieve genome-wide significance in the first-stage analysis. However, it is interesting to note that the association of rs3794087 was consistently stronger if only patients with the highest diagnostic certainty (definite ET) were included in the analysis and reached genome-wide significance in the smaller subgroup of 658 patients with definite ET after genotyping QC in the combined first- and second-stage samples. The SLC1A2 gene encodes the predominant glutamate reuptake transporter (excitatory amino acid transporter 2), which removes glutamate from the synaptic cleft. Glutamate is the major excitatory neurotransmitter in the CNS. Prolonged elevated glutamate concentrations in the synaptic cleft are neurotoxic (so-called “excitotoxicity”) and thought to contribute to neurodegenerative disorders such as Alzheimer disease and amyotrophic lateral sclerosis. Based on the acute harmaline animal model of ET, a widely recognized hypothesis assumes that the abnormal oscillations causing the tremor are generated in the inferior olive and transmitted through the cerebellum to other brain areas.18 The inferior olivary neurons but not the cerebellar Purkinje neurons that are involved contain abundant NMDA-type glutamate receptors.19 Harmaline-induced tremor is blocked by NMDA receptor antagonists, suggesting that NMDA receptor activation is necessary to generate the abnormal olivary oscillation.20 In addition, sustained glutamatergic excitation of climbing fiber terminals in the cerebellum might be toxic to Purkinje neurons.19 Interestingly, SLC1A2 is strongly expressed in the inferior olive but not in other structures of the brain stem.21 Ethanol, which acutely alleviates ET in many patients significantly increases SLC1A2 expression and glutamate uptake activity.22 Furthermore, elevated glutamate concentrations are present in the CSF and serum of patients with ET.23 For further progress in the genetics of sporadic ET, 2 prerequisites have to be met. First, sample sizes have to be increased, either in subsequent studies or by pooling data of current and future studies. Second, detailed clinical assessment using the same diagnostic criteria in all studies as well as the assessment of intermediary phenotypes, first and foremost alcohol responsiveness, might lead to genetically more homogeneous ET samples. A standardized test to assess alcohol responsiveness is currently under development in our group. In conclusion, this is the first report showing an association of the glutamatergic amino acid transporter SLC1A2 and ET.
Supplementary Material
ACKNOWLEDGMENT
The authors thank all individuals who participated in this study.
GLOSSARY
- ET
essential tremor
- GWAS
genome-wide association study
- OR
odds ratio
- QC
quality control
- SNP
single nucleotide polymorphism
- TRIG
Tremor Investigation Group
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
G.D., D.L., S.K., S.T., M.N., and G.K. developed the idea and study concept and study design. S.T. performed SNP selection, genotyping, data analysis, and prepared the figures and tables. S.K. and G.K. supervised data analysis and coordinated project contributions. D.L., S.K., G.D., K.C., C.P., F.P., S.P., and F.H. coordinated the recruitment and collected and collected phenotype data of the German and Danish sample from Kiel. S.A. helped with data analyses. K.C. provided the Danish samples. M.N. helped with the statistical analyses and interpretation of the results. A.-C.H., S.H., W.P., F.A., T.G., and L.S. provided the verification samples and the respective phenotypes from Tübingen and Austria. A.N. and S.S. provided the German controls for the verification sample. S.T., S.A., G.K., M.N. conducted statistical analysis. S.T., S.K., and G.K. drafted the manuscript. All other authors edited the manuscript.
DISCLOSURE
Dr. Thier reports no disclosures. Dr. Lorenz has received research support from the German Research Council (DFG). Dr. Nothnagel serves on the editorial board of Human Genomics; is listed as author on a patent re: Genetic markers for prognosis of fast or slow onset of progression to AIDS in female HIV-1 infected patients; and receives research support from BMBF (the Federal Ministry of Education and Research) and DFG. Dr. Poremba and Dr. Papengut report no disclosures. Dr. Appenzeller receives research support from the Christian-Albrechts-University Kiel. Dr. Paschen has received speaker honoraria from Medtronic, Inc. Dr. Hofschulte, Dr. Hussl, and Dr. Hering report no disclosures. Dr. Poewe has received consultancy and lecture fees from AstraZeneca, Teva Pharmaceutical Industries Ltd., Novartis, GlaxoSmithKline, Boehringer Ingelheim, UCB, Orion Corporation, Abbott, and Merck Serono; receives publishing royalties for Therapeutics of Parkinson's Disease and Other Movement Disorders (Wiley, 2008) and Non-motor Symptoms of Parkinson's Disease (Oxford University Press, 2009); and has received research support from the Michael J. Fox Foundation. Dr. Asmus has received funding for travel, speaker honoraria, and research support from Merz Pharmaceuticals, LLC, Ipsen, and Allergan, Inc. and is an employee of Merz Pharmaceuticals. Prof. Gasser serves on the editorial boards of Parkinsonism and Related Disorders, Journal of Parkinson's Disease, and Neurogenetics; holds a patent re: KASPP (LRRK2) gene, its production and use for the detection and treatment of neurodegenerative diseases; serves as a consultant for Cephalon, Inc. and Merck Serono; serves on speaker's bureaus of Novartis, Merck Serono, SCHWARZ PHARMA, Boehringer Ingelheim, and Valeant Pharmaceuticals International; and receives research support from Novartis, the European Union, BMBF, and the Helmholtz Association. Dr. Schöls has received research support from Santhera Pharmaceuticals, DFG, BMBF, the EU, and the HSP-Selbsthilfegruppe Deutschland eV. Dr. Christensen serves on editorial advisory boards for Demographic Research, Aging Cell Twin Research & Human Genetics, and Frontiers in Genetics of Aging and receives research support from the NIH (NIA, NIDCR), March of Dimes Birth Defects Foundation, the CDC, and the EU, and LIFESPAN. Dr. Nebel receives research support from the German Research Council (DFG) and the EU. Dr. Schreiber receives research support from NGFN and DFG. Dr. Klebe has received research support from the German Research Council (DFG). Dr. Deuschl serves on scientific advisory boards for Teva Pharmaceutical Industries Ltd. and Medtronic, Inc.; has received funding for travel or speaker honoraria from Orion Corporation, Teva Pharmaceutical Industries Ltd., Lundbeck, Inc., and Pfizer Inc.; receives publishing royalties for Referenzreihe Neurologie (Thieme, 2004−2011); receives research support from Medtronic, Inc., the German Research Foundation (DFG), the German Ministry of Education and Research; and serves as an expert witness for Merck Serono. Dr. Kuhlenbäumer receives research support from the German Research Council (DFG) and the Christian-Albrechts-University Kiel. Go to Neurology.org for full disclosures.
REFERENCES
- 1. Deuschl G, Bain P, Brin M. Consensus statement of the Movement Disorder Society on Tremor: Ad Hoc Scientific Committee. Mov Disord 1998; 13 (suppl 3): 2− 23 [DOI] [PubMed] [Google Scholar]
- 2. Boecker H, Wills AJ, Ceballos-Baumann A, et al. The effect of ethanol on alcohol-responsive essential tremor: a positron emission tomography study. Ann Neurol 1996; 39: 650– 658 [DOI] [PubMed] [Google Scholar]
- 3. Deuschl G. Differential diagnosis of tremor. J Neural Transm Suppl 1999; 56: 211– 220 [DOI] [PubMed] [Google Scholar]
- 4. Lorenz D, Frederiksen H, Moises H, Kopper F, Deuschl G, Christensen K. High concordance for essential tremor in monozygotic twins of old age. Neurology 2004; 62: 208– 211 [DOI] [PubMed] [Google Scholar]
- 5. Deuschl G, Elble R. Essential tremor: neurodegenerative or nondegenerative disease towards a working definition of ET. Mov Disord 2009; 24: 2033– 2041 [DOI] [PubMed] [Google Scholar]
- 6. Stefansson H, Steinberg S, Petursson H, et al. Variant in the sequence of the LINGO1 gene confers risk of essential tremor. Nat Genet 2009; 41: 277– 279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tan EK, Teo YY, Prakash KM, et al. LINGO1 variant increases risk of familial essential tremor. Neurology 2009; 73: 1161– 1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thier S, Lorenz D, Nothnagel M, et al. LINGO1 polymorphisms are associated with essential tremor in Europeans. Mov Disord 2010; 25: 709– 715 [DOI] [PubMed] [Google Scholar]
- 9. Vilarino-Guell C, Wider C, Ross OA, et al. LINGO1 and LINGO2 variants are associated with essential tremor and Parkinson disease. Neurogenetics 2010; 11: 401− 408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lao O, Lu TT, Nothnagel M, et al. Correlation between genetic and geographic structure in Europe. Curr Biol 2008; 18: 1241– 1248 [DOI] [PubMed] [Google Scholar]
- 11. Steffens M, Lamina C, Illig T, et al. SNP-based analysis of genetic substructure in the German population. Hum Hered 2006; 62: 20– 29 [DOI] [PubMed] [Google Scholar]
- 12. Lorenz D, Papengut F, Frederiksen H, et al. Evaluation of a screening instrument for essential tremor. Mov Disord 2008; 23: 1006– 1012 [DOI] [PubMed] [Google Scholar]
- 13. Krawczak M, Nikolaus S, von Eberstein H, Croucher PJ, El Mokhtari NE, Schreiber S. PopGen: population-based recruitment of patients and controls for the analysis of complex genotype-phenotype relationships. Community Genet 2006; 9: 55– 61 [DOI] [PubMed] [Google Scholar]
- 14. Nebel A, Croucher PJ, Stiegeler R, Nikolaus S, Krawczak M, Schreiber S. No association between microsomal triglyceride transfer protein (MTP) haplotype and longevity in humans. Proc Natl Acad Sci USA 2005; 102: 7906– 7909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559– 575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. R: A language and environment for statistical computing. [program]. Vienna, Austria: R Project for Statistical Computing; 2008 [Google Scholar]
- 17. Clayton D, Leung HT. An R package for analysis of whole-genome association studies. Hum Hered 2007; 64: 45– 51 [DOI] [PubMed] [Google Scholar]
- 18. Elble RJ. Central mechanisms of tremor. J Clin Neurophysiol 1996; 13: 133– 144 [DOI] [PubMed] [Google Scholar]
- 19. O'Hearn E, Molliver ME. Administration of a non-NMDA antagonist, GYKI 52466, increases excitotoxic Purkinje cell degeneration caused by ibogaine. Neuroscience 2004; 127: 373– 383 [DOI] [PubMed] [Google Scholar]
- 20. Du W, Aloyo VJ, Harvey JA. Harmaline competitively inhibits [3H]MK-801 binding to the NMDA receptor in rabbit brain. Brain Res 1997; 770: 26– 29 [DOI] [PubMed] [Google Scholar]
- 21. Berger UV, Hediger MA. Comparative analysis of glutamate transporter expression in rat brain using differential double in situ hybridization. Anat Embryol (Berl) 1998; 198: 13– 30 [DOI] [PubMed] [Google Scholar]
- 22. Wu J, Lee MR, Choi S, Kim T, Choi DS. ENT1 regulates ethanol-sensitive EAAT2 expression and function in astrocytes. Alcohol Clin Exp Res 2010; 34: 1110– 1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mally J, Baranyi M, Vizi ES. Change in the concentrations of amino acids in CSF and serum of patients with essential tremor. J Neural Transm 1996; 103: 555– 560 [DOI] [PubMed] [Google Scholar]
- 24. Altshuler DM, Gibbs RA, Peltonen L, et al. Integrating common and rare genetic variation in diverse human populations. Nature 2010; 467: 52– 58 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.