

Abstract
Multiprotein complexes and other protein-protein interactions play important roles in virtually all cellular processes. Analysis of co-immunoprecipitation of protein complexes by flow cytometry (IP-FCM, or “the fly-p” method) provides a sensitive means to measure these interactions in the native/non-denatured state. First, immunoprecipitating antibodies are covalently coupled to polystyrene latex beads whose low autofluorescence is compatible with flow cytometry. These antibody-coupled beads are used to immunoprecipitate a specific protein (primary analyte) present in cell lysates. Finally, the protein complexes associated with the beads are probed with fluorochrome-conjugated antibodies specific for interaction partners, or secondary analytes, that may be associated with the primary analyte. The use of quantitative FACS methodology can allow the semi-quantitative fluorescence data generated to be converted into estimated numbers of co-associated molecules on the beads. The method represents a robust technique to assess native protein-protein interactions without requiring genetic engineering or large sample sizes.
Unit Introduction
Analysis of co-immunoprecipitated proteins by flow cytometry (IP-FCM) provides a highly sensitive means of studying multiprotein complexes (MPC) and other protein-protein interactions (PPI). In casual laboratory jargon, we refer to this method as “the Fly-p”. First, immunoprecipitation (IP) antibodies (Ab) are covalently coupled to carboxylate-modified polystyrene latex (CML) beads (Basic Protocol 1). Next, the IP is performed by incubating cell lysates with the IP Ab-CML beads (Basic Protocol 2). The primary analyte is the protein directly bound by the IP Ab, while secondary analytes, other proteins that co-immunoprecipitate with the first, are measured with fluorochrome-conjugated probe Abs (Figure 1). A quantitative fluorescent bead set can provide a standard curve to translate experimental fluorescence values into known numbers of fluorchromes, allowing an estimation of the number of molecules in the complexes (Support Protocol 1). The instructions that follow outline this procedure using 20 × 106 primary T lymphocytes to generate IP samples sufficient for use with up to 10 different probes.
Figure 1.
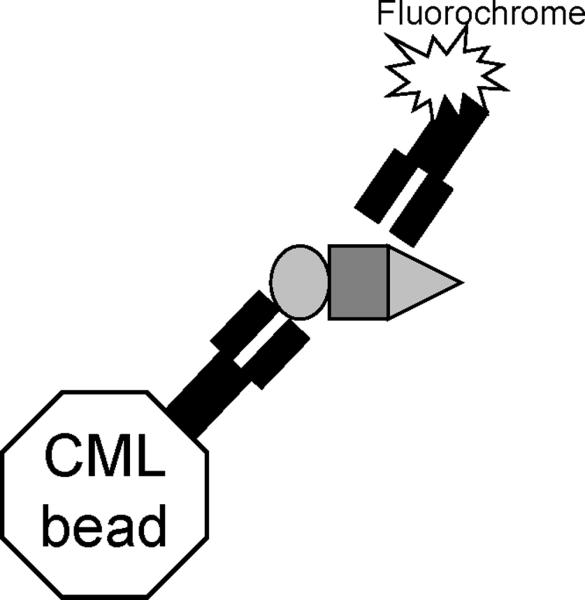
Principle of IP-FCM (“the fly-p”). Immunoprecipitation Abs are covalently coupled to CML polystyrene latex beads. When these beads are incubated with cell lysates, the protein for which the IP Ab is specific (the primary analyte, oval) can bind to the beads together with co-associated proteins (secondary analytes, rectangle and triangle). The primary and secondary analytes on the beads can be probed with fluorochrome-conjugated Abs and analyzed by flow cytometry.
Basic Protocol 1
Covalent coupling of Ab to CML beads
Introduction
A batch of IP beads is prepared by covalently coupling primary amino groups of a specific Ab to carboxyl groups on CML beads. At the end point of the assay during IP detection by FCM, the number of beads stained per tube can vary between 2.5 ×103 – 2.5 ×105. We include here conditions to make a batch starting with 18 × 106 beads, with an expected yield of approximately 12 × 106 beads post-coupling. Depending on the IP conditions, this batch will be enough for between 50–5000 FCM samples. Scale up the coupling reaction as needed.
Materials list
Hemacytometer (Neubauer chamber) for bead counting
Microscope capable of 100× magnification for bead counting
CML beads
PBS (see recipe)
MES coupling buffer (see recipe)
EDAC-MES solution (see recipe)
Antibody for IP, in PBS (see recipe)
Vibrating shaker, or Thermomixer (Eppendorf product 5436)
QBS buffer (see recipe)
Steps and Annotations
- Empirically determine the concentration of beads from the purchased stock. Insure that the beads are well suspended before diluting 1:10,000 in PBS and counting with a hemacytometer under a microscope. Alter the dilution as necessary in order to count 30–300 beads to achieve an accurate count.Alternatively, the beads can be counted using a Coulter Counter.
Pipette 18 × 106 beads (~30 μL of our lab's current stock bead suspension) into a 1.5-mL microcentrifuge tube.
Wash the beads 2–3× in MES coupling buffer. The wash volume should be 0.5–1.5-mL, centrifuging at 15,000g for 3 minutes in between washes (room temperature).
Resuspend the beads in 50 μL MES coupling buffer.
Activate the carboxyl groups on the beads by adding 20 μL of freshly prepared EDAC-MES.
- Mix gently for 15 min (room temperature).This is best achieved by manually pipetting up and down throughout the period.
Wash the activated beads 2–3× in 0.5–1.0 mL PBS, centrifuging at 15,000g for 3 minutes in between washes (room temperature).
Resuspend the activated beads in 50 μL PBS.
- Add 50 μL of the AbStock Ab should be ≥ 0.2 mg/mL, and must be in PBS.
- Mix for 3–4 hours (room temperature).We prefer to do this by placing the tube horizontally on a vibrating shaker. Shake sufficiently to prevent settling of the beads on the bottom of the tube.Alternatively, the tube can be shaken in the upright position in a thermomixer.
- Wash the IP beads 2–3× in 0.5–1.0 mL PBS, centrifuging at 15,000g for 3 minutes in between washes (room temperature).It is critical to discard all the supernatant volume possible between each wash in this step. Otherwise, soluble unconjugated Ab may be present in the IP bead prep. Such soluble Ab can bind the target antigen when the IP is performed, inhibiting the capture of the target antigen on the beads.
Resuspend the IP beads in 100 μL QBS buffer, and store overnight (or indefinitely) at 4°C.
- Count the IP beads as in step 1, and document their concentration.This must be done for each preparative batch, since the number of beads used per IP experiment is a critical parameter that must be precisely controlled.
Basic Protocol 2
Immunoprecipitation from cell lysates analyzed by flow cytometry (IP-FCM)
Introduction
Methods to harvest and prepare cells for lysis vary greatly depending on the cell type, and will not be covered here. In addition, optimal detergent and lysis conditions must be determined empirically, and can vary depending on the protein-protein interactions under study. Here, we follow the common practice of using 1% non-ionic detergent and isotonic lysis buffer to generate post-nuclear lysates containing native protein complexes. The goal is to completely solubilize the native cellular proteins while keeping the lysate as concentrated as possible. A general rule of thumb is to lyse 200 × 106 “small” cells (such as lymphocytes) per mL, or 50 × 106 “big” cells (such as macrophages or tumor cells) per mL of lysis buffer. After cell lysis, the IP is performed by adding 2.5 × 106 IP beads per mL of post-nuclear lysate. Finally, the IP beads are stained with fluorochrome-conjugated Abs and FCM analysis is performed. This protocol is outlined with the example of 20 × 106 primary lymphocytes lysed in 100 μL. Figure 2 shows an example of IP-FCM data obtained following this protocol.
Figure 2.

An example of data from IP-FCM. The method was performed according to the described protocol by lysing 50 × 106 human peripheral blood mononuclear cells in 1% Digitonin lysis buffer. The T cell antigen receptor (TCR)/CD3 multiprotein complex was immunoprecipitated with CML beads coupled with an anti-CD3ε Ab (clone APA1/1) specific for an intracellular (i.c.) epitope on the cytoplasmic tail. Flow cytometric analysis was subsequently performed. Identified by its Forward vs. Side Scatter profile, the homogeneous bead population is gated (top panel), and its fluorescence is displayed on a log-scale histogram (bottom panel). The histograms from several parallel samples are overlaid for comparison. The irrelevant Ig-PE control (anti-mouse CD11b) displayed low background staining (shaded gray histogram). The TCR αβ heterodimer was specifically co-immunoprecipitated in the predicted stoichiometric ratio: approximately 2-fold greater CD3ε (probe clone SK7, which binds an extracellular (e.c.) domain) was detected over TCRαβ (probe clone T10B9.1A-31). The mode fluorescence intensity (MoFI) was converted to the number of PE molecules (#PE) on the beads by following the instructions outlined in “Quantitative flow cytometry (qFCM)” (Support Protocol 1).
Materials list
Cells for lysis
IP beads (from Basic Protocol 1)
Lysis buffer (see recipe)
1.5-mL microcentrifuge tubes
Vertical rotating wheel
Post-IP wash buffer (see recipe)
FCM staining buffer (see recipe)
96-well 0.2 mL tall-chimney-bottom polypropylene plates (Fisher product 08-408-230)
Antibody probe(s), fluorochrome-conjugated
5-mL FACS tubes
Flow cytometer
Flow cytometry acquisition and analysis software, such as CellQuest (BD) and FlowJo (Treestar)
Steps and Annotations
Post-nuclear lysate preparation and IP
Lyse 20 × 106 primary lymphocytes in 100 μL ice-cold Lysis buffer (with freshly-added protease and phosphatase inhibitors) in a 1.5-mL microcentrifuge tube for 20 min on ice.
To remove nuclei and insoluble cellular debris, centrifuge the lysate at 20,000g for 10 min (4°C). Keep the supernatant as the post-nuclear lysate, and discard the pellet.
Place 2.5 × 105 IP beads in a 1.5-mL microcentrifuge tube. Wash the IP beads in 200 μL Lysis buffer, centrifuging at 15,000g for 3 minutes in between washes (4°C). Briefly keep the washed, pelleted IP beads on ice.
- Resuspend the IP beads in the post-nuclear lysate. The IP has begun.The wash in step 3 can be omitted if the bead aliquot represents a small proportion of the total volume in which the IP will take place. For example, 5 μL of unwashed stock IP beads can be added directly to 100 μL of post-nuclear lysate in order to begin the IP.
- Rock end-over-end if possible, 4hr-overnight (4°C).We prefer to place the tube on a vertical rotating wheel in a cold room, set to sufficient velocity to prevent the beads from settling. The small volume of the post-nuclear lysate usually remains at the bottom of the tube throughout the incubation, but the IP beads do not settle.
Staining the IP beads for FCM
Wash the IP beads 1–3× in 0.2 mL ice-cold Post-IP Wash buffer, centrifuging at 15,000g for 3 minutes in between washes (4°C).
- Wash the IP beads 2–3× in 0.2–1.0 mL ice-cold FCM staining buffer, centrifuging at 15,000g for 3 minutes in between washes (4°C).The number of washes, and whether the FCM staining buffer must vs. must not contain detergent are empirically determined. If the entire procedure including FCM is finished within a few hours, our experience has been that the wash in step 1 can be omitted, and the FCM staining buffer in step 2 may not need detergent, even when transmembrane proteins are being analyzed. It is possible that some detergents may not readily wash away from their hydrophobic interactions with transmembrane proteins over this short time course.
Resuspend the IP beads in 500 μL, and aliquot 2.5 × 104 beads in 50 μL into each of 10 wells of a 96-well chimney-bottom plate. (Alternatively, a separate 1.5-mL microcentrifuge tube or 5-mL FACS tube can be used for each sample to be stained.)
- Add fluorochrome-conjugated Abs to the samples to perform the FCM stain according to the vendor's instructions, and/or according to empirically derived dilutions. Incubate for 40 min (4°C).Aside from negative controls, the probe Abs should be specific for either the primary analyte itself, or for secondary analytes that potentially interact with the primary analyte.An example of a good starting point is 1:100 dilution of 0.2 mg/mL PE-conjugated Ab from BD Pharmingen or eBioscience. Usually, a staining dilution that is satisfactory for a similar number of cells (in the case of surface proteins) will work for the IP beads. Perform titration experiments to optimize.
If working in a 96-well chimney-bottom plate, wash 3× in 0.2 mL FCM staining buffer, centrifuging at 1000g for 5 minutes in between washes (4°C). (If working in microcentrifuge tubes, wash 2× in 0.5–1.0 mL FCM staining buffer, centrifuging at 15,000g for 3 minutes in between washes (4°C)).
Resuspend the IP beads in 0.2 mL FCM buffer per sample, transfer the samples to FACS tubes (or optionally, a 96-well plate compatible with a plate-reading flow cytometer), and acquire the samples by FCM.
At the Flow Cytometer
In most respects, acquiring the IP bead fluorescence data by flow cytometry is no different than acquiring fluorescence data from cells. However, there are a few points in setting up the cytometer to bear in mind.
The IP beads described in this protocol are 3–5 microns in diameter, approximately half the diameter of a quiescent mouse lymphocyte. Therefore, it can be necessary to manually increase the Forward Scatter amp gain and the Side Scatter voltage in order to see the population of bead events on the cytometer.
Make a gate around the small homogenous bead population, visualized on a dot plot showing Forward Scatter vs. Side Scatter. Apply this gate to the data to be visualized on the fluorescence channel(s). In doing this, the data from doublet-beads and non-bead debris is disregarded (see Figure 2A).
- The default collection criterion for many flow cytometers is 10,000 acquisition events. We suggest changing this criterion to 250–2500 gated acquisition events.The staining of the beads normally produces an easily identifiable mode fluorescence intensity (MoFI) when visualized on a histogram (Figure 2). After collecting very few events this pattern is quickly apparent. Sufficient events are only needed for statistical analyses and clear figures.
Select “log mode” for the fluorescence channel(s) to be collected (as is general practice for much of cellular flow cytometry).
If quantitative flow cytometry (qFCM) analysis is desired, we exclusively utilize the fluorescence channel assigned to phycoerythrin (PE)-conjugated Ab probes, and we turn off the other fluorescence channels.
- It is convenient to use unlabeled, unconjugated beads as the first sample in order to set the negative control photomultiplier tube (PMT) voltage. It is also advisable to have a bright positive control for the second sample, such as IP beads stained with fluorochrome-conjugated anti-immunoglobulin (Ig) Abs. If both of these controls can be visualized at the two extremes of the log scale, it is practically sure that the fluorescence from the experimental IP bead samples will be on-scale. Since our experiments use only 1 fluorescent probe per sample, no fluorescence compensation is necessary.In general, we currently tend to stain multiprotein complexes with a single fluorochrome-conjugated Ab per FCM sample, and we examine multiple subunits by staining parallel IP bead samples with Abs specific for the various subunits. An alternative is to stain IP beads for multiple subunits using different fluorochrome-conjugated Abs simultaneously. The former strategy facilitates qFCM when used in conjunction with PE-conjugated Abs, since steric binding problems resulting from multiple probe antibodies are avoided. Also avoided are potential alterations in fluorescence signals by FRET from adjacent fluorochromes, which can occur with common fluorochrome combinations, including FITC/PE and PE/APC.
The flow cytometer is now ready to acquire the IP bead samples.
Support Protocol 1
Quantitative Flow Cytometry (qFCM)
Introduction
Fluorescence data is often presented on a relative scale with arbitrary units because it is inherently semi-quantitative. However, a standard curve can be generated from commercially available fluorescent beads, where fluorescence values correspond to known numbers of fluorochromes. If the standard curve and the experimental data from IP-FCM beads are acquired with the same flow cytometer PMT voltage, compensation, and other settings, then the relative fluorescence values from the IP-FCM data can be translated into numbers of fluorochromes per IP bead. Figure 3 provides an example of a PE calibration curve and equation. PE is the fluorochrome of choice for qFCM analysis, and it is important to use PE-conjugated Abs where the PE:Ab ratio is known. The conjugation chemistry is normally set up with a 1:1 PE:Ab molar ratio, but the ratio of the final product can be most easily confirmed by: a) calling the vendor's technical department and asking what the PE:Ab ratio is for a particular batch of Ab probe; b) running the PE-Ab probe over a size exclusion column by FPLC and collecting the fraction corresponding to the molecular weight expected for 1:1 conjugation; c) paying extra for a vendor to provide a guaranteed 1:1 conjugation reagent.
Figure 3.

The fluorescence standard curve for qFCM. This is an example of a PE calibration curve using RCP-30-5A beads from Spherotech, Inc.. (A) During data acquisition, the Forward Scatter amp gain and Side Scatter voltage are adjusted to get the beads on-scale. All other flow cytometry parameters and settings must remain identical to those used during the acquisition of experimental samples. For data analysis, a gate is drawn to restrict fluorescence analysis to that of singlet beads. (B) The series of unimodal fluorescence peaks in the histogram represent known numbers of mean equivalent soluble PE molecules (MEPE). By inputting the fluorescence intensities into the vendor-provided spreadsheet (not shown), experimental relative fluorescence values can be converted into numbers of PE molecules. (C) The standard curve and its equation can be generated manually, by plotting the RCP-30-5A relative fluorescence values against the number of PE molecules associated with each peak. (D) Various software programs can extrapolate an equation for the calibration curve, which can then be used to input experimental fluorescence data and solve for the number of PE molecules. The equation displayed here was calculated using Prism GraphPad.
Materials list
Rainbow calibration particles (Spherotech product RCP-30-5A)
PBS (see recipe)
Flow cytometer
Flow cytometry acquisition and analysis software, such as CellQuest (BD) and FlowJo (Treestar)
Spreadsheet software (Microsoft Excel)
Curve generation and analysis software (such as Prism GraphPad)
Steps and Annotations
Acquisition of fluorescence standard curve
After the experimental samples have been acquired by FCM, maintain the cytometer settings unaltered and return to set-up mode, so that new data files will temporarily not be acquired.
- Run a sample of Spherotech beads RCP-30-5A (2 drops diluted in 300 °L PBS).Because these beads are smaller than the IP-FCM beads, it may be necessary to increase the Forward Scatter amp gain and the Side Scatter voltage to get the beads on-scale.
- Make a gate around the new small homogenous bead population, visualized on a dot plot showing Forward vs. Side Scatter (Figure 3A). Apply this gate to the data to be visualized on the PE fluorescence channel (Figure 3B). In doing this, the fluorescence from doublet-beads is disregarded.It is absolutely vital that all parameters of the flow cytometer's settings, except the Forward and Side Scatter, be identical between the RCP-30-5A beads and the experimental samples. Only this way is it valid to apply the experimental fluorescence data to the standard curve.
A series of clear, single-mode peaks should now appear in the PE fluorescence channel (Figure 3B).
Set the acquisition collection criterion to 10,000 gated events.
Remove the set-up mode selection, so that new data files can once again be acquired.
Acquire the RCP-30-5A sample.
qFCM data analysis
Each fluorescence peak from the RCP-30-5A sample represents a defined number of mean equivalent soluble PE molecules (MEPE). By plotting the relative fluorescence against the MEPE for each peak, the qFCM standard curve is generated and an equation for the curve can be calculated. Upon request, Spherotech, Inc., provides to users an Excel spreadsheet which carries out the standard curve set-up and converts user-input experimental fluorescence values into MEPE. Alternatively, the equation for the calibration curve can be determined using other software. Figure 3C–D shows a standard curve and calculated equation for the curve using Prism GraphPad. In the end, a quantitative estimate for the number of PE molecules associated with each IP-FCM sample is obtained, as reported in Figure 2.
Reagents and Solutions
Antibody (Ab) for IP
≥0.2 mg/mL (ideally ≥1 mg/mL) in PBS (see recipe)
optional addition of Sodium azide stock solution (see recipe; 1× (0.02%))
Store at 4°C (stability varies but is commonly 6 months)
Antibody (Ab) probes, fluorochrome-conjugated
≥0.2 mg/mL or as supplied by manufacturer
CML (carboxylate-modified latex) beads
~5 °m surfactant-free white carboxylate-modified polystyrene latex beads
Interfacial Dynamics Corporation, Product # 2-5000 or 2-6000
Store at 4°C (stable for at least 3 years)
EDAC (1-Ethyl-3-(3-dimethylaminopropl) carbodiimide HCl)
Pierce product 22980 or Sigma product E-6383
Store powder at −20°C (stable 6 months)
EDAC-MES solution
50 mg EDAC
1 mL MES coupling buffer (see recipe)
Always make fresh from EDAC powder stored at −20°C.
FCM (Flow Cytometry) staining buffer
50 mL Tris stock solution (1M Tris, pH 7.4 in dH20) (50 mM final)
20 mL NaCl stock solution (5M NaCl in dH20) (100 mM final)
25 mL Fetal Bovine Serum (FBS) (5% v/v final)
2 mL Sodium azide stock solution (see recipe; 1×, 0.02% final)
dH20 to 1L
Store at 4°C (stable for at least 6 months)
Optional ingredients
1× Phosphatase Inhibitors (add fresh from stock)
0–1% Detergent (the same one chosen for use in lysis buffer)
If phosphorylated proteins are of interest, be sure to add phosphatase inhibitors to the FCM staining buffer. Additionally, FBS (5%) can be replaced by BSA (1%) so that the blocking agent is free of phosphorylated proteins. For example, this can help if the common Ab cone 4G10 (anti-phosphotyrosine) is being used as a probe.
The addition of detergent to the FCM staining buffer can either help or hinder the detection of proteins by IP-FCM. This must be determined empirically. On the first attempt, try either adding 0.1–0.2% detergent, or excluding detergent from this buffer altogether.
Lysis buffer (non-ionic)
Core lysis buffer
50 mM Tris (diluted from 1M Tris, pH 7.4 in dH20)
150 mM NaCl (diluted from 5M NaCl in dH20)
1× Protease Inhibitors
Always add protease inhibitors fresh from stock.
1% nonionic detergent
such as Triton X-100, or NP-40, or Digitonin, etc.
The detergent used in a particular experiment must be determined empirically for the protein-protein interactions under study. However, for many protein complexes, published articles can provide an excellent starting point as to which detergent(s) is likely to best preserve the complexes of interest.
Digitonin must be prepared fresh from a 2% solution in dH2O. Boil the 2% solution until it becomes clear, and then cool the solution at room temperature or on ice. The 2% stock is good for up to 3 days when stored at 4°C.
Optional ingredients
1× Phosphatase Inhibitors (add fresh from stock)
0.5–5% Fetal Bovine Serum (FBS)
If phosphorylated proteins are of interest, be sure to add phosphatase inhibitors to the core lysis buffer.
For lysis of extremely low numbers of cells, pre-block the tube in which the lysis will take place with non-specific protein in order to minimize the amount of cellular proteins that stick to the tube and are lost from analysis. First coat the bottom of a tube with FBS and incubate 90 min (37°C). Then discard the FBS and place the tube on ice. Next add FBS to the core lysis buffer and proceed with cell lysis.
MES coupling buffer, pH 6.0
0.9762 g of 2-[N-Morpholino] ethanesulfonic acid (MES, Sigma product M-5287) (50 mM final)
0.2 mL of 0.5M EDTA stock solution (1 mM final)
dH20 to 100 mL
Store at room temperature (stable at least 1 year)
Phosphatase inhibitor stock solutions, 100×
100× Sodium orthovanadate
100 mg Sodium orthovanadate (200 mM final)
2.7 mL dH20
After adjusting to pH 10 with NaOH or HCl, the solution may turn yellow. Boil until the solution turns colorless, and then cool the solution to room temperature. Readjust the pH to 10 and repeat the process until the room temperature solution remains colorless.
Store in 50 μL aliquots (−20°C, stable at least 6 months).
100× Sodium fluoride
50 mg Sodium fluoride (100 mM final)
11.9 mL dH20
Store at room temperature or in 50 μL frozen aliquots (−20°C, stable at least 6 months).
Phosphate buffered saline solution, pH 7.4 (PBS)
Make as 10-fold dilution in dH20 of the following 10× stock solution:
16g KCl (final 10× = 27 mM; final 1× = 2.7 mM)
19.2g KH2PO4 (final 10× = 17.6 mM; final 1× = 1.76 mM)
640g NaCl (final 10× = 1.37 M; final 1× = 137 mM)
115.2g Na2HPO4 (final 10× = 53.7 mM; final 1× = 5.37 mM)
dH20 to 8L
Post-IP Wash buffer
50 mM Tris (diluted from 1M Tris, pH 7.4 stock solution in dH20)
150 mM NaCl stock solution (5M NaCl in dH20)
1× Protease Inhibitors
Always add protease inhibitors fresh from stock.
Optional ingredients
0–1% Detergent (the same one chosen for use in lysis buffer)
1× Phosphatase Inhibitors (add fresh from stock)
If phosphorylated proteins are of interest, be sure to add phosphatase inhibitors to the post-IP wash buffer.
Protease inhibitor stock solutions, 100×
100× general protease inhibitor cocktail
Dissolve protease inhibitor general use cocktail (Sigma, product P2714: contains AEBSF, Bestatin, Aprotinin, EDTA, E-64, and Leupeptin) in 1 mL dH20.
Store in 50 μL aliquots (−20°C, stable at least 1 year).
100× AEBSF
25 mg of 4-(2-Aminoethyl) Benzenesulfonyl Fluoride (AEBSF, Sigma product A8456) (25 mM final)
4.1 mL dH20
Store in 100 μL aliquots (−20°C, stable at least 1 year).
QBS (Quenching, blocking, and storage) buffer
1 g Fraction V, 96–99% Bovine Serum Albumin (BSA, Sigma product A-2153) (1% final)
0.2 mL Sodium azide stock solution (see recipe; 1×, 0.02% final)
10 mL PBS 10× stock solution (see recipe; 1× final)
dH20 to 100 mL
Store at room temperature or at 4°C (stable at least 1 year)
Sodium azide stock solution, 500×
10g Sodium azide (10% final)
100 mL dH20
Store at room temperature or at 4°C (stable at least 1 year)
Caution: this is an extremely potent poison. Handle with gloves and dispense under a fume hood.
Commentary
Background Information
There is a great deal of interest in documenting, mapping, and understanding the relationships between the many protein complexes involved in `the interactome' (Ghavidel et al., 2005; Stelzl and Wanker, 2006). Well established strategies by which to approach protein-protein interaction experiments include co-immunoprecipitation (IP) with subsequent Western blotting (Phizicky and Fields, 1995) or mass spectrometry-based identification of binding partners (Conrotto et al., 2007; Tu et al., 2007), glutathione S-transferase (GST)-fusion protein pull-down (PD) (Smith and Johnson, 1988), yeast two-hybrid (Parrish et al., 2006), fluorescence resonance energy transfer (FRET) (Jares-Erijman and Jovin, 2006), and blue native-polyacrylamide gel electrophoreses (BN-PAGE) (Swamy et al., 2006). Together, these and other methods aim to (a) identify the individual members and stoichiometry of both constitutive and transient multiprotein complexes, (b) understand what controls their interactions, and (c) determine their biochemical function and relationship to other complexes. Significant efforts are being invested in converting methodologies and analytical tools into a form compatible with the high throughput platforms required for large-scale studies. Currently, however, the vast majority of PPI network information is binary in nature, represented as either presence or absence of possible association between respective proteins. PPI maps that are based on binary data represent maps in the truest sense: lines indicating pathways that can be traveled on, without indicating the level of traffic.
IP-FCM (or “the fly-p”) offers the possibility of measuring the “PPI traffic” as a continuous quantitative variable. It has been previously used to study the stoichiometry of multiprotein complexes, and the modulation of protein-protein interactions due to receptor signal transduction and calcium flux (Lund-Johansen et al., 2000; Teixeiro et al., 2004; Treves et al., 2004; Gil et al., 2005). The method allows a robust, quantitative, biochemical assessment of native protein-protein interactions when very little biomaterial is available. No genetic engineering, epitope tagging, or radioactivity is required, giving the method potential applications involving samples from wild-type subjects including clinical patients. In addition, it is compatible with 96-well plate/high throughput formatting.
There are other noteworthy advantages of this method. First, it is capable of assessing transmembrane protein complexes via the use of common Abs that are more often used to stain intact cells for flow cytometry (Figure 2). Therefore, for many surface receptors, the existence of plentiful commercial reagents greatly extends the utility of IP-FCM, and makes it an attractive strategy that can be approached quickly. Second, although the method is illustrated here using IP Abs to capture primary analytes, other specific protein-binding reagent can also be used to bind protein complexes, as has been previously demonstrated using GST-fusion protein pull-downs (Schrum et al., 2007).
Critical Parameters
IP-FCM relies on a candidate approach for identifying interaction partners, since pre-defined probes determine whether specific proteins are detected in the immunoprecipitated complexes. Not only will the presence of undefined proteins fail to be detected, but also well-defined proteins that simply were not probed. Related to this, unlike Western blotting, IP-FCM does not show the molecular weight of the proteins being probed. Therefore, from one point of view, IP-FCM may be best suited for the study of protein-protein interactions that are already well-documented, while other methods would serve better for the initial discovery of those interactions. Another point of view, however, is that a 96-well plate of IP-FCM samples could be rapidly screened with 96 different probe Abs. So if one had access to many probes, IP-FCM could in fact be very useful as a screening technique that might potentially contribute information about novel protein binding partners. Therefore, the drawback of the candidate approach is real, but there may be a payback in the high-throughput applicability of IP-FCM. A current aim of the Human Proteome Organization (HUPO) and the European Community-funded consortium ProteomeBinders is to generate Abs specific for all open reading frames of the human genome (Tyers and Mann, 2003). Such efforts are likely to greatly expand the screening capabilities of antibody-based high-throughput assays like IP-FCM.
IP-FCM follows the principle of a sandwich ELISA assay, where the capture and detection antibodies used can be specific for epitopes on co-associated proteins, allowing analysis of both primary and secondary analytes. If a protein expresses a single motif that is specific for several different binding partners, then IP-FCM will likely display all of those partners to some extent. However, none of those single partners may yield a very bright signal alone. Imagine that a lysate contains ligands X, Y, and Z, all of which compete equally for binding to a single site of receptor W. If the ligands are expressed equally, the maximum signal of W:X that can be detected by immunoprecipitating W is 33% of the total bead binding capacity. Increasing the amount of lysate (containing X, Y, and Z) would not alter this maximum. IP-FCM signals are limited by concentration, more than amount. In contrast, a low W:X signal by IP-Western blotting could be overcome by increasing the total amount of IP Ab and lysate (containing X, Y, and Z). Western blotting is limited by amount, more than concentration. One can attempt to avoid this problem with capture-elution-recapture strategies (Schrum et al., 2007). However, these assay characteristics should be kept in mind when deciding whether to design experiments based on IP-FCM as compared to other methods.
We recommend the use of purified monoclonal Abs for IP and for fluorochrome-conjugated probes whenever possible. Purified polyclonal antibodies may or may not work well as IP capture reagents by IP-FCM, even if they are proven effective for IP-Western blot applications. The polyclonal antibodies of interest represent a portion of the total Ig in polyclonal preparations. When the entire preparation is coupled to CML beads, only a fraction of the coupled material has the specificity of interest, which can translate into relatively low binding capacity per IP bead. All antibodies need to be well purified and in PBS for the coupling reaction to be effective. Unpurified antibody sources such as antisera and ascites fluid do not tend to work well, because the many other proteins in these preparations inhibit the coupling of the specific antibodies of interest. If polyclonal antibodies must be used to IP, our first suggestion is simply to follow the protocol as normal to determine if the signal-to-noise ratio is good. If it is not, then one solution could be to affinity-purify the specific Ig from the polyclonal preparation using the immunizing antigen, and then couple this specificity-controlled preparation to CML beads. Some antibodies that might make good IP-FCM probes may be available in purified form only, and not directly fluorochrome-conjugated. One could use the purified probe followed by a secondary fluorochrome-conjugated antibody, and this has been successfully done in the past with IP-FCM (Lund-Johansen et al., 2000). However, be sure to carefully control for the fact that the IP-bead itself is covered with Ig, and therefore many anti-Ig reagents will directly bind the IP bead. Our experience with polyclonal secondary antibodies is that species-specific adsorption is often insufficient to prevent this.
Especially when working with transmembrane proteins, incomplete solubilization of the membrane during lysis can result in membrane “chunks” that can allow proteins to co-precipitate although they do not interact. To monitor this potential problem, it is helpful to define some negative controls which in fact have their own positive controls. For example, when T cell membranes are not thoroughly solubilized, non-associated membrane proteins such as Thy1, CD45, and MHC class I can be co-precipitated with the TCR complex. However, these proteins do not co-precipitate when cells are lysed properly. Since both of these results can be demonstrated by IP-FCM, the absence of these extraneous proteins on an IP-bead can be interpreted to mean that they are not present, and that proper solubilization was achieved.
The specificity of the fluorochrome-conjugated Ab probes can be verified by demonstrating (a) that they fail to bind IP beads for irrelevant antigens, and/or (b) that they fail to bind IP beads specific for the antigen of interest, when the IP is performed on cell lysates that do not contain the antigen.
Troubleshooting
Conjugation of Abs to CML beads can be checked by staining the beads with fluorochrome-conjugated anti-Ig antibodies, followed by FCM analysis. If conjugation to the CML beads is poor, make sure that the IP Ab was not degraded prior to coupling. This can be achieved by verifying Ab size via SDS-PAGE followed by Coomassie staining or Western blotting, or by FPLC size exclusion chromatography. If the protein is not degraded, increase its concentration during the conjugation procedure. Also, make sure there are no primary amine-containing molecules other than the Ab present during the conjugation (no Tris, glycine, glutathione, or bovine serum albumin, for example).
After IP, if no fluorescence signal is observed on the IP-FCM beads, even from what should serve as positive controls for protein-protein interaction, then it is probably necessary to re-evaluate the validity of the assumptions and conditions upon which the experiment is based. The cellular source of the complexes, function of IP and fluorochrome-conjugated probe Abs, and optimal lysis buffers and lysis detergents must all be determined empirically. Information as to whether Abs function well during IP or FCM staining is often available from vendors, published literature, or better yet from experience. It is possible that covalent conjugation to CML beads could involve a lysine residue important for the binding function of a particular IP Ab. To address this, the Ab can be biotinylated and bound to streptavidin-coupled beads to perform the IP. However, because IP-FCM is quite sensitive, our experience has been that most experiments (at least the positive controls) work to some extent the first time they are attempted, even if a “suboptimal” detergent or IP antibody is used.
Low fluorescence signals can be improved by several means. First, the concentration of the Ab can be increased during covalent coupling to the CML beads to improve their binding capacity. Second, preliminary experiments can be performed to determine the minimal volume in which cells can be lysed while still achieving optimal protein solubilization. The lysate contents should be as concentrated as possible without increasing the detection of non-specific proteins. Third, various different fluorochrome-conjugated probe Abs can be tried. Sometimes, capture is excellent on the IP beads, but the IP Ab sterically crossblocks a particular probe Ab, leading to low fluorescence signals. Changing the probe-Ab to one that is not crossblocked by the IP Ab can fix the problem. Fourth, secondary probe reagents can be used to amplify the fluorescence signal. For example, a biotinylated probe Ab can be used, and then re-probed with streptavidin-PE.
When the IP and probe Ab combination is working well, a low fluorescence signal can be due to limited capture of a low-abundance protein. In this case, the signal can be increased by decreasing the number of IP beads incubated with the lysate. This exemplifies a critical difference in IP-FCM methodology, and is counter-intuitive for investigators accustomed to doing the opposite to improve IP-Western blot experiments. Decreasing the number of IP beads increases the amount of antigen captured on each individual bead, since low-abundance antigens become distributed across fewer beads. The goal is not to capture the maximum amount of antigen from the lysate; the goal is to obtain high average capture on each individual bead of the IP bead population. This principle can be used to maximize the IP-FCM signal obtainable when the amount of biomaterial available is quite low. For example, if one were to lyse 0.5 × 106 cells in 20 μL, it would be best to IP with a small number of beads, like 2.5 × 103, in order to maximize the signal-to-noise ratio seen when bead fluorescence is analyzed.
Anticipated Results
IP-FCM allows the measurement of interacting proteins via co-IP from lysates (Figure 1) in either a semi-quantitative or quantitative fashion. Because of the high signal-to-noise ratio of this method, the first attempt at an experiment often works to a fair extent, even before precise optimization has been performed. Beyond assessing the presence or absence of an interaction, IP-FCM can allow relative quantitative comparisons between experimental groups. Furthermore, experiments can be designed and reagents can be chosen such that the stoichiometry of interaction can be estimated. Semi-quantitative data can be converted to a quantitative estimate of the number of co-associated molecules on beads (Figure 3). The type of data generated is exemplified in Figure 2.
Time Considerations
Covalent coupling of IP Abs to beads (Basic Protocol 1) requires approximately 5–6 hours maximum. However, 3–4 hours of this time involves the bead coupling step during which no other procedure is performed. Also, the final step involves an overnight (or indefinitely longer) incubation in QBS buffer, followed by a bead counting step that requires 15 minutes. The IP from cell lysates (Basic Protocol 2) requires 1–2 hours maximum on the first day, assuming one cell type and 1–4 IP bead sets. This does not include the time required to harvest and prepare cells, which will vary depending on the experiment. The IP conditions described here are usually carried out overnight, but can be decreased to 1 hour as determined empirically. The time required the second day is more variable, as it depends on how many probe Abs will be used, which determines ultimately how many FACS tubes or plate samples will be generated. If 4 IPs have been performed, and 10 probe Abs will be used, then 40 experimental FACS samples will be generated. An experiment of this size can require 2–3 hours of washing and probing, followed by 1 hour at the flow cytometer. For the first experiments, several hours may be required for data analysis. However, in a more routine setting, analysis templates can be generated for both cytometry data and spreadsheet calculations.
Acknowledgements
The author is supported by start-up funds from the Mayo Foundation, and by an Eagles Innovation Award from the Fraternal Order of Eagles.
Literature Cited
- Conrotto P, Yakymovych I, Yakymovych M, Souchelnytskyi S. Interactome of transforming growth factor-beta type I receptor (TbetaRI): inhibition of TGFbeta signaling by Epac1. J Proteome Res. 2007;6:287–97. doi: 10.1021/pr060427q. [DOI] [PubMed] [Google Scholar]
- Ghavidel A, Cagney G, Emili A. A skeleton of the human protein interactome. Cell. 2005;122:830–2. doi: 10.1016/j.cell.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Gil D, Schrum AG, Alarcon B, Palmer E. T cell receptor engagement by peptide-MHC ligands induces a conformational change in the CD3 complex of thymocytes. J Exp Med. 2005;201:517–22. doi: 10.1084/jem.20042036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jares-Erijman EA, Jovin TM. Imaging molecular interactions in living cells by FRET microscopy. Curr Opin Chem Biol. 2006;10:409–16. doi: 10.1016/j.cbpa.2006.08.021. [DOI] [PubMed] [Google Scholar]
- Lund-Johansen F, Davis K, Bishop J, de Waal Malefyt R. Flow cytometric analysis of immunoprecipitates: high-throughput analysis of protein phosphorylation and protein-protein interactions. Cytometry. 2000;39:250–9. doi: 10.1002/(sici)1097-0320(20000401)39:4<250::aid-cyto2>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Parrish JR, Gulyas KD, Finley RL., Jr. Yeast two-hybrid contributions to interactome mapping. Curr Opin Biotechnol. 2006;17:387–93. doi: 10.1016/j.copbio.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Phizicky EM, Fields S. Protein-protein interactions: methods for detection and analysis. Microbiol Rev. 1995;59:94–123. doi: 10.1128/mr.59.1.94-123.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrum AG, Gil D, Dopfer EP, Wiest DL, Turka LA, Schamel WW, Palmer E. High-sensitivity detection and quantitative analysis of native protein-protein interactions and multiprotein complexes by flow cytometry. Sci STKE. 2007;2007:12. doi: 10.1126/stke.3892007pl2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DB, Johnson KS. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- Stelzl U, Wanker EE. The value of high quality protein-protein interaction networks for systems biology. Curr Opin Chem Biol. 2006;10:551–8. doi: 10.1016/j.cbpa.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Swamy M, Siegers GM, Minguet S, Wollscheid B, Schamel WW. Blue native polyacrylamide gel electrophoresis (BN-PAGE) for the identification and analysis of multiprotein complexes. Sci STKE. 2006;2006:14. doi: 10.1126/stke.3452006pl4. [DOI] [PubMed] [Google Scholar]
- Teixeiro E, Daniels MA, Hausmann B, Schrum AG, Naeher D, Luescher I, Thome M, Bragado R, Palmer E. T cell division and death are segregated by mutation of TCRbeta chain constant domains. Immunity. 2004;21:515–26. doi: 10.1016/j.immuni.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Treves S, Franzini-Armstrong C, Moccagatta L, Arnoult C, Grasso C, Schrum A, Ducreux S, Zhu MX, Mikoshiba K, Girard T, Smida-Rezgui S, Ronjat M, Zorzato F. Junctate is a key element in calcium entry induced by activation of InsP3 receptors and/or calcium store depletion. J Cell Biol. 2004;166:537–48. doi: 10.1083/jcb.200404079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu LC, Yan X, Hood L, Lin B. Proteomics analysis of the interactome of N-myc downstream regulated gene 1 and its interactions with the androgen response program in prostate cancer cells. Mol Cell Proteomics. 2007 doi: 10.1074/mcp.M600249-MCP200. [DOI] [PubMed] [Google Scholar]
- Tyers M, Mann M. From genomics to proteomics. Nature. 2003;422:193–7. doi: 10.1038/nature01510. [DOI] [PubMed] [Google Scholar]
Key References with Annotations
- Lund-Johansen F, Davis K, Bishop J, de Waal Malefyt R. Flow cytometric analysis of immunoprecipitates: high-throughput analysis of protein phosphorylation and protein-protein interactions. Cytometry. 2000;39:250–9. doi: 10.1002/(sici)1097-0320(20000401)39:4<250::aid-cyto2>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]; The first report of IP-FCM.
- Schrum AG, Gil D, Dopfer EP, Wiest DL, Turka LA, Schamel WW, Palmer E. High-sensitivity detection and quantitative analysis of native protein-protein interactions and multiprotein complexes by flow cytometry. Sci STKE. 2007;2007:12. doi: 10.1126/stke.3892007pl2. [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes the combination of IP-FCM with quantitative flow cytometry for (1) analysis of stoichiometric ratios within multiprotein complexes, and (2) co-IP data from rare primary cell subsets.