

Abstract
Purpose
Haplo-insufficiency at the paired box gene 6 (PAX6) locus causes aniridia,which is characterized by iris hypoplasia and other anterior and posterior eye defects leading to poor vision. This study aimed to identify novel PAX6 mutations that lead to familial and sporadic aniridia in northeastern China.
Methods
Two aniridia patients from a family and a sporadic patient underwent full ophthalmologic examinations. Genomic DNA was isolated from the affected individuals, 5 noncarriers in the family and 100 healthy normal controls. The coding regions and the adjacent intronic sequence of PAX6 were amplified by polymerase chain reaction (PCR) and direct bidirectional sequencing.
Results
A nonsense mutation in exon 9 (c.718C>T) was identified in the patients but not in any other unaffected families. A C>T substitution at codon 240 converts an arginine codon (CGA) to a termination codon (TGA).The same mutation was detected in the sporadic patient by chance.
Conclusions
A mutation in the PAX6 gene was confirmed to be capable of causing the classic aniridia phenotype. This is the first report on the “hotspot” c.718C>T transition from northeastern Chinese families.
Introduction
Aniridia (OMIM 106210) is a panocular and bilateral disease with a prevalence of 1 in 64,000 to 96,000 in the general population. In addition to the absence of the iris, other ocular disorders include corneal opacity, lens dislocation or cataracts, glaucoma, retina foveal dysplasia, and optic nerve hypoplasia, among others [1]. Approximately two thirds of all cases are familial, as they follow an autosomal dominant inheritance with variable expressivity, whereas the remaining one third is sporadic [2]. Aniridia can occur isolated or as part of the WAGR (Wilms tumor, aniridia, genitourinary disorders, and mental retardation) syndrome [1].
Various mutations in the paired box gene 6 (PAX6) gene are primarily responsible for aniridia [3,4]. Encoding a transcription factor,the highly conserved PAX6 gene plays a major role in developmental processes in several organs, including the eye [5,6]. Human PAX6 spans 22 kb on chromosome 11p13 and consists of 14 exons that encode 422 amino acids as transcriptional regulators. It has two DNA-binding domains, a bipartite paired domain (PD) and a paired-type home domain (HD), as well as a transactivation domain-rich proline, serine, and threonine (PST) at the COOH-terminal end. The PD and the HD, which are separated by a linker region (LNK), are the structural bases for the binding activity of the PAX6 protein [7,8]. The identification of a large number of mutations confirmed the role of PAX6 in human eye disease,and the features of aniridia also reflect the wide expression of PAX6 in the developing eye, including the neurectoderm, the surface ectoderm, and their derivatives. Up to now, more than 300 PAX6 mutations have been identified in aniridic subjects worldwide [9-11], and mutations in the PAX6 gene have been reported in patients with various ethnicities. In the present study, the same mutation (c.718 C>T) was described in the PAX6 gene in one northeastern Chinese family and in one sporadic patient with aniridia.
Methods
One family and one sporadic patient diagnosed with aniridia at the 1st affiliated hospital of Harbin Medical University were from Heilongjiang Province in northeastern China. Two aniridia patients from the family,five non-carrier family members, one sporadic patient and one hundred healthy controls (34.79±7.51 years old, 46 males) were recruited for the study. Experimental protocols were approved by the Institutional Review Board and complied with the tenets of the Declaration of Helsinki. The patients and relatives were evaluated by an ophthalmologist after a careful clinical ocular evaluation to exclude congenital anomalies other than aniridia, such as Axenfeld Rieger syndrome, iridocorneal endothelial syndromes, Peter’s anomaly, or sclerocornea. The possibility of the relatives of the sporadic patient having aniridia was excluded by full ophthalmologic examination. All patients underwent full ophthalmologic examination, including visual acuity, slit lamp biomicroscopy, and measurement of intraocular pressure (IOP) by applanation tonometry. Pentacam (Oculus, Wetzlar, Germany) was used to assess the cornea and anterior segment. Fundus photograph was taken with the use of Heidelberg retina angiograph (Heidelberg Engineering, Heidelberg, Germany). Ophthalmoscope and eye-ground photography were used to estimate the retina and the optic nerve. Systemic examinations were performed to exclude associated anomalies (e.g., Wilms’ tumor, urogenital anomalies, and mental retardation) in all subjects included in this study.
Molecular methods
Venous blood samples from 3 aniridia patients, 5 noncarriers in the family, and 100 healthy normal controls were collected for genomic DNA drawn from peripheral blood leucocytes with the use of standard protocols. Total genomic DNA was extracted from peripheral blood with the use of the QIAmp Blood kit (Qiagen, Hilden, Germany). Eleven coding exons (exon 4 to exon 13 and an extra exon, 5a) and the flanking regions of PAX6 were amplified by polymerase chain reaction (PCR) with eight pairs of primers (Table 1) [12]. The DNA was subsequently purified with the use of the help of the Qiaex II kit (Qiagen). The purified DNA was sequenced with ABI BigDye Terminator Cycle Sequencing kit v3.1, (ABI Applied Biosystems, Foster City, CA); the sequencing results were bidirectional and compared with the reference sequences in the database at the National Center for Biotechnology Information (NCBI; NC_000011.9). Mutation descriptions follow the new nomenclature system recommended by the Human Genomic Variation Society (HGVS) [13]. Any detected variation in PAX6 was further evaluated in 7 family members,in 1 sporadic patient, and in 100 normal controls with the use of heteroduplex PCR-single strand conformational polymorphism (SSCP) analysis as previously described in the literature [14]. One additional pair of primers for heteroduplex PCR-SSCP analysis [15] was synthesized for the amplification of exon 9 (Table 1).
Table 1. Primers used for PCR.
| Exon | Forward (5′-3′) | Reverse (5′-3′) | Product size (bp) | Annealing temperature (°C) |
|---|---|---|---|---|
| 4 |
GGACTTAGGGTTTGATGACAG |
CCAGAAAGACCAGAGGCAC |
684 |
50 |
| 5 |
GAGATTGGCACAGGTTGG |
CATAAGTAGCATCGTTTACAGT |
423 |
60 |
| 5 a,6 |
AACGCCACTTTAAGCAAGGT |
GGAGGGCAGATGTTCTCAA |
620 |
52 |
| 7 |
AATCCACCCACTGTCCCG |
CCAGCCACCTTCATACCG |
542 |
60 |
| 8 |
TCAGGTAACTAACATCGCA |
GTTGACTGTACTTGGAAGAA |
719 |
53 |
| 9,10,11 |
GAGGTGGGAACCAGTTTGATG |
CAAGCCAATCTCTGTAGTGCG |
890 |
52 |
| 12 |
GAGGCTTGATACATAGGC |
CCATAAGACCAGGAGATT |
452 |
58 |
| 13(1) |
GTTTCTGAGGGTGCTACT |
TTGAATGGCTAACTGGG |
1519 |
48 |
| 13(2) |
CAGTTTCTGAAGGTGCTA |
TCCATCCAGTCTACATTG |
578 |
48 |
| 9 | GTAGTTCTGGCACAATATGG | GAGGTGCTTGTACAGAGTAC | 83 | 55 |
Sequence of oligonucleotides and annealing temperatures used for exon-by-exon PAX6 amplification.
Results
Clinical findings
Three individuals in three successive generations (Figure 1) were found to have similar congenital ocular disease. In the family, two living patients had bilateral aniridia and horizontal tremor, and they had subnormal vision even from early childhood. Another similar phenotype is characterized by clear and normal-sized corneas and anterior chamber (Figure 2C) by Pentacam photo, and normal IOP and foveal hypoplasia with absence of foveolar and perifoveal reflexes were observed (Figure 2D) in both eyes. No other abnormalities were found in the retina, choroids, or optic nerve head in the family. Patient II:1 in the family had a congenital cataract (Figure 2A), whereas no abnormalities were detected in the lens in patient III:1 (Figure 2B). The sporadic patient is a female aged 30 years old. The defining characteristics of the patient were subnormal vision nystagmus, irregular cornea (Figure 3B) as shown by Pentacam, bilateral aniridia, and congenital cataract (Figure 3A). The anterior chamber depth for the sporadic patient was 1.87 mm (OD) and 1.96 mm (OS), which is shorter than normal during her first evaluation (Figure 3C). She demonstrated an IOP of approximately 28 mmHg and incipient glaucomatous changes in the optic cup in both eye.
Figure 1.
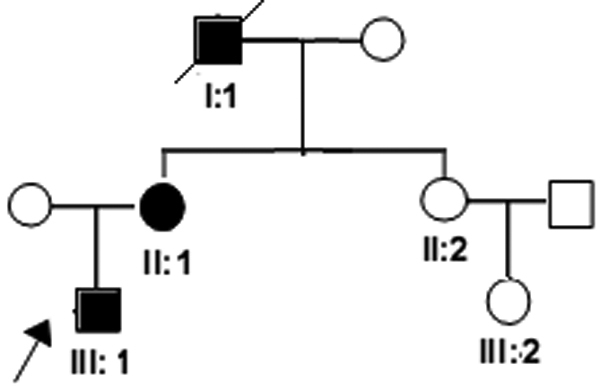
Pedigree of the family. Males and females are represented by squares and circles, respectively, and affected family members as confirmed by an ophthalmologist are represented by darkened symbols. The arrow points to the proband. The inheritance pattern in the family appears to be autosomal dominant.
Figure 2.
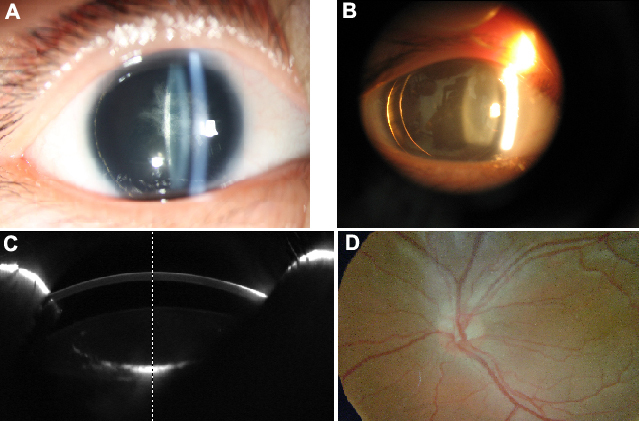
Photographic demonstration of aniridia expression in family members. The eyes of patient II:1 exhibit iris hypoplasia and lenses opacity (A), whereas those of patient III:1 had aniridia and transparent lenses (B). Pentacam photo shows the normal anterior segment picture of the patient II:1 from the family (C), and that of patient III:1 shows the same. The eyes of the family exhibited foveal hypoplasia by eyeground photography, including II:1 and III:1 (D).
Figure 3.
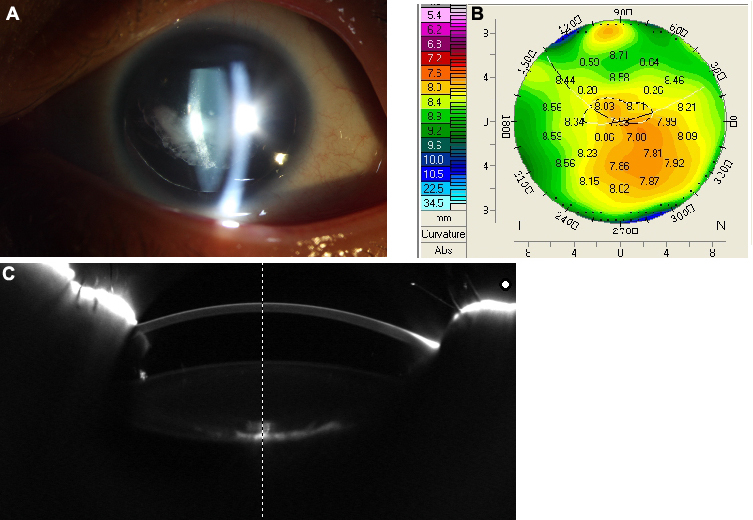
Photographic demonstration of aniridia expression in the sporadic patient. The sporadic patient suffered complete bilateral defects of the iris and a cataract (A). Pentacam photo shows that the cornea of the sporadic patient is irregular (B). The anterior segment is shorter than normal (C), as measured by Pentacam.
Mutation screening
The family exhibited a nonsense mutation (Figure 4) in exon 9 (c.718 C>T) but not in any other coding exons in PAX6. Mutations were confirmed by sequencing both sense and antisense strands in the patients and in the unaffected families. The same mutation c.718 C>T in exon 9 was unexpectedly detected in the sporadic patient. C>T substitution at codon 240 converts an arginine codon (CGA) to a termination codon (TGA), which is R240X. The mutation in three patients was also detected by SSCP analysis. No mutations were detected in any of the unaffected family members or control samples. This study is the first to report on c.718 C>T transition in northeastern China.
Figure 4.
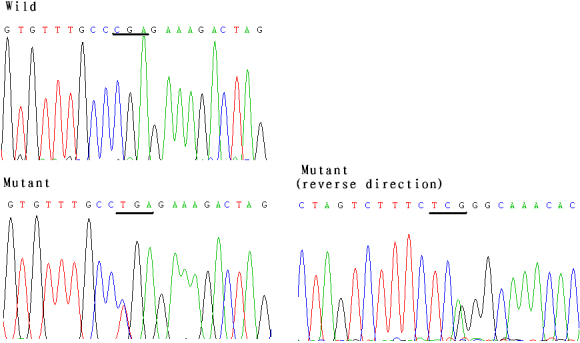
Nonsense mutation (c.718C>T) in the families and in the sporadic patient in northeastern China. DNA sequence of a part of PAX6 in the affected patients and unaffected individuals. The wild shows the corresponding normal sequence from the unaffected families and the normal controls. The mutant presents mutation sequence in two patients of the family and in the sporadic patient as confirmed by sense and antisense strands. It is a nonsense mutation(c.718 C>T) in exon 9.
Discussion
Aniridia is a human eye malformation caused by heterozygous null mutations of PAX6, which is extraordinarily conserved throughout evolution. The molecular bases for aniridia are derived from mutations of PAX6 that lead to premature protein termination [16]. It is assumed to cause loss of function of one allele and thus result in 50% reduction in overall activity rather than accumulation of dominant or dominant-negative forms of the protein [17]. Therefore, haplo-insufficiency of the PAX6 gene has been suggested to underlie the aniridia phenotype.
In this study, a mutation (c.718 C>T, p.R240X) in a Chinese family and in a sporadic patient with aniridia, which is not a polymorphism in the normal population was described. The nonsense mutation in exon 9 has been reported as a mutation hotspot for PAX6 in other ethnic pedigrees. Only one report in a Chinese family from Zhongshan Ophthalmic Center in south China existed [18]. However, it is the first mutation reported in northeastern Chinese patients. From the family’s data, we could determine that the affected members from the same family with the same mutation cause different phenotypes. Patient II:1 had a congenital cataract, whereas no abnormalities were detected in the lens in patient III:1. Patient II: 1in the family and the sporadic patient were found to have similar phenotypes in lens. Therefore, PAX6 plays a pleiotropic role in variable phenotypes among individuals [19]. The reason for this varying phenotype among individuals with the same mutation is worth exploring [20]. Some hypomorphic alleles with reduced activity probably generate less severe or variant phenotypes [21]. In such case, nonsense mutations are deemed to be mostly related to variant phenotypes,a result indicating that the altered function of the protein may trigger distinct or milder phenotypes compared with complete loss of function. c.718 C>T in exon 9 has been previously reported several times in other ethnic groups and appears to represent a hotspot of PAX6 mutation, the mutation was predicted to be attributed to methylated cytosine deamination in PAX6 [20].
In summary, this study identified one mutation of PAX6 first reported in northeastern Chinese patients with aniridia. Our genetic analysis provides further examples of haplo-insufficiency of PAX6 in aniridia. It is also valuable for genetic counseling and prenatal diagnosis in families where aniridia appears.
Acknowledgments
The authors are grateful to all patients, their families, and the normal volunteers who participated in the research. This work was supported by the Special Fund Research Project for Technological Innovation Talent of Harbin (RFXYS051).
References
- 1.Nelson LB, Spaeth GL, Nowinski TS, Margo CE, Jackson L. Aniridia. A review. Surv Ophthalmol. 1984;28:621–42. doi: 10.1016/0039-6257(84)90184-x. [DOI] [PubMed] [Google Scholar]
- 2.Ton CCT, Hirvonen J, Mioa H. Positional cloning and characterization of a paired box and homeobox containing gene from the aniridia region. Cell. 1991;67:1059–74. doi: 10.1016/0092-8674(91)90284-6. [DOI] [PubMed] [Google Scholar]
- 3.Chien YH, Huang HP, Hwu WL, Chien YH, Chang TC, Lee NC. Eye anomalies and neurological manifestations in patients with PAX6 mutations. Mol Vis. 2009;15:2139–45. [PMC free article] [PubMed] [Google Scholar]
- 4.Jordan T, Hanson I, Zaletayav D, Hodgson S, Prosser J, Seawright A, Hastie N, van Heyningen V. The human PAX6 gene is mutated in two patients with aniridia. Nat Genet. 1992;1:328–32. doi: 10.1038/ng0892-328. [DOI] [PubMed] [Google Scholar]
- 5.van Heyningen V, Williamson KA. PAX6 in sensory development. Hum Mol Genet. 2002;11:1161–7. doi: 10.1093/hmg/11.10.1161. [DOI] [PubMed] [Google Scholar]
- 6.Simpson TI, Price DJ. Pax6:a pleiotropic player in development. Bioessays. 2002;24:1041–51. doi: 10.1002/bies.10174. [DOI] [PubMed] [Google Scholar]
- 7.Mishra R, Gorlov IP, Chao LY, Singh S, Saunders GF. PAX6, paired domain influences sequence recognition by the homeodomain. J Biol Chem. 2002;277:49488–94. doi: 10.1074/jbc.M206478200. [DOI] [PubMed] [Google Scholar]
- 8.Grzeskowiak R, Amin J, Oetjen E, Knepel W. Insulin responsiveness of the glucagon gene conferred by interactions between proximal promoter and more distal enhancer-like elements involving the paired-domain transcription factor Pax6. J Biol Chem. 2000;275:30037–45. doi: 10.1074/jbc.M000984200. [DOI] [PubMed] [Google Scholar]
- 9.http://pax6.hgu.mrc.ac.uk
- 10.Beby F, Dieterich K, Calvas P. A [c.566–2A>G] heterozygous mutation in the PAX6 gene causes aniridia with mild visual impairment. Eye (Lond) 2011;25:657–8. doi: 10.1038/eye.2010.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang X, Zhang Q, Tong Y, Dai H, Zhao X, Bai F, Xu L, Li Y. Large novel deletions detected in Chinese families with aniridia: correlation between genotype and phenotype. Mol Vis. 2011;17:548–57. [PMC free article] [PubMed] [Google Scholar]
- 12.Yuan H, Kang Y, Shao Z, Li Y, Yang G, Xu N. Two novel PAX6 mutations identified in northeastern Chinese patients with aniridia. Mol Vis. 2007;13:1555–61. [PubMed] [Google Scholar]
- 13.den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat. 2000;15:7–12. doi: 10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Q, Minoda K. Detection of congenital color vision defects using heteroduplex-SSCP analysis. Jpn J Ophthalmol. 1996;40:79–85. [PubMed] [Google Scholar]
- 15.Love J, Axton R, Churchill A. A new set of primers for mutation analysis of the human PAX6 gene. Hum Mutat. 1998;12:128–34. doi: 10.1002/(SICI)1098-1004(1998)12:2<128::AID-HUMU8>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 16.Vincent MC, Pujo AL, Olivier D, Calvas P. Screening for PAX6 gene mutations is consistent with haploinsufficiency as the main mechanism leading to various ocular defects. Eur J Hum Genet. 2003;11:163–9. doi: 10.1038/sj.ejhg.5200940. [DOI] [PubMed] [Google Scholar]
- 17.Tzoulaki I, White IM, Hanson IM. PAX6 mutations: genotype-phenotype correlations. BMC Genet. 2005;6:27. doi: 10.1186/1471-2156-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang X, Li S, Xiao X, Jia X, Wang P, Shen H, Guo X, Zhang Q. Mutational screening of 10 genes in Chinese patients with microphthalmia and/or coloboma. Mol Vis. 2009;15:2911–8. [PMC free article] [PubMed] [Google Scholar]
- 19.Kokotas H, Petersen MB. Clinical and molecular aspects of aniridia. Clin Genet. 2010;77:409–20. doi: 10.1111/j.1399-0004.2010.01372.x. [DOI] [PubMed] [Google Scholar]
- 20.Neethirajan G, Nallathambi J, Krishnadas SR, Vijayalakshmi P, Shashikanth S, Collinson JM, Sundaresan P. Identification of novel mutant PAX6 alleles in Indian cases of familial aniridia. BMC Ophthalmol. 2006;6:28. doi: 10.1186/1471-2415-6-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang P, Guo X, Jia X, Li S, Xiao X, Zhang Q. Novel mutations of the PAX6 gene identified in Chinese patients with aniridia. Mol Vis. 2006;12:644–8. [PubMed] [Google Scholar]