

Abstract
Acetylation has recently emerged as an important mechanism for controlling a broad array of proteins mediating cellular adaptation to metabolic fuels. Acetylation is governed, in part, by SIRTs (sirtuins), class III NAD+-dependent deacetylases that regulate lipid and glucose metabolism in liver during fasting and aging. However, the role of acetylation or SIRTs in pathogenic hepatic fuel metabolism under nutrient excess is unknown. In the present study, we isolated acetylated proteins from total liver proteome and observed 193 preferentially acetylated proteins in mice fed on an HFD (high-fat diet) compared with controls, including 11 proteins not previously identified in acetylation studies. Exposure to the HFD led to hyperacetylation of proteins involved in gluconeogenesis, mitochondrial oxidative metabolism, methionine metabolism, liver injury and the ER (endoplasmic reticulum) stress response. Livers of mice fed on the HFD had reduced SIRT3 activity, a 3-fold decrease in hepatic NAD+ levels and increased mitochondrial protein oxidation. In contrast, neither SIRT1 nor histone acetyltransferase activities were altered, implicating SIRT3 as a dominant factor contributing to the observed phenotype. In Sirt3−/− mice, exposure to the HFD further increased the acetylation status of liver proteins and reduced the activity of respiratory complexes III and IV. This is the first study to identify acetylation patterns in liver proteins of HFD-fed mice. Our results suggest that SIRT3 is an integral regulator of mitochondrial function and its depletion results in hyperacetylation of critical mitochondrial proteins that protect against hepatic lipotoxicity under conditions of nutrient excess.
Keywords: cellular metabolism, mitochondrial metabolism, NAD, non-alcoholic fatty liver disease, obesity, proteomics, sirtuin
INTRODUCTION
Acetylation has recently emerged as an important mechanism for controlling a broad array of proteins governing cellular adaptation to different metabolic fuels. In eukaryotes [1] and in human liver [2], acetylation profiles have been shown to control gluconeogenesis, glycolysis and the TCA (tricarboxylic acid) cycle coupled to the urea cycle. Schwer et al. [3] reported that acetylation of up to 70 mitochondrial proteins in liver changed by at least 2.5-fold in response to caloric restriction, suggesting the importance of extranuclear acetylation in events influencing metabolic fuel utilization.
Acetylation is controlled, in part, by SIRTs (sirtuins), which are class III NAD+-dependent deacetylases [HDACs (histone deacetylases)]. SIRTs are now recognized to function as nutrient and redox sensors that modulate cellular function via deacetylation of lysine residues [1]. There are seven mammalian SIRT isoforms: SIRTs 1, 2, 6 and 7 are located in the nucleus, whereas SIRTs 3, 4 and 5 are mitochondrial (although SIRT3 has been reported to shuttle from the cytosol to mitochondria with cell stress [4]). Under conditions of caloric restriction, SIRTs influence a variety of key metabolic pathways, including fat mobilization [5], insulin secretion [6], gluconeogenesis [7], muscle fatty acid oxidation [8] and mitochondrial regulation [9]. A proteomic study found 277 acetylation sites in 133 mitochondrial proteins in mouse liver [10]. However, relatively little is known about the role of SIRTs and acetylation under conditions of caloric excess, where many organs, including the liver, play a critical role in diseases such as Type 2 diabetes, NAFLD (non-alcoholic fatty liver disease) and inflammation.
SIRT3 appears to be the primary mediator of mitochondrial acetylation, since no significant changes in acetylation status of mitochondrial proteins were detectable in mice lacking SIRT4 and SIRT5 [11]. A number of studies have suggested that SIRT3 plays an important role in regulating mitochondrial function [12–14]. SIRT3 overexpression increases cellular respiration and decreases mitochondrial membrane potential and ROS (reactive oxygen species) production [14–17], and mice lacking SIRT3 have impaired ATP production and mitochondrial respiration during starving [11,15]. SIRT3 regulates mitochondrial protein synthesis through acetylation of MRPL10 (mitochondrial ribosomal protein L10) [18,19] and controls mitochondrial activity through acetylation of succinate dehydrogenase [20], long-chain acyl-CoA dehydrogenase [13], acetyl-CoA synthetase 2 [21] and isocitrate dehydrogenase 2, an enzyme that promotes regeneration of antioxidants and catalyses a key regulation point in the TCA cycle [22]. A decrease in Sirt3 mRNA has been described in brown adipose tissue of genetically obese animals [14]; however, its role in the liver has not been studied under conditions of caloric excess.
In the present study, in response to a chronic HFD (high-fat diet), we reveal a diverse set of hyperacetylated proteins important for controlling pathological responses in the mitochondria and cytosol. In addition, we find that exposure to the HFD suppresses SIRT3 activity concomitant with NAD+ levels, and that Sirt3−/− mice have marked hyperacetylation of gluconeogenic and mitochondrial proteins under HFD conditions. Sirt3-deficient mice have a disruption in mitochondrial complex assembly when fed on the HFD. Taken together, these results suggest that SIRT3 and hyperacetylation may play an important role in the regulation of cellular and mitochondrial metabolism induced by high-fat feeding.
EXPERIMENTAL
Reagents
Polyclonal anti-(acetylated-lysine) and AP (alkaline phosphatase)-conjugated anti-biotin antibodies were purchased from Cell Signaling Technology, and the anti-(total OXPHOS) (oxidative phosphorylation) antibody was from MitoSciences. All other antibodies were obtained from Santa Cruz Biotechnology. HRP (horseradish peroxidase)-conjugated goat anti-(rabbit IgG) and anti-(mouse IgG) antibodies were purchased from Bio-Rad Laboratories. Protease inhibitors were obtained from Sigma-Aldrich. Sequencing-grade trypsin was purchased from Promega, and iodoacetamide and DTT (dithiothreitol) were from Sigma-Aldrich. SIRT1 and SIRT3 fluorimetric assays were obtained from Enzo Life Sciences, NAD+ quantification was performed with a kit from BioVision, and an HAT (histone acetyltransferase) activity assay kit from Millipore. Control and high-fat mouse chows were purchased from Research Diets and Harlan Laboratories. Reagents used for measurement of respiratory enzyme activities were purchased from Sigma-Aldrich and Fisher Scientific.
Animals and sample extraction for proteomic analysis
Animal protocols were approved by the University of Colorado Institutional Animal Care and Use Committee in accordance with National Institutes of Health guidelines. Male C57BL/6 SVJ mice at 5-6-weeks of age were housed separately and fed on either CH (control chow) or an HFD (45 kcal% fat; where 1 kcal ≈ 4.184 J) for 16 weeks. WT (wild-type) and Sirt3−/− mice [23] were fed on either CH or an HFD (60 kcal% fat) for 16–24 weeks. Livers used for the respiratory enzyme activity assay were from mice fed on the HFD from Harlan (42 kcal% fat) for 24 weeks after weaning. Following overnight starving, mice were killed by an overdose of pentobarbital, and livers were harvested immediately and snap frozen at −70 °C in liquid nitrogen before analysis. Livers (approx. 200 mg) were homogenized and proteins extracted as described previously [24]. Briefly, whole-cell lysates were obtained by incubating with buffer A [10 mM Hepes (pH 7.9), 1.5 mM KCl, 10 mM MgCl2, 0.5 mM DTT, 0.1 % Igepal CA-630 and 0.5 mM PMSF] for 30 min and centrifuging. The supernatants were stored at −70 °C and were used for the initial proteomic analysis (see Supplementary Figure S1 at http://www.BiochemJ.org/bj/433/bj4330505add.htm). The extraction of mitochondrial/nuclear fractions was modified slightly from the original protocol [24]. After addition of buffer B [20 mM Hepes (pH 7.9), 25 % glycerol, 1.5 mM MgCl2, 420 mM NaCl, 0.5 mM DTT, 0.2 mM EDTA, 0.5 mM PMSF and 4 μM leupeptin], samples were incubated for 30 min to retain both nuclear and mitochondrial fractions in the lysates. The mitochondrial/nuclear fraction was stored at −70 °C and used for proteomics and all other analyses.
IP (immunoprecipitation), one-dimensional electrophoresis and Western blot analysis
Samples (500 μg of total protein) for IP were incubated overnight with 2 μl of the anti-(acetylated-lysine) antibody or, in the case of Hsp70 (heat-shock protein 70), with 2 μl of the anti-Hsp70 antibody, at 4 °C. Subsequently, 40 μl of agarose beads was added and incubated with the sample for 2 h at room temperature (25 °C). Beads were washed four times with ice-cold RIPA buffer. Elution of immunoprecipitated proteins with Laemmli sample buffer and heat-denaturation were performed immediately prior to one-dimensional gel electrophoresis. Proteins were separated by SDS/PAGE (10 % gels) and either transferred on to a PVDF membrane for Western blot analysis or visualized by staining overnight with a fluorescent gel stain (Lava Purple; Fluorotechnics). Gels were imaged using a Typhoon 9600 imager (GE Healthcare) and bands exhibiting significant differential staining between CH- and HFD-fed mice were processed further as described below. Western blot analysis was performed as described previously [11,25]. At least five separate IPs followed by Western blotting were typically performed. Western blots for total protein expression were assayed using 20 μg of total protein per sample and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used as a loading control.
In-gel digestion and MS analysis
Bands of interest were excised and diced, destained (1:1 acetonitrile in 50 mM ammonium bicarbonate), and dried in a vacuum centrifuge for 1 h. Dried slices were rehydrated and proteins were reduced with DTT (1.5 mg/ml), alkylated with iodoacetamide (10 mg/ml) and digested overnight at 37 °C with trypsin (0.2 μg/μl). The resultant peptides were extracted and analysed by reverse-phase nanoscale LC (liquid chromatography)-MS/MS (tandem MS). Approx. 50 % of each sample was loaded on to a column (Agilent 1100 nano-HPLC, 75 μm internal diameter × 15 cm column, Zorbax C18). Peptides were eluted into the mass spectrometer using a gradient of increasing buffer B (90 % acetonitrile and 0.1 % formic acid) at a flow rate of 300 nl/min. Nanospray was induced using a capillary voltage of 1550 V applied to a fused silica emitter (PicoTip; New Objective) with an 8 μm aperture. Spectra were collected over an m/z range of 350 to 1800 Da (Agilent LC/MSD Ultra Trap). Over 10 000 MS/MS spectra were typically obtained during a run. SpectrumMill (Agilent) was used to search processed spectra against the Swiss-Prot database (UniProtKB/Swiss-Prot Release 57.6; 495 880 entries). Each spectrum utilized for protein assignment was manually confirmed and only accepted if the signal-to-noise ratio of fragment ions was at least five. Additionally, reversed database searching was used to minimize false-positive results. At least two confirmed peptide hits were required for protein identification.
NAD+ quantification and activity assays
Quantification of NAD+ levels was carried out using a colorimetric assay, according to the manufacturer’s protocol. Livers were snap frozen in liquid nitrogen, pulverized and extracted at 4 °C with extraction buffer provided with the NAD+ quantification kit. NAD+ values are expressed as ng/mg of tissue. The activity of HATs was measured using a colorimetric HAT activity assay as described previously [26]. SIRT1 and SIRT3 enzymatic activities were assayed following the manufacturer’s instructions with slight modifications [27]. Protein (40 μg) was incubated at 37 °C for 45 min with specific substrates, 25 μl of developer was added and, following 45 min of incubation, SIRT activity was measured using a fluorimetric microplate reader at 350 nm/450 nm.
Isolation, purification and assessment of mitochondrial oxidized proteins from livers of CH- and HFD-fed animals
Selective isolation and purification of oxidized or S-nitrosylated mitochondrial proteins were adapted from the method described by Moon et al. [28]. Liver mitochondrial protein extracts were suspended in a buffer containing a final concentration of 40 mM Hepes (pH 7.4), 50 mM NaCl, 1 mM EGTA and 1 mM EDTA with the protease inhibitor cocktail and NEM (N-ethylmaleimide) (6 μg/μl), and were incubated for 30 min at room temperature to block residual thiols in the protein samples. Samples were eluted through PD MiniTrap G-25 columns (GE Healthcare) with the same buffer containing 1 % CHAPS (elution buffer). Biotin-NEM was added to the eluted proteins to a final concentration of 6 mM and samples were incubated for 3 h at 4 °C. Excess biotin-NEM was removed from the protein samples using G-25 columns. Protein-NEM-biotin complexes were affinity-purified using streptavidin-agarose beads. Complexes were washed twice in elution buffer, centrifuged at 4000 g for 10 s and the supernatant decanted. Pelleted complexes were resuspended in 2× Laemmli sample buffer and boiled; the supernatants were then subjected to SDS/PAGE (12 % gels), transferred on to a PVDF membrane and Western blotted using an AP-conjugated anti-biotin antibody and CDP-Star (New England Biolabs) for detection of oxidized protein-NEM-biotin complexes.
Measurement of respiratory chain enzymatic activity in livers of WT and Sirt3−/− animals fed on the HFD
Post 600 g mitochondrial supernatants were isolated from WT and Sirt3−/− mouse livers. The supernatants were used to assay activity of respiratory chain enzyme complexes I, II, II+III, III, IV and CS (citrate synthase) spectrophotometrically on a Cary 300 spectrophotometer as described by Rahman et al. [29] with some modifications. For complex II, the reaction was started with the addition of 20 μM CoQ1 (coenzyme Q1). The activity of complex II+III was performed in the presence and absence of antimycin A because liver has a substantial antimycin-A-insensitive rate. For complex III, the sample was incubated with buffer and then the reaction was initiated by the addition of both decylbenzylquinol and oxidized cytochrome c. For complex IV, the sample was incubated with buffer and then the reaction was generated by the addition of reduced cytochrome c. The protein content of each sample was determined using a Bradford assay. For complexes I, II and II+III, and CS, enzyme activities were calculated as initial rates (nmol/min). For complexes III and IV, enzyme activities were calculated as the first-order rate constants derived within 1 min of the reaction [30,31]. All activities were normalized to the total protein content of each sample and are expressed as ratios over the activity of CS.
Statistical analysis
Data were analysed using an unpaired Student’s t test, a Mann-Whitney U test and/or a one-way ANOVA. Differences with P < 0.05 were considered significant.
RESULTS
Proteomics reveals a broad array of hyperacetylated proteins central to metabolism in livers from obese mice
As expected [32], mice fed on the HFD were approx. 50 % heavier and had 4-fold greater hepatic triacylglycerol (triglyceride) levels compared with CH-fed controls (results not shown). A proteomic approach was used to identify liver proteins with altered acetylation status in the HFD-fed animals. As shown in Figure 1(A), multiple hyperacetylated proteins were present in livers from HFD-fed mice compared with CH-fed mice. Of the 193 proteins identified in bands from three separate experiments (Supplementary Table S1 at http://www.BiochemJ.org/bj/433/bj4330505add.htm), approx. 90 %of the hyperacetylated proteins regulate metabolic pathways and are known to contain acetylated lysine residues [10]. Functional classification of 35 representative hyperacetylated proteins of interest (listed in Table 1), based on their central role in metabolism, is shown in Figure 1(B). These classes include proteins implicated in amino acid metabolism, fatty acid metabolism, glycolysis, gluconeogenesis, TCA cycle enzymes, redox regulation and the stress response. Eleven of the proteins observed have not been reported previously to be acetylated.
Figure 1. Functional classification of hyperacetylated proteins in the livers of HFD-fed mice.

C57BL/6 SVJ mice were fed on either CH or an HFD (45 % fat) for 16 weeks and starved overnight before sample extraction (see details in the Experimental section; n = 3 per group). Lysine-acetylated liver proteins were immunoprecipitated from mitochondrial/nuclear extracts. (A) Proteins were separated by one-dimensional SDS/PAGE (10 % gel) and visualized with fluorescence staining (see Experimental section). Proteins from differentially stained bands were excised, digested and identified by MS/MS and database searching. The molecular mass in kDa is shown. (B) The functional classification of 35 high-scoring proteins of interest identified via MS across a variety of important metabolic pathways. Proteins were selected and separated into functional groups based on information contained in the UniProtKB database.
Table 1. Hyperacetylated proteins in livers from HFD-fed mice compared with CH-fed mice.
A subset of 35 high-scoring proteins of interest were identified from differentially stained gel bands.
| Distinct hits* | Summed MS/MS scores† | Amino acid coverage (%)‡ | Molecular mass (Da)§ | Accession number|| | Protein name |
|---|---|---|---|---|---|
| 35 | 535.2 | 28 | 164618.9 | Q8C196 | Carbamoyl-phosphate synthase [ammonia] |
| 16 | 247.49 | 18 | 129685.3 | Q05920 | Pyruvate carboxylase |
| 16 | 226.96 | 36 | 59752.9 | Q03265 | ATP synthase subunit α |
| 8 | 137.97 | 36 | 45020.8 | O35490 | Betaine-homocysteine S-methyltransferase 1 |
| 6 | 97.92 | 13 | 72422.4 | P20029 | Glucose-regulated protein of 78 kDa |
| 6 | 90.54 | 13 | 56823.1 | P54869 | Hydroxymethylglutaryl-CoA synthase |
| 6 | 98.51 | 19 | 44186.4 | Q8QZT1 | Acetyl-CoA acetyltransferase |
| 5 | 76.9 | 9 | 73528.7 | P38647 | Stress-70 protein, mitochondrial |
| 5 | 86.01 | 11 | 60955.8 | P63038 | Heat-shock protein 60 kDa, mitochondrial |
| 5 | 73.88 | 12 | 59765.6 | P24270 | Catalase |
| 5 | 68.63 | 11 | 62515.7 | P34914 | Epoxide hydrolase 2¶ |
| 5 | 79.43 | 11 | 50114.1 | P10126 | Elongation factor 1-α1 |
| 4 | 49.1 | 18 | 22176.6 | P35700 | Peroxiredoxin-1 |
| 3 | 41.18 | 6 | 70871.4 | P63017 | Heat-shock cognate 71 kDa protein |
| 3 | 48.21 | 6 | 59126 | P32020 | Non-specific lipid-transfer protein |
| 3 | 50.77 | 10 | 43508.9 | Q91X83 | S-Adenosylmethionine synthetase isoform type-1¶ |
| 3 | 46.52 | 17 | 41793.1 | P63260 | Actin, cytoplasmic 2 |
| 3 | 51.83 | 10 | 42119.9 | P15105 | Glutamine synthetase |
| 3 | 36.2 | 11 | 44930.8 | O88986 | 2-Amino-3-ketobutyrate CoA ligase |
| 2 | 34.55 | 17 | 15840.3 | P02088 | Haemoglobin subunit β-1¶ |
| 2 | 28.85 | 4 | 63318.1 | Q63880 | Liver carboxylesterase 31¶ |
| 2 | 28.58 | 6 | 54371.1 | P97807 | Fumarate hydratase, mitochondrial |
| 2 | 28.49 | 5 | 49990.2 | O09173 | Homogentisate 1,2-dioxygenase |
| 2 | 22.48 | 5 | 46584.7 | P16460 | Argininosuccinate synthase |
| 2 | 31.62 | 7 | 41858.2 | Q8BWT1 | 3-Ketoacyl-CoA thiolase, mitochondrial |
| 2 | 32.77 | 9 | 39507.2 | Q91Y97 | Fructose bisphosphate aldolase B |
Total number of fragmentation spectra identified as belonging to a protein.
Number of non-redundant fragmentation spectra identified as belonging to a protein.
Sum of peptide scores from distinct spectra correlating to a protein.
All proteins reported had at least two distinct peptides whose sequence assignments were validated manually.
UniProtKB/Swiss-Prot database accession number.
Proteins not reported previously as acetylated, on the basis of UniProtKB/Swiss-Prot database information.
Validation of the acetylated proteins in livers from HFD-fed mice by Western blotting
To validate a cross-section of hyperacetylated proteins identified by MS, we performed IPs with acetylated-lysine affinity matrix antibodies followed by Western blot analysis using commercially available antibodies against candidate proteins. We confirmed the hyperacetylation of selected metabolic proteins, including the following: the urea cycle regulator CPS1 (carbamoyl-phosphate synthase 1); the gluconeogenic enzyme PC (pyruvate carboxylase); the glycolysis/gluconeogenic mediator fructose-bisphosphate aldolase B; and the metabolic stress-response factor Hsp70 (Figure 2). With the exception of Hsp70, the levels of acetylation, rather than the protein levels, were all increased in livers of HFD-fed mice, confirming the proteomic analysis. Mitochondrial protein levels were unchanged in both the CH- and HFD-fed groups (Supplementary Figure S3 at http://www.BiochemJ.org/bj/433/bj4330505add.htm)
Figure 2. HFD increases acetylation of proteins involved in key metabolic pathways in mouse liver.

Equivalent amounts of liver protein homogenates from C57BL/6 SVJ mice fed on either CH or an HFD (45 kcal % fat) for 16 weeks (n = 3/group) and starved overnight prior to sample extraction were subjected to IP and Western blot analysis (see the Experimental section). Total protein expression for (A) CPS1, (B) PC, (C) aldolase B and (D) Hsp70 was assessed by immunoblotting with an appropriate antibody. GAPDH was used as a loading control. The significant decrease in CPS1 protein in HFD-fed mice is shown in (A). Values are means ± S.E.M., ##P < 0.01. In a separate experiment (E), acetylation (Ac) status was assessed by IP followed by Western blotting. Representative blots are shown. The representative Western blot (C) is a composite of a larger gel in which all of the samples were run simultaneously together. The composite was made by splicing complete lanes together for presentation purposes.
SIRT activity, not expression, is suppressed in livers of obese animals
We postulated that either decreased SIRT expression and/or activity could account for protein hyperacetylation in livers from HFD-fed mice. Figures 3(A), 3(C) and 3(E) demonstrate that protein expression of SIRT1, SIRT3 and SIRT4 were unchanged in the livers of HFD-fed mice. Although SIRT1 activity remained unchanged, SIRT3 activity was decreased by 38 % (P < 0.01) in liver samples from HFD-fed mice (Figures 3B and 3D).
Figure 3. HFD suppresses SIRT3 activity with no change in SIRT1, SIRT3 or HAT activity in livers of HFD-fed mice.

C57BL/6 SVJ mice were fed on either CH or an HFD (45 kcal % fat) for 16 weeks. Mice were killed following overnight starvation and lysates from homogenized livers were used to detect SIRT1, SIRT3 and SIRT4 protein expression and activity. HAT activity was also assayed as described in the Experimental section. Equivalent amounts of protein extracts were subjected to Western blot analysis. (A, C and E) PVDF membranes were probed with anti-SIRT1, anti-SIRT3, anti-SIRT4 and anti-GAPDH antibodies and monitored using ECL (enhanced chemiluminescence) detection. Three separate experiments in different mice (n = 5/group) were performed with similar results. (B and D) The effect of the HFD on SIRT1 and SIRT3 activity were examined using a fluorimetric method. Assays were performed according to the manufacturer’s instructions with slight modifications (see the Experimental section). Values are means ± S.E.M., n = 4/group, and represent a fold change over the control group. #P < 0.05. (F) An ELISA was used to investigate the effect of the HFD on HAT activity. Extracts were co-incubated with histone H3 peptide and an HAT assay cocktail (as described in the Experimental section). Values are means ± S.E.M., n = 4/group. The representative Western blots (A and C) are a composite of larger gels in which all the samples were run simultaneously together. The composite was made by splicing complete lanes together for presentation purposes.
Protein hyperacetylation may be caused by both increased HAT activity and/or reduced HDAC enzyme activity. Figure 3(F) reveals that HAT activity was unchanged in the livers of HFD-fed mice. These findings suggest that a reduction in deacetylase activity, most probably that of SIRT3, is primarily responsible for the observed protein hyperacetylation in HFD-fed mice.
NAD+ levels decrease with increasing body weight
Because SIRT3 activity requires NAD+ as a cofactor [33] and this nucleotide is reduced in the liver of obese mice [34], we postulated that reduced SIRT activity might be linked to a decrease in hepatic NAD+ levels in HFD-fed mice. Overall, NAD+ levels were significantly decreased in HFD-fed mice by 35 % (Figure 4A) and appeared to decrease linearly when plotted against increasing weight gain (Figure 4B).
Figure 4. Reduced hepatic NAD+ concentration correlates with increasing weight gain in HFD-fed mice.

C57BL/6 SVJ mice were fed on either CH or an HFD (45 kcal % fat) for 16 weeks (n = 4/group) and starved overnight prior to killing. (A) NAD+ levels were quantified using a colorimetric assay according to the manufacturer’s protocol. Values are means ± S.E.M., n = 6/group; #P < 0.05. (B) Hepatic NAD+ concentrations were compared with the net increase in body weight in mice fed on the HFD. NAD+ levels negatively correlated with the body weight increase (R2 = 0.88).
Increased oxidation of mitochondrial proteins in livers from HFD-fed mice
SIRT3 activity is primarily associated with mitochondria [10]. Given that the HFD was associated with hyperacetylation of predominantly mitochondrial proteins (Table 1), we next examined whether the HFD might increase protein oxidation in liver mitochondria. We isolated and purified mitochondrial proteins and tested for their oxidized or S-nitrosylated state in CH-fed compared with HFD-fed mice, according to the methods described by Moon et al. [28]. The results shown in Figure 5 indeed demonstrate that the HFD increased the levels of oxidized proteins in liver mitochondria.
Figure 5. HFD increases mitochondrial protein oxidation in liver.
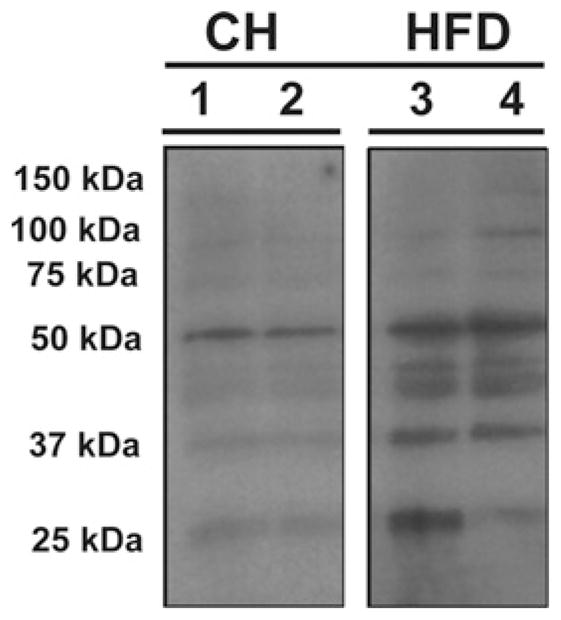
Selective isolation and purification of oxidized or S-nitrosylated mitochondrial proteins from C57BL/6 SVJ mice fed on either CH or an HFD (45 kcal % fat) for 16 weeks were performed as adapted from the methods described by Moon et al. [28]. Equivalent amounts of protein extracts from CH- and HFD-fed mice were assayed. Purified biotin-NEM-labelled mitochondrial proteins were separated by SDS/PAGE (12 % gels), transferred on to PVDF membranes and subjected to Western blot analysis using a biotin-AP-conjugated antibody. The blot shown is a composite gel and displayed are two lanes per group from the same gel and same exposure. The complete gel is provided in Supplementary Figure S2 at (http://www.BiochemJ.org/bj/433/bj4330505add.htm).
Reduced SIRT3 activity augments lysine acetylation under HFD conditions
To demonstrate that the acetylated proteins identified in the livers of WT HFD-fed mice were SIRT3 targets, we obtained liver extracts from Sirt3−/− mice under CH- and HFD-fed conditions. Proteins were immunoprecipitated using anti-(acetylated-lysine) antibodies and imunoblotted for CPS1, PC and aldolase B. Similar to previous results [11,13,15], Sirt3−/− mice had evidence of protein hyperacetylation (results not shown). However, following HFD feeding, a relative increase in acetylation of CPS1, PC and aldolase B was observed in livers of HFD-fed Sirt3−/− mice compared with WT HFD-fed mice (Figure 6), demonstrating that reduced SIRT3 activity augments protein acetylation under HFD conditions.
Figure 6. HFD increases acetylation of selected liver proteins in Sirt3−/− mice.

WT and Sirt3−/− mice were fed on the HFD (60 kcal % fat) for 8–9 weeks and starved overnight prior to killing and protein extraction. Western blots of liver homogenates were probed to obtain total protein expression levels of (A) CPS1, (B) PC and (C) aldolase B. GAPDH was used as a loading control. In a separate experiment (D), the acetylation (Ac) status was assessed by IP of equivalent amounts of sample with the anti-(acetylated-lysine) antibody, followed by Western blotting for the proteins of interest. Analysis was carried out in n = 4/genotype. Representative Western blots are shown. In each case, the blot is a composite of a larger gel in which all of the samples were run simultaneously. The composite was made by splicing complete lanes together for presentation purposes.
Reduced mitochondrial respiratory capacity and supercomplex formation in Sirt3−/− mice fed on the HFD
Given that Sirt3−/− mice are susceptible to increased cellular levels of ROS [12,17] and considering the well-established role between obesity and impaired mitochondrial metabolism [35,36], we hypothesized that Sirt3−/− mice would have evidence of reduced mitochondrial integrity under HFD conditions. As shown in Figure 7(A), under CH-fed conditions, there was no change in mitochondrial complex protein levels in Sirt3−/− mice compared with WT mice. However, under HFD conditions, there was a striking reduction in the amounts of subunits from mitochondrial complexes II, III, IV and V in livers from Sirt3−/− mice compared with WT HFD-fed mice, supporting the association of HFD conditions with reduced SIRT3 activity and mitochondrial dysfunction. Additionally, we measured the activities of respiratory chain complexes in livers of WT and Sirt3−/− animals fed on the HFD (Figures 7B-7F). The activities of complex III and IV were significantly decreased by 74 and 60 % respectively in livers from Sirt3−/− animals fed on the HFD compared with WT animals on the same diet (P < 0.02). Detailed raw data are shown in Table 2. The present study is the first to report combined defects in complexes III and IV in Sirt3-deficient mice. The respiratory activity was normalized to CS activity, which remained unchanged in WT compared with Sirt3−/− mice (results not shown).
Figure 7. HFD is associated with reduced mitochondrial activity and supercomplex assembly in livers from Sirt3−/− mice.

WT and Sirt3−/− mice were fed on either CH or the HFD (60 kcal % fat) for 8–9 weeks (n = 3/group) were starved overnight prior to killing and lysates of homogenized livers were prepared. (A) Western blot analysis with total OXPHOS antibody cocktail (complex I subunit NDUFB8, approx. 20 kDa; complex II subunit, approx. 30 kDa; complex III subunit core 2, approx. 47 kDa; complex IV subunit II, approx. 39 kDa; and complex V subunit ATP synthase α, approx. 53 kDa). Bands were detected using ECL (enhanced chemiluminescence). GAPDH provided a loading control. Representative blots are presented. The representative Western blot (A) is a composite of a larger gel in which all of the samples were run simultaneously together and the composite was made by splicing complete lanes together for presentation purposes. Separately, the activities of OXPHOS enzymes were assayed: (B) complex I, (C) complex II, (D) complex III, (E) complex II+III and (F) complex IV. Mitochondrial supernatants were isolated from WT and Sirt3−/− mice fed on the HFD and respiratory chain complex proteins were assayed for enzymatic activity (see the Experimental section). The activity for each complex is expressed as the fold change compared with the WT relative to CS activity. Values are means ± S.E.M., n = 4–5/group; #P < 0.02.
Table 2.
Individual activity of respiratory chain enzyme complexes and ratio over CS
| Parameter | HFD-fed WT mice
|
HFD-fed Sirt3−/− mice
|
||
|---|---|---|---|---|
| Activity | Activity/CS ratio | Activity | Activity/CS ratio | |
| Complex I | 15.7 (2.7–21.2) | 83 (16–124) | 10.7 (2.4–12.9) | 58 (14–65) |
| Complex II | 85.8 (49.3–118.5) | 436 (290–703) | 65.5 (54.8–123.4) | 387 (326–554) |
| Complex III | 3.0 (1.7–5.3)† | 14 (10–35)† | 0.7 (0.6–1.2) | 4 (3–7) |
| Complex II+III | 5.1 (4.1–7.8) | 33 (23–46) | 4.0 (3.3–6.5) | 23 (15–39) |
| Complex IV | 5.3 (4.3–10.3)† | 31 (23–67)† | 2.7 (2.4–3.3) | 15 (14–19) |
| CS | 170.1 (154.6–221.9) | NA | 171.6 (166.3–222.9) | NA |
Values are medians (range).
P < 0.01 compared with Sirt3−/−.
NA, not applicable.
DISCUSSION
The present study is the first to identify acetylation patterns in liver proteins of HFD-fed mice. The results show a remarkably broad spectrum of hyperacetylated proteins in the liver of HFD-fed mice that are potentially linked to mitochondrial dysfunction. In WT mice, the HFD was associated with decreased NAD+ levels and correlated with decreased SIRT3 activity, increased oxidation of mitochondrial proteins and fatty liver. Because SIRT3 is in the mitochondria and requires NAD+ as a cofactor, reduced SIRT3 activity may play a major function in mitochondrial fidelity and, by extension, loss of SIRT3 activity while on the HFD could result in hyperacetylation and reduced mitochondrial function. Low levels of NAD+ in obesity are consistent with recent findings showing reduced NAD+ in the liver of db/db and ob/ob mice and in insulin-resistant hepatic Irs1 (insulin receptor substrate 1)/Irs2-double knockout mice [34]. In these studies, PGC-1α (peroxisome-proliferator-activated receptor-γ coactivator-1α) was strongly acetylated and contributed to the dysregulation of mitochondrial function in obese mice [34]. Our present analysis of Sirt3-knockout mice fed on an HFD showed even further hyperacetylation of several mitochondrial proteins and disruption of mitochondrial complexes III, IV and V respiratory capacity. Sirt3−/− mice have normal levels of superoxide under unstressed conditions, but exhibit a stress-induced increase in superoxide levels and an age-related decrease in mtDNA (mitochondrial DNA) integrity in the liver [12]. This combination of findings suggests that reduced SIRT3 activity and hyperacetylation following exposure to an HFD may play a pivotal role in the mechanisms underlying mitochondrial dysfunction in the livers of obese mice.
ROS and oxidative stress are commonly increased in obesity [37] and both increase further in Sirt3−/− mice [15]. Notably, Sirt3-deficient mice fed on the HFD had significantly decreased mitochondrial respiratory chain complex integrity and a general loss of mitochondrial respiratory capacity without a concomitant change in mitochondrial abundance (Supplementary Figure S3). Combined with an increase in mitochondrial oxidized proteins, these results point to a generalized defect in mitochondrial capacity and increased oxidative stress in mice lacking SIRT3 fed on an HFD [38]. The disruption of mitochondrial respiratory chain complexes suggests that a decrease in SIRT3 may lead to the accumulation of cellular ROS following exposure to an HFD [23]. The disruption of OXPHOS supercomplexes shown in the present study in HFD-fed Sirt3−/− mice suggests that SIRT3 is important for the maintenance of mitochondrial integrity, which supports substrate channelling between associated OXPHOS complexes [39,40]. We identified the ROS-scavenging antioxidase enzymes catalase, uricase, peroxiredoxin and glutathione peroxidase as being hyperacetylated in livers from obese mice. Our present results showing increased mitochondrial protein oxidation and decreased OXPHOS enzyme activities in the livers of HFD-fed mice are consistent with higher levels of production of mitochondrial ROS under stress conditions [12,17], and suggest that hyperacetylation plays an important role in suppressing the effective function of oxidative stress defence enzymes and contributing to HFD-induced liver injury.
Several enzymes important for maintaining hepatic energy metabolism were identified as being hyperacetylated in HFD-fed mice. Among these, adenosylhomocysteinase is an NAD+-dependent enzyme that generates homocysteine from methionine during amino acid metabolism. Defects in methionine metabolism induce a complex set of cellular responses that disrupt hepatocyte lipid metabolism and, in some instances, activate apoptotic or non-apoptotic cell death pathways. S-Adenosylmethionine synthase (MAT or Ado) is key in the generation of AdoMet (S-adenosylmethionine), and the synthesis and breakdown of AdoMet is implicated in the methylation of phospholipids, proteins, DNA, RNA and small molecules [41]. Disruption in the balance of this process by hyperacetylation could therefore potentially have a myriad of downstream epigenetic consequences influencing the progression of fatty liver disease.
In addition to the heat-shock proteins Hsp60, Hsp70 and Hsp71, we identified GRP78 (glucose-regulated protein of 78 kDa), also known as BiP (immunoglobulin heavy-chain-binding protein), as a hyperacetylated target in fatty liver. GRP78 is the ER (endoplasmic reticulum) homologue of Hsp70 and contains conserved ATPase and peptide-binding domains [42]. As a chaperone protein, GRP78 directly interacts with all three ER stress sensors {PERK [PKR (double-stranded-RNA-dependent protein kinase)-like ER kinase], ATF6 (activating transcription factor 6) and IRE1 (inositol-requiring enzyme 1)} and maintains them in inactive forms in non-stressed cells. Stable overexpression of GRP78 inhibits the activation of SREBPs (sterol-regulatory-element-binding proteins) and the genes under their regulation [43], suggesting that hyperacetylation of GRP78 may be a potential factor affecting lipid metabolism as well as the ER stress response in fatty livers [43].
We observed hyperacetylation of important rate-limiting enzymes involved in gluconeogenesis: aldolase B and PC. Aldolase B stimulates gluconeogenesis by virtue of its strong association with fructose-1,6-bisphosphatase [44], whereas PC is an anaplerotic enzyme that participates in both gluconeogenesis and in refilling the TCA cycle. Importantly, PC is induced in obesity and diabetes [45,46], suggesting that its acetylation could potentially increase its activity during exposure to an HFD. Although not detected in the present study, recent results show that the key gluconeogenic enzyme PEPCK (phosphoenolpyruvate carboxykinase) is acetylated, which affects its protein stability and activity in human liver cells [1]. Given that gluconeogenesis is increased in obesity and Type 2 diabetic liver associated with insulin resistance [47], our present results showing hyperacetylation of PC and aldolase B in livers from HFD-fed mice suggest that acetylation may play an important role in the regulation of gluconeogenic activity in the liver.
Some subunits of complex V (e.g. ATP synthase subunit α and β) and complex III (cytochrome b-c1 complex subunit 1 and subunit 2) were hyperacetylated, and the amount of these protein products in the mature complex was also decreased. No subunits of complex IV were in the compendium of acetylated proteins, yet its function was clearly decreased in Sirt3-deficient mice fed on an HFD. However, several mitochondrial translation factors (elongation factor 1-α, elongation factor 1-γ and elongation factor Tu) were found to be acetylated. A deficiency in these factors is associated with mitochondrial dysfunction [48,49]. Other hyperacetylated proteins that can affect mitochondrial transcription and translation include certain components of the mitochondrial ribosome and subunits of succinyl-CoA ligase, the latter associated with reduced mtDNA [50].
Obesity and fatty liver disease are characterized by the paradoxical accumulation of triacylglycerols in the livers of insulin-resistant mice [34]. Strong evidence suggests that maintenance of NAD+ concentrations is required for normal mitochondrial fatty acid oxidation [8,34]. It has been shown that pharmacological stimulation of mitochondrial NADH oxidation dramatically promotes β-oxidation and ameliorates dyslipidaemia, adiposity and fatty liver in obese mice [34]. Escande et al. [51] recently showed that HFD-induced hepatic steatosis was associated with reduced SIRT1 activity with no change in NAD+ levels or SIRT1 expression, due to an association between SIRT1 and DBC-1 (deleted in breast cancer-1), a nuclear-localized protein that reduced SIRT1 activity. These investigators used a 60 kcal %HFD and did not measure SIRT3 or HAT activity. Our present results demonstrated that, although SIRT1 and SIRT3 levels did not change in the livers of HFD-fed mice, there was a significant decline in NAD+ levels with increasing obesity and a reduction in SIRT3 activity. Because SIRT3 requires NAD+ as a co-factor, it is ideally suited as a fast-acting metabolic sensor of the cells’ energy needs. Indeed, SIRT3 has emerged as a mediator of global lysine acetylation in mitochondria [11], and is ideally situated to function as a mitochondrial fidelity protein. Given the extensive association between obesity, oxidative stress [52,53] and fatty liver, these findings suggest that reduced SIRT3 may play a pivotal role in the mechanisms that govern hepatic HFD-induced mitochondrial dysfunction and will be the subject for future investigation.
Supplementary Material
Acknowledgments
FUNDING
This work was supported by the National Institutes of Health [grant numbers DK59767, P30-DK48520 (to J.E.F.)]; a Pilot & Feasibility Award from the University of Colorado, Center for Human Nutrition [grant number P30-DK048520-09 to K.R.J.)]; the National Institutes of Health Office of Research in Women’s Health BIRCWH Program [grant number K12 HD057022 (to C.E.M.)]; a Beginning Grant in Aid from the American Heart Association [grant number 09BGIA2060705 (to S.M.R.)]; and an American Diabetes Mentor-based Post-Doctoral Fellowship [grant number 7-08-MN-17 (to M.C.)]. P.W. was supported by Veterans Administration Merit Awards. J.B. and M.N.S. were funded by the National Heart, Lung, and Blood Institute. D.G. was supported by the Center for Cancer Research, National Cancer Institute.
Abbreviations used
- AdoMet
S-adenosylmethionine
- AP
alkaline phosphatase
- CH
control chow
- CPS1
carbamoyl-phosphate synthase 1
- CS
citrate synthase
- DTT
dithiothreitol
- ER
endoplasmic reticulum
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GRP78
glucose-regulated protein of 78 kDa
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- HFD
high-fat diet
- HRP
horseradish peroxidase
- Hsp70
heat-shock protein 70
- IP
immunoprecipitation
- IRS
insulin receptor substrate
- MS/MS
tandem MS
- mtDNA
mitochondrial DNA
- NAFLD
non-alcoholic fatty liver disease
- NEM
N-ethylmaleimide
- OXPHOS
oxidative phosphorylation
- PC
pyruvate carboxylase
- ROS
reactive oxygen species
- SIRT
sirtuin
- TCA
tricarboxylic acid
- WT
wild-type
Footnotes
AUTHOR CONTRIBUTION
Karen Jonscher performed the proteomic analysis; Shaikh Rahman and Carrie McCurdy provided the WT HFD-fed mice; Marisa Friederich, Mahua Choudhury and Johan Van Hove performed the mitochondrial assays; Peter Watson, Mahua Choudhury and Nicholas Birdsey performed the oxyblot and provided technical help on mitochondrial preparation; Jianjun Bao, David Gius, Michael Sack, Enxuan Jing and Ronald Kahn provided the Sirt3−/− mice, and Mahua Choudhury and Agnieszka Kendrick carried out the HFD experiments; Mahua Choudhury and Agnieszka Kendrick performed the SIRT analysis; and Agnieszka Kendrick, Mahua Choudhury, Karen Jonscher and Jacob Friedman wrote the paper. All authors contributed to and have approved the final manuscript.
References
- 1.Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, et al. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science. 2010;327:1004–1007. doi: 10.1126/science.1179687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwer B, Eckersdorff M, Li Y, Silva JC, Fermin D, Kurtev MV, Giallourakis C, Comb MJ, Alt FW, Lombard DB. Calorie restriction alters mitochondrial protein acetylation. Aging Cell. 2009;8:604–606. doi: 10.1111/j.1474-9726.2009.00503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scher MB, Vaquero A, Reinberg D. SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev. 2007;21:920–928. doi: 10.1101/gad.1527307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R, Leid M, McBurney MW, Guarente L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-γ. Nature. 2004;429:771–776. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- 7.Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z, Puigserver P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α. EMBO J. 2007;26:1913–1923. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, Kitamura Y, Altomonte J, Dong H, Accili D, Spiegelman BM. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1α interaction. Nature. 2003;423:550–555. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- 9.Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390–392. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- 10.Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell. 2006;23:607–618. doi: 10.1016/j.molcel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 11.Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol. 2007;27:8807–8814. doi: 10.1128/MCB.01636-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim HS, Patel K, Muldoon-Jacobs K, Bisht KS, Aykin-Burns N, Pennington JD, van der Meer R, Nguyen P, Savage J, Owens KM, et al. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell. 2010;17:41–52. doi: 10.1016/j.ccr.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464:121–125. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi T, Wang F, Stieren E, Tong Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J Biol Chem. 2005;280:13560–13567. doi: 10.1074/jbc.M414670200. [DOI] [PubMed] [Google Scholar]
- 15.Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, Finkel T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci USA. 2008;105:14447–14452. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacobs KM, Pennington JD, Bisht KS, Aykin-Burns N, Kim HS, Mishra M, Sun L, Nguyen P, Ahn BH, Leclerc J, Deng CX, et al. SIRT3 interacts with the daf-16 homolog FOXO3a in the mitochondria, as well as increases FOXO3a dependent gene expression. Int J Biol Sci. 2008;4:291–299. doi: 10.7150/ijbs.4.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest. 2009;119:2758–2771. doi: 10.1172/JCI39162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koc EC, Yang Y, Cimen H, Han M-J, Tong S, Deng J-H, Koc H, Palacios OM, Montier L, Bai Y. NAD+-dependent deacetylase SIRT3 regulates mitochondrial protein synthesis by deacetylation of mitochondrial ribosomal protein L10. FASEB J. 2010;24:467.462. doi: 10.1074/jbc.M109.053421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pillai VB, Sundaresan NR, Kim G, Gupta M, Rajamohan SB, Pillai JB, Samant S, Ravindra PV, Isbatan A, Gupta MP. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMP-activated kinase pathway. J Biol Chem. 2010;285:3133–3144. doi: 10.1074/jbc.M109.077271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cimen H, Han MJ, Yang Y, Tong Q, Koc H, Koc EC. Regulation of succinate dehydrogenase activity by SIRT3 in mammalian mitochondria. Biochemistry. 2010;49:304–311. doi: 10.1021/bi901627u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc Natl Acad Sci USA. 2006;103:10224–10229. doi: 10.1073/pnas.0603968103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schlicker C, Gertz M, Papatheodorou P, Kachholz B, Becker CF, Steegborn C. Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J Mol Biol. 2008;382:790–801. doi: 10.1016/j.jmb.2008.07.048. [DOI] [PubMed] [Google Scholar]
- 23.Bao J, Lu Z, Joseph JJ, Carabenciov D, Dimond CC, Pang L, Samsel L, McCoy JP, Jr, Leclerc J, Nguyen P, et al. Characterization of the murine SIRT3 mitochondrial localization sequence and comparison of mitochondrial enrichment and deacetylase activity of long and short SIRT3 isoforms. J Cell Biochem. 2010;110:238–247. doi: 10.1002/jcb.22531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Banerjee S, Bueso-Ramos C, Aggarwal BB. Suppression of 7,12-dimethylbenz(a)anthracene-induced mammary carcinogenesis in rats by resveratrol: role of nuclear factor-κB, cyclooxygenase 2, and matrix metalloprotease 9. Cancer Res. 2002;62:4945–4954. [PubMed] [Google Scholar]
- 25.Choudhury M, Shukla SD. Surrogate alcohols and their metabolites modify histone H3 acetylation: involvement of histone acetyl transferase and histone deacetylase. Alcohol Clin Exp Res. 2008;32:829–839. doi: 10.1111/j.1530-0277.2008.00630.x. [DOI] [PubMed] [Google Scholar]
- 26.Trapp J, Jung M. The role of NAD+ dependent histone deacetylases (sirtuins) in ageing. Curr Drug Targets. 2006;7:1553–1560. doi: 10.2174/1389450110607011553. [DOI] [PubMed] [Google Scholar]
- 27.Yang Y, Fu W, Chen J, Olashaw N, Zhang X, Nicosia SV, Bhalla K, Bai W. SIRT1 sumoylation regulates its deacetylase activity and cellular response to genotoxic stress. Nat Cell Biol. 2007;9:1253–1262. doi: 10.1038/ncb1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moon KH, Hood BL, Mukhopadhyay P, Rajesh M, Abdelmegeed MA, Kwon YI, Conrads TP, Veenstra TD, Song BJ, Pacher P. Oxidative inactivation of key mitochondrial proteins leads to dysfunction and injury in hepatic ischemia reperfusion. Gastroenterology. 2008;135:1344–1357. doi: 10.1042/BJ20100791. Received 27 May 2010/30 September 2010; accepted 2 November 2010 Published as BJ Immediate Publication 2 November 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rahman S, Blok RB, Dahl HH, Danks DM, Kirby DM, Chow CW, Christodoulou J, Thorburn DR. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann Neurol. 1996;39:343–351. doi: 10.1002/ana.410390311. [DOI] [PubMed] [Google Scholar]
- 30.Fersht A. Enzyme Structure and Mechanism. W.H. Freeman & Company; New York: 1985. [Google Scholar]
- 31.Smith L. Spectrophotometric assay of cytochrome c oxidase. Methods Biochem Anal. 1955;2:427–434. doi: 10.1002/9780470110188.ch13. [DOI] [PubMed] [Google Scholar]
- 32.Buettner R, Scholmerich J, Bollheimer LC. High-fat diets: modeling the metabolic disorders of human obesity in rodents. Obesity. 2007;15:798–808. doi: 10.1038/oby.2007.608. [DOI] [PubMed] [Google Scholar]
- 33.Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- 34.Cheng Z, Guo S, Copps K, Dong X, Kollipara R, Rodgers JT, Depinho RA, Puigserver P, White MF. Foxo1 integrates insulin signaling with mitochondrial function in the liver. Nat Med. 2009;15:1307–1311. doi: 10.1038/nm.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hojlund K, Mogensen M, Sahlin K, Beck-Nielsen H. Mitochondrial dysfunction in type 2 diabetes and obesity. Endocrinol Metab Clin North Am. 2008;37:713–731. doi: 10.1016/j.ecl.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 36.Civitarese AE, Ravussin E. Mitochondrial energetics and insulin resistance. Endocrinology. 2008;149:950–954. doi: 10.1210/en.2007-1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nagae A, Fujita M, Kawarazaki H, Matsui H, Ando K, Fujita T. Sympathoexcitation by oxidative stress in the brain mediates arterial pressure elevation in obesity-induced hypertension. Circulation. 2009;119:978–986. doi: 10.1161/CIRCULATIONAHA.108.824730. [DOI] [PubMed] [Google Scholar]
- 38.Bao J, Scott I, Lu Z, Pang L, Dimond CC, Gius D, Sack MN. SIRT3 is regulated by nutrient excess and modulates hepatic susceptibility to lipotoxicity. Free Radical Biol Med. 2010;49:1230–1237. doi: 10.1016/j.freeradbiomed.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heinemeyer J, Braun HP, Boekema EJ, Kouril R. A structural model of the cytochrome C reductase/oxidase supercomplex from yeast mitochondria. J Biol Chem. 2007;282:12240–12248. doi: 10.1074/jbc.M610545200. [DOI] [PubMed] [Google Scholar]
- 40.Schagger H. Respiratory chain supercomplexes. IUBMB Life. 2001;52:119–128. doi: 10.1080/15216540152845911. [DOI] [PubMed] [Google Scholar]
- 41.Chiang PK, Gordon RK, Tal J, Zeng GC, Doctor BP, Pardhasaradhi K, McCann PP. S-Adenosylmethionine and methylation. FASEB J. 1996;10:471–480. [PubMed] [Google Scholar]
- 42.Ni M, Lee AS. ER chaperones in mammalian development and human diseases. FEBS Lett. 2007;581:3641–3651. doi: 10.1016/j.febslet.2007.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Werstuck GH, Lentz SR, Dayal S, Hossain GS, Sood SK, Shi YY, Zhou J, Maeda N, Krisans SK, Malinow MR, Austin RC. Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J Clin Invest. 2001;107:1263–1273. doi: 10.1172/JCI11596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yanez AJ, Ludwig HC, Bertinat R, Spichiger C, Gatica R, Berlien G, Leon O, Brito M, Concha II, Slebe JC. Different involvement for aldolase isoenzymes in kidney glucose metabolism: aldolase B but not aldolase A colocalizes and forms a complex with FBPase. J Cell Physiol. 2005;202:743–753. doi: 10.1002/jcp.20183. [DOI] [PubMed] [Google Scholar]
- 45.Jitrapakdee S, Walker ME, Wallace JC. Functional expression, purification, and characterization of recombinant human pyruvate carboxylase. Biochem Biophys Res Commun. 1999;266:512–517. doi: 10.1006/bbrc.1999.1846. [DOI] [PubMed] [Google Scholar]
- 46.Jitrapakdee S, Wallace JC. Structure, function and regulation of pyruvate carboxylase. Biochem J. 1999;340:1–16. doi: 10.1042/bj3400001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gastaldelli A, Cusi K, Pettiti M, Hardies J, Miyazaki Y, Berria R, Buzzigoli E, Sironi AM, Cersosimo E, Ferrannini E, Defronzo RA. Relationship between hepatic/visceral fat and hepatic insulin resistance in nondiabetic and type 2 diabetic subjects. Gastroenterology. 2007;133:496–506. doi: 10.1053/j.gastro.2007.04.068. [DOI] [PubMed] [Google Scholar]
- 48.Coenen MJ, Antonicka H, Ugalde C, Sasarman F, Rossi R, Heister JG, Newbold RF, Trijbels FJ, van den Heuvel LP, Shoubridge EA, Smeitink JA. Mutant mitochondrial elongation factor G1 and combined oxidative phosphorylation deficiency. N Engl J Med. 2004;351:2080–2086. doi: 10.1056/NEJMoa041878. [DOI] [PubMed] [Google Scholar]
- 49.Shutt TE, Shadel GS. A compendium of human mitochondrial gene expression machinery with links to disease. Environ Mol Mutagen. 2010;51:360–379. doi: 10.1002/em.20571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Van Hove JL, Saenz MS, Thomas JA, Gallagher RC, Lovell MA, Fenton LZ, Shanske S, Myers SM, Wanders RJ, Ruiter J, et al. Succinyl-CoA ligase deficiency: a mitochondrial hepatoencephalomyopathy. Pediatr Res. 2010;68:159–164. doi: 10.1203/PDR.0b013e3181e5c3a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Escande C, Chini CC, Nin V, Dykhouse KM, Novak CM, Levine J, van Deursen J, Gores GJ, Chen J, Lou Z, Chini EN. Deleted in breast cancer-1 regulates SIRT1 activity and contributes to high-fat diet-induced liver steatosis in mice. J Clin Invest. 2010;120:545–558. doi: 10.1172/JCI39319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Urakawa H, Katsuki A, Sumida Y, Gabazza EC, Murashima S, Morioka K, Maruyama N, Kitagawa N, Tanaka T, Hori Y, et al. Oxidative stress is associated with adiposity and insulin resistance in men. J Clin Endocrinol Metab. 2003;88:4673–4676. doi: 10.1210/jc.2003-030202. [DOI] [PubMed] [Google Scholar]
- 53.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.