

Abstract
With fast development and wide applications of next-generation sequencing (NGS) technologies, genomic sequence information is within reach to aid the achievement of goals to decode life mysteries, make better crops, detect pathogens, and improve life qualities. NGS systems are typically represented by SOLiD/Ion Torrent PGM from Life Sciences, Genome Analyzer/HiSeq 2000/MiSeq from Illumina, and GS FLX Titanium/GS Junior from Roche. Beijing Genomics Institute (BGI), which possesses the world's biggest sequencing capacity, has multiple NGS systems including 137 HiSeq 2000, 27 SOLiD, one Ion Torrent PGM, one MiSeq, and one 454 sequencer. We have accumulated extensive experience in sample handling, sequencing, and bioinformatics analysis. In this paper, technologies of these systems are reviewed, and first-hand data from extensive experience is summarized and analyzed to discuss the advantages and specifics associated with each sequencing system. At last, applications of NGS are summarized.
1. Introduction
(Deoxyribonucleic acid) DNA was demonstrated as the genetic material by Oswald Theodore Avery in 1944. Its double helical strand structure composed of four bases was determined by James D. Watson and Francis Crick in 1953, leading to the central dogma of molecular biology. In most cases, genomic DNA defined the species and individuals, which makes the DNA sequence fundamental to the research on the structures and functions of cells and the decoding of life mysteries [1]. DNA sequencing technologies could help biologists and health care providers in a broad range of applications such as molecular cloning, breeding, finding pathogenic genes, and comparative and evolution studies. DNA sequencing technologies ideally should be fast, accurate, easy-to-operate, and cheap. In the past thirty years, DNA sequencing technologies and applications have undergone tremendous development and act as the engine of the genome era which is characterized by vast amount of genome data and subsequently broad range of research areas and multiple applications. It is necessary to look back on the history of sequencing technology development to review the NGS systems (454, GA/HiSeq, and SOLiD), to compare their advantages and disadvantages, to discuss the various applications, and to evaluate the recently introduced PGM (personal genome machines) and third-generation sequencing technologies and applications. All of these aspects will be described in this paper. Most data and conclusions are from independent users who have extensive first-hand experience in these typical NGS systems in BGI (Beijing Genomics Institute).
Before talking about the NGS systems, we would like to review the history of DNA sequencing briefly. In 1977, Frederick Sanger developed DNA sequencing technology which was based on chain-termination method (also known as Sanger sequencing), and Walter Gilbert developed another sequencing technology based on chemical modification of DNA and subsequent cleavage at specific bases. Because of its high efficiency and low radioactivity, Sanger sequencing was adopted as the primary technology in the “first generation” of laboratory and commercial sequencing applications [2]. At that time, DNA sequencing was laborious and radioactive materials were required. After years of improvement, Applied Biosystems introduced the first automatic sequencing machine (namely AB370) in 1987, adopting capillary electrophoresis which made the sequencing faster and more accurate. AB370 could detect 96 bases one time, 500 K bases a day, and the read length could reach 600 bases. The current model AB3730xl can output 2.88 M bases per day and read length could reach 900 bases since 1995. Emerged in 1998, the automatic sequencing instruments and associated software using the capillary sequencing machines and Sanger sequencing technology became the main tools for the completion of human genome project in 2001 [3]. This project greatly stimulated the development of powerful novel sequencing instrument to increase speed and accuracy, while simultaneously reducing cost and manpower. Not only this, X-prize also accelerated the development of next-generation sequencing (NGS) [4]. The NGS technologies are different from the Sanger method in aspects of massively parallel analysis, high throughput, and reduced cost. Although NGS makes genome sequences handy, the followed data analysis and biological explanations are still the bottle-neck in understanding genomes.
Following the human genome project, 454 was launched by 454 in 2005, and Solexa released Genome Analyzer the next year, followed by (Sequencing by Oligo Ligation Detection) SOLiD provided from Agencourt, which are three most typical massively parallel sequencing systems in the next-generation sequencing (NGS) that shared good performance on throughput, accuracy, and cost compared with Sanger sequencing (shown in Table 1(a)). These founder companies were then purchased by other companies: in 2006 Agencourt was purchased by Applied Biosystems, and in 2007, 454 was purchased by Roche, while Solexa was purchased by Illumina. After years of evolution, these three systems exhibit better performance and their own advantages in terms of read length, accuracy, applications, consumables, man power requirement and informatics infrastructure, and so forth. The comparison of these three systems will be focused and discussed in the later part of this paper (also see Tables 1(a), 1(b), and 1(c)).
Table 1.
(a) Advantage and mechanism of sequencers. (b) Components and cost of sequencers. (c) Application of sequencers.
(a)
| Sequencer | 454 GS FLX | HiSeq 2000 | SOLiDv4 | Sanger 3730xl |
|---|---|---|---|---|
| Sequencing mechanism | Pyrosequencing | Sequencing by synthesis | Ligation and two-base coding | Dideoxy chain termination |
| Read length | 700 bp | 50SE, 50PE, 101PE | 50 + 35 bp or 50 + 50 bp |
400 ~ 900 bp |
| Accuracy | 99.9%* | 98%, (100PE) | 99.94% *raw data | 99.999% |
| Reads | 1 M | 3 G | 1200~1400 M | — |
| Output data/run | 0.7 Gb | 600 Gb | 120 Gb | 1.9~84 Kb |
| Time/run | 24 Hours | 3~10 Days | 7 Days for SE 14 Days for PE |
20 Mins~3 Hours |
| Advantage | Read length, fast | High throughput | Accuracy | High quality, long read length |
| Disadvantage | Error rate with polybase more than 6, high cost, low throughput | Short read assembly | Short read assembly | High cost low throughput |
(b)
| Sequencers | 454 GS FLX | HiSeq 2000 | SOLiDv4 | 3730xl |
|---|---|---|---|---|
| Instrument price | Instrument $500,000, $7000 per run | Instrument $690,000, $6000/(30x) human genome | Instrument $495,000, $15,000/100 Gb | Instrument $95,000, about $4 per 800 bp reaction |
| CPU | 2* Intel Xeon X5675 | 2* Intel Xeon X5560 | 8* processor 2.0 GHz | Pentium IV 3.0 GHz |
| Memory | 48 GB | 48 GB | 16 GB | 1 GB |
| Hard disk | 1.1 TB | 3 TB | 10 TB | 280 GB |
| Automation in library preparation | Yes | Yes | Yes | No |
| Other required device | REM e system | cBot system | EZ beads system | No |
| Cost/million bases | $10 | $0.07 | $0.13 | $2400 |
(c)
| Sequencers | 454 GS FLX | HiSeq 2000 | SOLiDv4 | 3730xl |
|---|---|---|---|---|
| Resequencing | Yes | Yes | ||
| De novo | Yes | Yes | Yes | |
| Cancer | Yes | Yes | Yes | |
| Array | Yes | Yes | Yes | Yes |
| High GC sample | Yes | Yes | Yes | |
| Bacterial | Yes | Yes | Yes | |
| Large genome | Yes | Yes | ||
| Mutation detection | Yes | Yes | Yes | Yes |
(1) All the data is taken from daily average performance runs in BGI. The average daily sequence data output is about 8 Tb in BGI when about 80% sequencers (mainly HiSeq 2000) are running.
(2) The reagent cost of 454 GS FLX Titanium is calculated based on the sequencing of 400 bp; the reagent cost of HiSeq 2000 is calculated based on the sequencing of 200 bp; the reagent cost of SOLiDv4 is calculated based on the sequencing of 85 bp.
(3) HiSeq 2000 is more flexible in sequencing types like 50SE, 50PE, or 101PE.
(4) SOLiD has high accuracy especially when coverage is more than 30x, so it is widely used in detecting variations in resequencing, targeted resequencing, and transcriptome sequencing. Lanes can be independently run to reduce cost.
2. Roche 454 System
Roche 454 was the first commercially successful next generation system. This sequencer uses pyrosequencing technology [5]. Instead of using dideoxynucleotides to terminate the chain amplification, pyrosequencing technology relies on the detection of pyrophosphate released during nucleotide incorporation. The library DNAs with 454-specific adaptors are denatured into single strand and captured by amplification beads followed by emulsion PCR [6]. Then on a picotiter plate, one of dNTP (dATP, dGTP, dCTP, dTTP) will complement to the bases of the template strand with the help of ATP sulfurylase, luciferase, luciferin, DNA polymerase, and adenosine 5′ phosphosulfate (APS) and release pyrophosphate (PPi) which equals the amount of incorporated nucleotide. The ATP transformed from PPi drives the luciferin into oxyluciferin and generates visible light [7]. At the same time, the unmatched bases are degraded by apyrase [8]. Then another dNTP is added into the reaction system and the pyrosequencing reaction is repeated.
The read length of Roche 454 was initially 100–150 bp in 2005, 200000+ reads, and could output 20 Mb per run [9, 10]. In 2008 454 GS FLX Titanium system was launched; through upgrading, its read length could reach 700 bp with accuracy 99.9% after filter and output 0.7 G data per run within 24 hours. In late 2009 Roche combined the GS Junior a bench top system into the 454 sequencing system which simplified the library preparation and data processing, and output was also upgraded to 14 G per run [11, 12]. The most outstanding advantage of Roche is its speed: it takes only 10 hours from sequencing start till completion. The read length is also a distinguished character compared with other NGS systems (described in the later part of this paper). But the high cost of reagents remains a challenge for Roche 454. It is about $12.56 × 10−6 per base (counting reagent use only). One of the shortcomings is that it has relatively high error rate in terms of poly-bases longer than 6 bp. But its library construction can be automated, and the emulsion PCR can be semiautomated which could reduce the manpower in a great extent. Other informatics infrastructure and sequencing advantages are listed and compared with HiSeq 2000 and SOLiD systems in Tables 1(a), 1(b), and 1(c).
2.1. 454 GS FLX Titanium Software
GS RunProcessor is the main part of the GS FLX Titanium system. The software is in charge of picture background normalization, signal location correction, cross-talk correction, signals conversion, and sequencing data generation. GS RunProcessor would produce a series of files including SFF (standard flowgram format) files each time after run. SFF files contain the basecalled sequences and corresponding quality scores for all individual, high-quality reads (filtered reads). And it could be viewed directly from the screen of GS FLX Titanium system. Using GS De Novo Assembler, GS Reference Mapper and GS Amplicon Variant Analyzer provided by GS FLX Titanium system, SFF files can be applied in multiaspects and converted into fastq format for further data analyzing.
3. AB SOLiD System
(Sequencing by Oligo Ligation Detection) SOLiD was purchased by Applied Biosystems in 2006. The sequencer adopts the technology of two-base sequencing based on ligation sequencing. On a SOLiD flowcell, the libraries can be sequenced by 8 base-probe ligation which contains ligation site (the first base), cleavage site (the fifth base), and 4 different fluorescent dyes (linked to the last base) [10]. The fluorescent signal will be recorded during the probes complementary to the template strand and vanished by the cleavage of probes' last 3 bases. And the sequence of the fragment can be deduced after 5 round of sequencing using ladder primer sets.
The read length of SOLiD was initially 35 bp reads and the output was 3 G data per run. Owing to two-base sequencing method, SOLiD could reach a high accuracy of 99.85% after filtering. At the end of 2007, ABI released the first SOLiD system. In late 2010, the SOLiD 5500xl sequencing system was released. From SOLiD to SOLiD 5500xl, five upgrades were released by ABI in just three years. The SOLiD 5500xl realized improved read length, accuracy, and data output of 85 bp, 99.99%, and 30 G per run, respectively. A complete run could be finished within 7 days. The sequencing cost is about $40 × 10−9 per base estimated from reagent use only by BGI users. But the short read length and resequencing only in applications is still its major shortcoming [13]. Application of SOLiD includes whole genome resequencing, targeted resequencing, transcriptome research (including gene expression profiling, small RNA analysis, and whole transcriptome analysis), and epigenome (like ChIP-Seq and methylation). Like other NGS systems, SOLiD's computational infrastructure is expensive and not trivial to use; it requires an air-conditioned data center, computing cluster, skilled personnel in computing, distributed memory cluster, fast networks, and batch queue system. Operating system used by most researchers is GNU/LINUX. Each solid sequencer run takes 7 days and generates around 4 TB of raw data. More data will be generated after bioinformatics analysis. This information is listed and compared with other NGS systems in Tables 1(a), 1(b), and 1(c). Automation can be used in library preparations, for example, Tecan system which integrated a Covaris A and Roche 454 REM e system [14].
3.1. SOLiD Software
After the sequencing with SOLiD, the original sequence of color coding will be accumulated. According to double-base coding matrix, the original color sequence can be decoded to get the base sequence if we knew the base types for one of any position in the sequence. Because of a kind of color corresponding four base pair, the color coding of the base will directly influence the decoding of its following base. It said that a wrong color coding will cause a chain decoding mistakes. BioScope is SOLiD data analysis package which provides a validated, single framework for resequencing, ChIP-Seq, and whole transcriptome analysis. It depends on reference for the follow-up data analysis. First, the software converts the base sequences of references into color coding sequence. Second, the color-coding sequence of references is compared with the original sequence of color-coding to get the information of mapping with newly developed mapping algorithm MaxMapper.
4. Illumina GA/HiSeq System
In 2006, Solexa released the Genome Analyzer (GA), and in 2007 the company was purchased by Illumina. The sequencer adopts the technology of sequencing by synthesis (SBS). The library with fixed adaptors is denatured to single strands and grafted to the flowcell, followed by bridge amplification to form clusters which contains clonal DNA fragments. Before sequencing, the library splices into single strands with the help of linearization enzyme [10], and then four kinds of nucleotides (ddATP, ddGTP, ddCTP, ddTTP) which contain different cleavable fluorescent dye and a removable blocking group would complement the template one base at a time, and the signal could be captured by a (charge-coupled device) CCD.
At first, solexa GA output was 1 G/run. Through improvements in polymerase, buffer, flowcell, and software, in 2009 the output of GA increased to 20 G/run in August (75PE), 30 G/run in October (100PE), and 50 G/run in December (Truseq V3, 150PE), and the latest GAIIx series can attain 85 G/run. In early 2010, Illumina launched HiSeq 2000, which adopts the same sequencing strategy with GA, and BGI was among the first globally to adopt the HiSeq system. Its output was 200 G per run initially, improved to 600 G per run currently which could be finished in 8 days. In the foreseeable future, it could reach 1 T/run when a personal genome cost could drop below $1 K. The error rate of 100PE could be below 2% in average after filtering (BGI's data). Compared with 454 and SOLiD, HiSeq 2000 is the cheapest in sequencing with $0.02/million bases (reagent counted only by BGI). With multiplexing incorporated in P5/P7 primers and adapters, it could handle thousands of samples simultaneously. HiSeq 2000 needs (HiSeq control software) HCS for program control, (real-time analyzer software) RTA to do on-instrument base-calling, and CASAVA for secondary analysis. There is a 3 TB hard disk in HiSeq 2000. With the aid of Truseq v3 reagents and associated softwares, HiSeq 2000 has improved much on high GC sequencing. MiSeq, a bench top sequencer launched in 2011 which shared most technologies with HiSeq, is especially convenient for amplicon and bacterial sample sequencing. It could sequence 150PE and generate 1.5 G/run in about 10 hrs including sample and library preparation time. Library preparation and their concentration measurement can both be automated with compatible systems like Agilent Bravo, Hamilton Banadu, Tecan, and Apricot Designs.
4.1. HiSeq Software
HiSeq control system (HCS) and real-time analyzer (RTA) are adopted by HiSeq 2000. These two softwares could calculate the number and position of clusters based on their first 20 bases, so the first 20 bases of each sequencing would decide each sequencing's output and quality. HiSeq 2000 uses two lasers and four filters to detect four types of nucleotide (A, T, G, and C). The emission spectra of these four kinds of nucleotides have cross-talk, so the images of four nucleotides are not independent and the distribution of bases would affect the quality of sequencing. The standard sequencing output files of the HiSeq 2000 consist of *bcl files, which contain the base calls and quality scores in each cycle. And then it is converted into *_qseq.txt files by BCL Converter. The ELAND program of CASAVA (offline software provided by Illumina) is used to match a large number of reads against a genome.
In conclusion, of the three NGS systems described before, the Illumina HiSeq 2000 features the biggest output and lowest reagent cost, the SOLiD system has the highest accuracy [11], and the Roche 454 system has the longest read length. Details of three sequencing system are list in Tables 1(a), 1(b), and 1(c).
5. Compact PGM Sequencers
Ion Personal Genome Machine (PGM) and MiSeq were launched by Ion Torrent and Illumina. They are both small in size and feature fast turnover rates but limited data throughput. They are targeted to clinical applications and small labs.
5.1. Ion PGM from Ion Torrent
Ion PGM was released by Ion Torrent at the end of 2010. PGM uses semiconductor sequencing technology. When a nucleotide is incorporated into the DNA molecules by the polymerase, a proton is released. By detecting the change in pH, PGM recognized whether the nucleotide is added or not. Each time the chip was flooded with one nucleotide after another, if it is not the correct nucleotide, no voltage will be found; if there is 2 nucleotides added, there is double voltage detected [15]. PGM is the first commercial sequencing machine that does not require fluorescence and camera scanning, resulting in higher speed, lower cost, and smaller instrument size. Currently, it enables 200 bp reads in 2 hours and the sample preparation time is less than 6 hours for 8 samples in parallel.
An exemplary application of the Ion Torrent PGM sequencer is the identification of microbial pathogens. In May and June of 2011, an ongoing outbreak of exceptionally virulent Shiga-toxin- (Stx) producing Escherichia coli O104:H4 centered in Germany [16, 17], there were more than 3000 people infected. The whole genome sequencing on Ion Torrent PGM sequencer and HiSeq 2000 helped the scientists to identify the type of E. coli which would directly apply the clue to find the antibiotic resistance. The strain appeared to be a hybrid of two E. coli strains—entero aggregative E. coli and entero hemorrhagic E. coli—which may help explain why it has been particularly pathogenic. From the sequencing result of E. coli TY2482 [18], PGM shows the potential of having a fast, but limited throughput sequencer when there is an outbreak of new disease.
In order to study the sequencing quality, mapping rate, and GC depth distribution of Ion Torrent and compare with HiSeq 2000, a high GC Rhodobacter sample with high GC content (66%) and 4.2 Mb genome was sequenced in these two different sequencers (Table 2). In another experiment, E. coli K12 DH10B (NC_010473.1) with GC 50.78% was sequenced by Ion Torrent for analysis of quality value, read length, position accuracies, and GC distribution (Figure 1).
Table 2.
Comparison in alignment between Ion Torrent and HiSeq 2000.
| Ion Torrenta | HiSeq 2000b | |
|---|---|---|
| Total reads num | 165518 | 205683 |
| Total bases num | 18574086 | 18511470 |
| Max read length | 201 | 90 |
| Min read length | 15 | 90 |
| Map reads num | 157258 | 157511 |
| Map rate | 95% | 76.57% |
| Covered rate | 96.50% | 93.11% |
| Total map length | 15800258 | 14176420 |
| Total mismatch base | 53475 | 142425 |
| Total insertion base | 109550 | 1397 |
| Total insertion num | 95740 | 1332 |
| Total deletion base | 152495 | 431 |
| Total deletion num | 139264 | 238 |
| Ave mismatch rate | 0.338% | 1.004% |
| Ave insertion rate | 0.693% | 0.009% |
| Ave deletion rate | 0.965% | 0.003% |
a: use TMAP to align; b: use SOAP2 to align.
Figure 1.
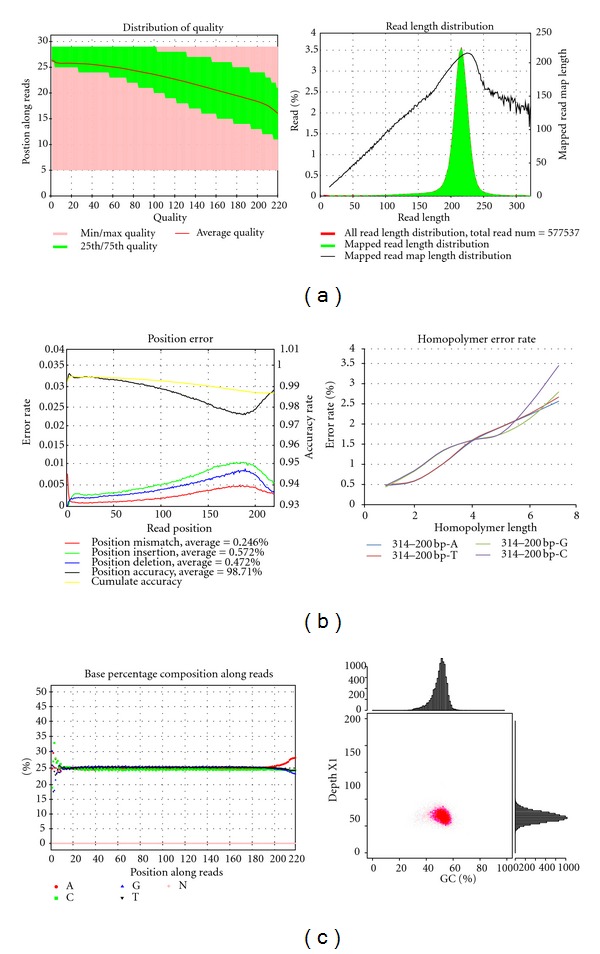
Ion Torrent sequencing quality. E. coli K12 DH10B (NC_010473.1) with GC 50.78% was used for this experiment. (a) is 314–200 bp from Ion Torrent. The left figure is quality value: pink range represents quality minimum and maximum values each position has. Green area represents the top and bottom quarter (1/4) reads of quality. Red line represents the average quality value in the position. The right figure is read length analysis: colored histogram represents the real read length. The black line represents the mapped length, and because it allows 3′ soft clipping, the length is different from the real read length. (b) is accuracy analysis. In each position, accuracy type including mismatch, insertion, and deletion is shown on the left y-axis. The average accuracy is shown the right y-axis. Accuracy of 200 bp sequencing could reach 99%. (c) is base composition along reads (left) and GC distribution analysis (right). The left figure is base composition in each position of reads. Base line splits after about 95 cycles indicating an inaccurate sequencing. The right one uses 500 bp window and the GC distribution is quite even. The data using high GC samples also indicates a good performance in Ion Torrent (data not shown).
5.1.1. Sequencing Quality
The quality of Ion Torrent is more stable, while the quality of HiSeq 2000 decreases noticeably after 50 cycles, which may be caused by the decay of fluorescent signal with increasing the read length (shown in Figure 1).
5.1.2. Mapping
The insert size of library of Rhodobacter was 350 bp, and 0.5 Gb data was obtained from HiSeq. The sequencing depth was over 100x, and the contig and scaffold N50 were 39530 bp and 194344 bp, respectively. Based on the assembly result, we used 33 Mb which is obtained from ion torrent with 314 chip to analyze the map rate. The alignment comparison is Table 2.
The map rate of Ion Torrent is higher than HiSeq 2000, but it is incomparable because of the different alignment methods used in different sequencers. Besides the significant difference on data including mismatch rate, insertion rate, and deletion rate, HiSeq 2000 and Ion Torrent were still incomparable because of the different sequencing principles. For example, the polynucleotide site could not be indentified easily in Ion Torrent. But it is shown that Ion Torrent has a stable quality along sequencing reads and a good performance on mismatch accuracies, but rather a bias in detection of indels. Different types of accuracy are analyzed and shown in Figure 1.
5.1.3. GC Depth Distribution
The GC depth distribution is better in Ion Torrent from Figure 1. In Ion Torrent, the sequencing depth is similar while the GC content is from 63% to 73%. However in HiSeq 2000, the average sequencing depth is 4x when the GC content is 60%, while it is 3x with 70% GC content.
Ion Torrent has already released Ion 314 and 316 and planned to launch Ion 318 chips in late 2011. The chips are different in the number of wells resulting in higher production within the same sequencing time. The Ion 318 chip enables the production of >1 Gb data in 2 hours. Read length is expected to increase to >400 bp in 2012.
5.2. MiSeq from Illumina
MiSeq which still uses SBS technology was launched by Illumina. It integrates the functions of cluster generation, SBS, and data analysis in a single instrument and can go from sample to answer (analyzed data) within a single day (as few as 8 hours). The Nextera, TruSeq, and Illumina's reversible terminator-based sequencing by synthesis chemistry was used in this innovative engineering. The highest integrity data and broader range of application, including amplicon sequencing, clone checking, ChIP-Seq, and small genome sequencing, are the outstanding parts of MiSeq. It is also flexible to perform single 36 bp reads (120 MB output) up to 2 × 150 paired-end reads (1–1.5 GB output) in MiSeq. Due to its significant improvement in read length, the resulting data performs better in contig assembly compared with HiSeq (data not shown). The related sequencing result of MiSeq is shown in Table 3. We also compared PGM with MiSeq in Table 4.
Table 3.
MiSeq 150PE data.
| Sample | GC | Q20 | Q30 |
|---|---|---|---|
| Human HPV | 33.57; 33.62 | 98.26; 95.52 | 93.64; 88.52 |
| Bacteria | 61.33; 61.43 | 90.84; 83.86 | 78.46; 69.04 |
(1) The data in the table includes both read 1 and read 2 from paired-end sequencing.
(2) GC represents the GC content of libraries.
(3) Q20 value is the average Q20 of all bases in a read, which represents the ratio of bases with probability of containing no more than one error in 100 bases. Q30 value is the average Q30 of all bases in a read, which represents the ratio of bases with probability of containing no more than one error in 1,000 bases.
Table 4.
The comparison between PGM and MiSeq.
| PGM | MiSeq | |
|---|---|---|
| Output | 10 MB–100 MB | 120 MB–1.5 GB |
| Read length | ~200 bp | Up to 2 × 150 bp |
| Sequencing time | 2 hours for 1 × 200 bp | 3 hours for 1 × 36 single read |
| 27 hours for 2 × 150 bp pair end read | ||
| Sample preparation time | 8 samples in parallel, less than 6 hrs | As fast as 2 hrs, with 15 minutes hand on time |
| Sequencing method | semiconductor technology with a simple sequencing chemistry |
Sequencing by synthesis (SBS) |
| Potential for development | Various parameters (read length, cycle time, accuracy, etc.) |
Limited factors, major concentrate in flowcell surface size, insert sizes, and how to pack cluster in tighter |
| Input amount | μg | Ng (Nextera) |
| Data analysis | Off instrument | On instrument |
5.3. Complete Genomics
Complete genomics has its own sequencer based on Polonator G.007, which is ligation-based sequencer. The owner of Polonator G.007, Dover, collaborated with the Church Laboratory of Harvard Medical School, which is the same team as SOLiD system, and introduced this cheap open system. The Polonator could combine a high-performance instrument at very low price and the freely downloadable, open-source software and protocols in this sequencing system. The Polonator G.007 is ligation detection sequencing, which decodes the base by the single-base probe in nonanucleotides (nonamers), not by dual-base coding [19]. The fluorophore-tagged nonamers will be degenerated by selectively ligate onto a series of anchor primers, whose four components are labeled with one of four fluorophores with the help of T4 DNA ligase, which correspond to the base type at the query position. In the ligation progress, T4 DNA ligase is particularly sensitive to mismatches on 3′-side of the gap which is benefit to improve the accuracy of sequencing. After imaging, the Polonator chemically strips the array of annealed primer-fluorescent probe complex; the anchor primer is replaced and the new mixture are fluorescently tagged nonamers is introduced to sequence the adjacent base [20]. There are two updates compared with Polonator G.007, DNA nanoball (DNB) arrays, and combinatorial probe-anchor ligation (cPAL). Compared with DNA cluster or microsphere, DNA nanoball arrays obtain higher density of DNA cluster on the surface of a silicon chip. As the seven 5-base segments are discontinuous, so the system of hybridization-ligation-detection cycle has higher fault-tolerant ability compared with SOLiD. Complete genomics claim to have 99.999% accuracy with 40x depth and could analyze SNP, indel, and CNV with price 5500$–9500$. But Illumina reported a better performance of HiSeq 2000 use only 30x data (Illumina Genome Network). Recently some researchers compared CG's human genome sequencing data with Illumina system [21], and there are notable differences in detecting SNVs, indels, and system-specific detections in variants.
5.4. The Third Generation Sequencer
While the increasing usage and new modification in next generation sequencing, the third generation sequencing is coming out with new insight in the sequencing. Third-generation sequencing has two main characteristics. First, PCR is not needed before sequencing, which shortens DNA preparation time for sequencing. Second, the signal is captured in real time, which means that the signal, no matter whether it is fluorescent (Pacbio) or electric current (Nanopore), is monitored during the enzymatic reaction of adding nucleotide in the complementary strand.
Single-molecule real-time (SMRT) is the third-generation sequencing method developed by Pacific Bioscience (Menlo Park, CA, USA), which made use of modified enzyme and direct observation of the enzymatic reaction in real time. SMRT cell consists of millions of zero-mode waveguides (ZMWs), embedded with only one set of enzymes and DNA template that can be detected during the whole process. During the reaction, the enzyme will incorporate the nucleotide into the complementary strand and cleave off the fluorescent dye previously linked with the nucleotide. Then the camera inside the machine will capture signal in a movie format in real-time observation [19]. This will give out not only the fluorescent signal but also the signal difference along time, which may be useful for the prediction of structural variance in the sequence, especially useful in epigenetic studies such as DNA methlyation [22].
Comparing to second generation, PacBio RS (the first sequencer launched by PacBio) has several advantages. First the sample preparation is very fast; it takes 4 to 6 hours instead of days. Also it does not need PCR step in the preparation step, which reduces bias and error caused by PCR. Second, the turnover rate is quite fast; runs are finished within a day. Third, the average read length is 1300 bp, which is longer than that of any second-generation sequencing technology. Although the throughput of the PacBioRS is lower than second-generation sequencer, this technology is quite useful for clinical laboratories, especially for microbiology research. A paper has been published using PacBio RS on the Haitian cholera outbreak [19].
We have run a de novo assembly of DNA fosmid sample from Oyster with PacBio RS in standard sequencing mode (using LPR chemistry and SMRTcells instead of the new version FCR chemistry and SMRTcells). An SMRT belt template with mean insert size of 7500 kb is made and run in one SMRT cell and a 120-minute movie is taken. After Post-QC filter, 22,373,400 bp reads in 6754 reads (average 2,566 bp) were sequenced with the average Read Score of 0.819. The Coverage is 324x with mean read score of 0.861 and high accuracy (~99.95). The result is exhibited in Figure 2.
Figure 2.
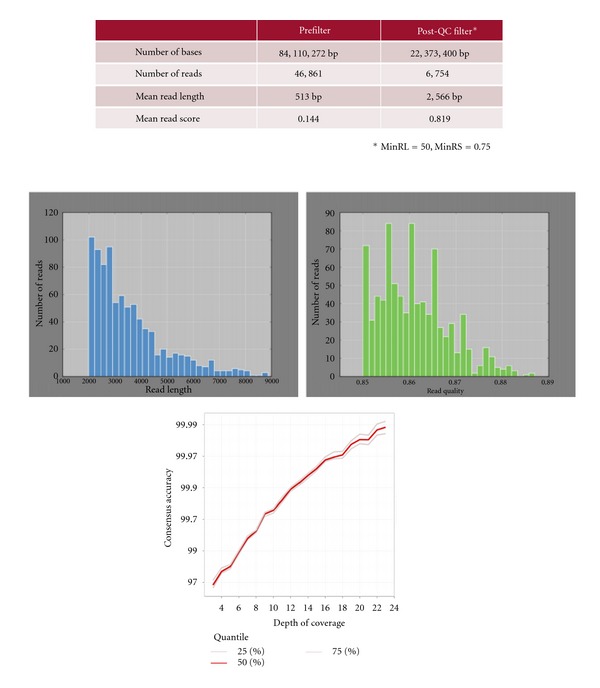
Sequencing of a fosmid DNA using Pacific Biosciences sequencer. With coverage, the accuracy could be above 97%. The figure was constructed by BGI's own data.
Nanopore sequencing is another method of the third generation sequencing. Nanopore is a tiny biopore with diameter in nanoscale [23], which can be found in protein channel embedded on lipid bilayer which facilitates ion exchange. Because of the biological role of nanopore, any particle movement can disrupt the voltage across the channel. The core concept of nanopore sequencing involves putting a thread of single-stranded DNA across α-haemolysin (αHL) pore. αHL, a 33 kD protein isolated from Staphylococcus aureus [20], undergoes self-assembly to form a heptameric transmembrane channel [23]. It can tolerate extraordinary voltage up to 100 mV with current 100 pA [20]. This unique property supports its role as building block of nanopore. In nanopore sequencing, an ionic flow is applied continuously. Current disruption is simply detected by standard electrophysiological technique. Readout is relied on the size difference between all deoxyribonucleoside monophosphate (dNMP). Thus, for given dNMP, characteristic current modulation is shown for discrimination. Ionic current is resumed after trapped nucleotide entirely squeezing out.
Nanopore sequencing possesses a number of fruitful advantages over existing commercialized next-generation sequencing technologies. Firstly, it potentially reaches long read length >5 kbp with speed 1 bp/ns [19]. Moreover, detection of bases is fluorescent tag-free. Thirdly, except the use of exonuclease for holding up ssDNA and nucleotide cleavage [24], involvement of enzyme is remarkably obviated in nanopore sequencing [22]. This implies that nanopore sequencing is less sensitive to temperature throughout the sequencing reaction and reliable outcome can be maintained. Fourthly, instead of sequencing DNA during polymerization, single DNA strands are sequenced through nanopore by means of DNA strand depolymerization. Hence, hand-on time for sample preparation such as cloning and amplification steps can be shortened significantly.
6. Discussion of NGS Applications
Fast progress in DNA sequencing technology has made for a substantial reduction in costs and a substantial increase in throughput and accuracy. With more and more organisms being sequenced, a flood of genetic data is inundating the world every day. Progress in genomics has been moving steadily forward due to a revolution in sequencing technology. Additionally, other of types-large scale studies in exomics, metagenomics, epigenomics, and transcriptomics all become reality. Not only do these studies provide the knowledge for basic research, but also they afford immediate application benefits. Scientists across many fields are utilizing these data for the development of better-thriving crops and crop yields and livestock and improved diagnostics, prognostics, and therapies for cancer and other complex diseases.
BGI is on the cutting edge of translating genomics research into molecular breeding and disease association studies with belief that agriculture, medicine, drug development, and clinical treatment will eventually enter a new stage for more detailed understanding of the genetic components of all the organisms. BGI is primarily focused on three projects. (1) The Million Species/Varieties Genomes Project, aims to sequence a million economically and scientifically important plants, animals, and model organisms, including different breeds, varieties, and strains. This project is best represented by our sequencing of the genomes of the Giant panda, potato, macaca, and others, along with multiple resequencing projects. (2) The Million Human Genomes Project focuses on large-scale population and association studies that use whole-genome or whole-exome sequencing strategies. (3) The Million Eco-System Genomes Project has the objective of sequencing the metagenome and cultured microbiome of several different environments, including microenvironments within the human body [25]. Together they are called 3 M project.
In the following part, each of the following aspects of applications including de novo sequencing, mate-pair, whole genome or target-region resequencing, small RNA, transcriptome, RNA seq, epigenomics, and metagenomics, is briefly summarized.
In DNA de novo sequencing, the library with insert size below 800 bp is defined as DNA short fragment library, and it is usually applied in de novo and resequencing research. Skovgaard et al. [26] have applied a combination method of WGS (whole-genome sequencing) and genome copy number analysis to identify the mutations which could suppress the growth deficiency imposed by excessive initiations from the E. coli origin of replication, oriC.
Mate-pair library sequencing is significant beneficial for de novo sequencing, because the method could decrease gap region and extend scaffold length. Reinhardt et al. [27] developed a novel method for de novo genome assembly by analyzing sequencing data from high-throughput short read sequencing technology. They assembled genomes into large scaffolds at a fraction of the traditional cost and without using reference sequence. The assembly of one sample yielded an N50 scaffold size of 531,821 bp with >75% of the predicted genome covered by scaffolds over 100,000 bp.
Whole genome resequencing sequenced the complete DNA sequence of an organism's genome including the whole chromosomal DNA at a single time and alignment with the reference sequence. Mills et al. [28] constructed a map of unbalanced SVs (genomic structural variants) based on whole genome DNA sequencing data from 185 human genomes with SOLiD platform; the map encompassed 22,025 deletions and 6,000 additional SVs, including insertions and tandem duplications [28]. Most SVs (53%) were mapped to nucleotide resolution, which facilitated analyzing their origin and functional impact [28].
The whole genome resequencing is an effective way to study the functional gene, but the high cost and massive data are the main problem for most researchers. Target region sequencing is a solution to solve it. Microarray capture is a popular way of target region sequencing, which uses hybridization to arrays containing synthetic oligo-nucleotides matching the target DNA sequencing. Gnirke et al. [29] developed a captured method that uses an RNA “baits” to capture target DNA fragments from the “pond” and then uses the Illumina platform to read out the sequence. About 90% of uniquely aligning bases fell on or near bait sequence; up to 50% lay on exons proper [29].
Fehniger et al. used two platforms, Illumina GA and ABI SOLiD, to define the miRNA transcriptomes of resting and cytokine-activated primary murine NK (natural killer) cells [30]. The identified 302 known and 21 novel mature miRNAs were analyzed by unique bioinformatics pipeline from small RNA libraries of NK cell. These miRNAs are overexpressed in broad range and exhibit isomiR complexity, and a subset is differentially expressed following cytokine activation, which were the clue to identify the identification of miRNAs by the Illumina GA and SOLiD instruments [30].
The transcriptome is the set of all RNA molecules, including mRNA, rRNA, tRNA, and other noncoding RNA produced in one or a population of cells. In these years, next-generation sequencing technology is used to study the transcriptome compares with DNA microarray technology in the past. The S. mediterranea transcriptome could be sequenced by an efficient sequencing strategy which designed by Adamidi et al. [31]. The catalog of assembled transcripts and the identified peptides in this study dramatically expand and refine planarian gene annotation, which is demonstrated by validation of several previously unknown transcripts with stem cell-dependent expression patterns.
RNA-seq is a new method in RNA sequencing to study mRNA expression. It is similar to transcriptome sequencing in sample preparation, except the enzyme. In order to estimate the technical variance, Marioni et al. [32] analyzed a kidney RNA samples on both Illumina platform and Affymetrix arrays. The additional analyses such as low-expressed genes, alternative splice variants, and novel transcripts were found on Illumina platform. Bradford et al. [33] compared the data of RNA-seq library on the SOLiD platform and Affymetrix Exon 1.0ST arrays and found a high degree of correspondence between the two platforms in terms of exon-level fold changes and detection. And the greatest detection correspondence was seen when the background error rate is extremely low in RNA-seq. The difference between RNA-seq and transcriptome on SOLiD is not so obvious as Illumina.
There are two kinds of application of epigenetic, Chromatin immunoprecipitation and methylation analysis. Chromatin immunoprecipitation (ChIP) is an immunoprecipitation technique which is used to study the interaction between protein and DNA in a cell, and the histone modifies would be found by the specific location in genome. Based on next-generation sequencing technology, Johnson et al. [34] developed a large-scale chromatin immunoprecipitation assay to identify motif, especially noncanonical NRSF-binding motif. The data displays sharp resolution of binding position (±50 bp), which is important to infer new candidate interaction for the high sensitivity and specificity (ROC (receiver operator characteristic) area ≥0.96) and statistical confidence (P < 10–4). Another important application in epigenetic is DNA methylation analysis. DNA methylation exists typically in vertebrates at CpG sites; the methylation caused the conversion of the cytosine to 5-methylcytosine. Chung presented a whole methylome sequencing to study the difference between two kinds of bisulfite conversion methods (in solution versus in gel) by SOLiD platform [35].
The world class genome projects include the 1000 genome project, and the human ENCODE project, the human Microbiome (HMP) project, to name a few. BGI takes an active role in these and many more ongoing projects like 1000 Animal and Plant Genome project, the MetaHIT project, Yanhuang project, LUCAMP (Diabetes-associated Genes and Variations Study), ICGC (international cancer genome project), Ancient human genome, 1000 Mendelian Disorders Project, Genome 10 K Project, and so forth [25]. These internationally collaborated genome projects greatly enhanced genomics study and applications in healthcare and other fields.
To manage multiple projects including large and complex ones with up to tens of thousands of samples, a superior and sophisticated project management system is required handling information processing from the very beginning of sample labeling and storage to library construction, multiplexing, sequencing, and informatics analysis. Research-oriented bioinformatics analysis and followup experiment processed are not included. Although automation techniques' adoption has greatly simplified bioexperiment human interferences, all other procedures carried out by human power have to be managed. BGI has developed BMS system and Cloud service for efficient information exchange and project management. The behavior management mainly follows Japan 5S onsite model. Additionally, BGI has passed ISO9001 and CSPro (authorized by Illumina) QC system and is currently taking (Clinical Laboratory Improvement Amendments) CLIA and (American Society for Histocompatibility and Immunogenetics) AShI tests. Quick, standard, and open reflection system guarantees an efficient troubleshooting pathway and high performance, for example, instrument design failure of Truseq v3 flowcell resulting in bubble appearance (which is defined as “bottom-middle-swatch” phenomenon by Illumina) and random N in reads. This potentially hazards sequencing quality, GC composition as well as throughput. It not only effects a small area where the bubble locates resulting in reading N but also effects the focus of the place nearby, including the whole swatch, and the adjacent swatch. Filtering parameters have to be determined to ensure quality raw data for bioinformatics processing. Lead by the NGS tech group, joint meetings were called for analyzing and troubleshooting this problem, to discuss strategies to best minimize effect in terms of cost and project time, to construct communication channel, to statistically summarize compensation, in order to provide best project management strategies in this time. Some reagent QC examples are summaried in Liu et al. [36].
BGI is establishing their cloud services. Combined with advanced NGS technologies with multiple choices, a plug-and-run informatics service is handy and affordable. A series of softwares are available including BLAST, SOAP, and SOAP SNP for sequence alignment and pipelines for RNAseq data. Also SNP calling programs such as Hecate and Gaea are about to be released. Big-data studies from the whole spectrum of life and biomedical sciences now can be shared and published on a new journal GigaSicence cofounded by BGI and Biomed Central. It has a novel publication format: each piece of data links to a standard manuscript publication with an extensive database which hosts all associated data, data analysis tools, and cloud-computing resources. The scope covers not just omic type data and the fields of high-throughput biology currently serviced by large public repositories but also the growing range of more difficult-to-access data, such as imaging, neuroscience, ecology, cohort data, systems biology, and other new types of large-scale sharable data.
References
- 1.Church GM, Gilbert W. Genomic sequencing. Proceedings of the National Academy of Sciences of the United States of America. 1984;81(7):1991–1995. doi: 10.1073/pnas.81.7.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. http://en.wikipedia.org/wiki/DNA_sequencing/
- 3.Collins FS, Morgan M, Patrinos A. The Human Genome Project: lessons from large-scale biology. Science. 2003;300(5617):286–290. doi: 10.1126/science.1084564. [DOI] [PubMed] [Google Scholar]
- 4. http://genomics.xprize.org/
- 5. http://my454.com/products/technology.asp.
- 6.Berka J, Chen YJ, Leamon JH, et al. Bead emulsion nucleic acid amplification. U.S. Patent Application, 2005.
- 7.Foehlich T, et al. High-throughput nucleic acid analysis. U.S. Patent, 2010.
- 8. http://www.pyrosequencing.com/DynPage.aspx.
- 9. http://www.roche-applied-science.com/
- 10.Mardis ER. The impact of next-generation sequencing technology on genetics. Trends in Genetics. 2008;24(3):133–141. doi: 10.1016/j.tig.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 11.Huse SM, Huber JA, Morrison HG, Sogin ML, Welch DM. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biology. 2007;8(7, article R143) doi: 10.1186/gb-2007-8-7-r143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.The new GS junior sequencer. http://www.gsjunior.com/instrument-workflow.php.
- 13.SOLiD system accuray. http://www.appliedbiosystems.com/absite/us/en/home/applications-technologies/solid-next-generation-sequencing.html.
- 14. http://www.tecan.com/platform/apps/product/index.asp?MenuID=3465&ID=7191&Menu=1&Item=33.52.2.
- 15.Flusberg BA, Webster DR, Lee JH, et al. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nature Methods. 2010;7(6):461–465. doi: 10.1038/nmeth.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mellmann A, Harmsen D, Cummings CA, et al. Prospective genomic characterization of the german enterohemorrhagic Escherichia coli O104:H4 outbreak by rapid next generation sequencing technology. PLoS ONE. 2011;6(7) doi: 10.1371/journal.pone.0022751. Article ID e22751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rohde H, Qin J, Cui Y, et al. Open-source genomic analysis of Shiga-toxin-producing E. coli O104:H4. New England Journal of Medicine. 2011;365(8):718–724. doi: 10.1056/NEJMoa1107643. [DOI] [PubMed] [Google Scholar]
- 18.Chin CS, Sorenson J, Harris JB, et al. The origin of the Haitian cholera outbreak strain. New England Journal of Medicine. 2011;364(1):33–42. doi: 10.1056/NEJMoa1012928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Timp W, Mirsaidov UM, Wang D, Comer J, Aksimentiev A, Timp G. Nanopore sequencing: electrical measurements of the code of life. IEEE Transactions on Nanotechnology. 2010;9(3):281–294. doi: 10.1109/TNANO.2010.2044418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deamer DW, Akeson M. Nanopores and nucleic acids: prospects for ultrarapid sequencing. Trends in Biotechnology. 2000;18(4):147–151. doi: 10.1016/s0167-7799(00)01426-8. [DOI] [PubMed] [Google Scholar]
- 21.Performance comparison of whole-genome sequencing systems. Nature Biotechnology. 2012;30:78–82. doi: 10.1038/nbt.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Branton D, Deamer DW, Marziali A, et al. The potential and challenges of nanopore sequencing. Nature Biotechnology. 2008;26(10):1146–1153. doi: 10.1038/nbt.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE. Structure of staphylococcal α-hemolysin, a heptameric transmembrane pore. Science. 1996;274(5294):1859–1866. doi: 10.1126/science.274.5294.1859. [DOI] [PubMed] [Google Scholar]
- 24.Clarke J, Wu HC, Jayasinghe L, Patel A, Reid S, Bayley H. Continuous base identification for single-molecule nanopore DNA sequencing. Nature Nanotechnology. 2009;4(4):265–270. doi: 10.1038/nnano.2009.12. [DOI] [PubMed] [Google Scholar]
- 25. Website of BGI, http://www.genomics.org.cn.
- 26.Skovgaard O, Bak M, Løbner-Olesen A, et al. Genome-wide detection of chromosomal rearrangements, indels, and mutations in circular chromosomes by short read sequencing. Genome Research. 2011;21(8):1388–1393. doi: 10.1101/gr.117416.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reinhardt JA, Baltrus DA, Nishimura MT, Jeck WR, Jones CD, Dangl JL. De novo assembly using low-coverage short read sequence data from the rice pathogen Pseudomonas syringae pv. oryzae. Genome Research. 2009;19(2):294–305. doi: 10.1101/gr.083311.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mills RE, Walter K, Stewart C, et al. Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470(7332):59–65. doi: 10.1038/nature09708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gnirke A, Melnikov A, Maguire J, et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nature Biotechnology. 2009;27(2):182–189. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fehniger TA, Wylie T, Germino E, et al. Next-generation sequencing identifies the natural killer cell microRNA transcriptome. Genome Research. 2010;20(11):1590–1604. doi: 10.1101/gr.107995.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adamidi C, Wang Y, Gruen D, et al. De novo assembly and validation of planaria transcriptome by massive parallel sequencing and shotgun proteomics. Genome Research. 2011;21(7):1193–1200. doi: 10.1101/gr.113779.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Research. 2008;18(9):1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bradford JR, Hey Y, Yates T, Li Y, Pepper SD, Miller CJ. A comparison of massively parallel nucleotide sequencing with oligonucleotide microarrays for global transcription profiling. BMC Genomics. 2010;11(1, article 282) doi: 10.1186/1471-2164-11-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316(5830):1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 35.Gu H, Smith ZD, Bock C, Boyle P, Gnirke A, Meissner A. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nature Protocols. 2011;6(4):468–481. doi: 10.1038/nprot.2010.190. [DOI] [PubMed] [Google Scholar]
- 36.Liu L, Hu N, Wang B, et al. A brief utilization report on the Illumina HiSeq 2000 sequencer. Mycology. 2011;2(3):169–191. [Google Scholar]