

Abstract
Gliomas are uniformly fatal forms of primary brain neoplasms that vary from low-grade to high-grade (glioblastoma). While low-grade gliomas are weakly angiogenic, glioblastomas are among the most angiogenic of tumors. Thus, interactions between glioma cells and their tissue microenvironment may play an important role in aggressive tumor formation and progression. To quantitatively explore how tumor cells interact with their tissue microenvironment, we incorporated the interactions of normoxic glioma cells, hypoxic glioma cells, vascular endothelial cells, diffusible angiogenic factors, and necrosis formation into a first-generation, biologically-based mathematical model for glioma growth and invasion. Model simulations quantitatively described the spectrum of in vivo dynamics of gliomas visualized with medical imaging. Further, we investigated how proliferation and dispersal of glioma cells combine to induce increasing degrees of cellularity, mitoses, hypoxia-induced neo-angiogenesis and necrosis, features that characterize increasing degrees of “malignancy”, and we found that changes in the net rates of proliferation ({lower case rho}) and invasion (D) are not always necessary for malignant progression. Thus, although other factors, including the accumulation of genetic mutations, can change cellular phenotype (e.g. proliferation and invasion rates), this study suggests that these are not required for malignant progression. Simulated results are placed in the context of the current clinical World Health Organization grading scheme for studying specific patient examples. This study suggests that through the application of the proposed model for tumor-microenvironment interactions, predictable patterns of dynamic changes in glioma histology distinct from changes in cellular phenotype (e.g. proliferation and invasion rates) may be identified, thus providing a powerful clinical tool.
Keywords: glioma, malignant progression, microenvironment, angiogenesis, mathematical model
Introduction
One of the key outstanding questions in the clinical management of gliomas lies in the unpredictable pattern of progression from low-grade, with its associated modest survival time measured in decades, to high-grade, with its aggressive growth and short survival time measured in months. According to the WHO grading scheme for gliomas low-grade and high-grade are differentiated by features of cellularity, mitoses and vascular proliferation. The characteristic vascular morphology of gliomas, specifically high-grade glioblastomas, has led many to the hypothesis that the formation of new blood vessels, angiogenesis, is essential to their growth (1). The angiogenic property of high-grade gliomas was first revealed in a study by Brem et al (2) where a glioblastoma fragment was implanted into a rabbit cornea (a well-known angiogenesis assay). The endothelial-cell mitogen and permeability factor VEGF was found to be present in the hypoxic pseudo-palisading tumor cells lying adjacent to necrotic regions and hyperplastic vessels, identifying their role in glioma angiogenesis (3). Angiogenic features therefore are considered a key to the differentiation between low and high-grade gliomas.
We have previously developed a mathematical model for quantifying the heterogeneity of glioma proliferation and invasion kinetics across patients. This proliferation-invasion (PI) model has been successful in predicting untreated growth and invasion kinetics for each patient (4), providing predictions related to surgical resection (5), chemotherapy (6, 7), and radiation therapy (8). Further, the model allowed for direct estimation of the extent of diffuse glioma invasion (beyond that visible on MRI) (4). However, this model neglects the complex interplay between tumor and its environment (e.g., the blood vessels and stroma) and, thus, does not incorporate the angiogenic cascade.
In the present paper, we will develop the PI model to produce a new model that incorporates distinct cellular and microenvironmental changes related to the angiogenic cascade. Specifically, we will incorporate three distinct glioma cellular compartments (normoxic, hypoxic and necrotic) with a vascular compartment and diffusible angiogenic factors (e.g. VEGF). This new model will allow for an investigation of malignant progression by studying the dynamics of tumor grade as a function of these cellular and microenvironmental compartments. In summary, by comparing imaging, histologic and molecular characteristics of gliomas with in silico results, we identify predictable patterns of malignant progression, extent of hypoxic burden and the size of the necrotic region in three glioblastoma patients. Overall, we find that the accumulation of genetic mutations is not necessarily required for malignant progression but rather interactions of tumor cells with a fixed net invasion rate (D) and net proliferation rate (ρ) alone can stimulate malignant progression seen on histology.
Quick Guide
The proliferation invasion (PI) model
| (1) |
The PI model is the foundation for our expanded angiogenesis model and describes the density of glioma cells c = c(x,t) at time t and location x in units cells/mm3 in terms of two net rates: motility (D) and proliferation (ρ). D is the net invasion rate (mm2/year) (possibly spatially varying), ρ is the net proliferation rate (1/year), and k is the tumor cell carrying capacity of the tissue (cells/mm3), supposing a 10 μm diameter tumor cell. From left to right the terms signify the temporal rate of change of glioma cell density, the diffuse motility of the cancer cells, and the net cell proliferation rate, respectively. The net proliferation rate ρ includes both birth and death rates and assumes logistic growth with tumor cell carrying capacity K. The PI model assumes that tumor cells proliferate at a constant net rate independent of the availability of nutrients. The model further assumes that routine MRI modalities correspond to tumor cell densities, which can be mapped to model parameters D and ρ (4, 8).
The proliferation invasion hypoxia necrosis angiogenesis (PIHNA) model
The PIHNA model builds upon the PI model by incorporating the angiogenic cascade based net rates and concentrations of cell populations which interact, proliferate, decay and migrate:
| (2) |
where V= v/(c+h+v) and T = (c+h+v+n)/K.
Figure 1 provides a diagram defining the interacting elements of the PIHNA model. The normoxic (c) glioma cells are assumed to diffuse in the tissue at a rate D and to proliferate at a rate ρ (as described by our original model). These cells also compete for space with other neighboring cells. If the oxygen supply (determined by the relative amount of vasculature supplying the tissue) is insufficient, they convert to a hypoxic state (h) at a rate β. If oxygen continues to be inadequate, the hypoxic cells will become necrotic at a rate αh. As both of these processes should correlate positively with the metabolic demands of the tumor, we have made β and αh proportional to ρ. On the other hand, if the oxygen supply increases at a later time and becomes sufficient, then hypoxic glioma cells will convert back to a normoxic state at a constant rate γ. Necrosis that comes into contact with other cells induces those viable cells to also become necrotic at a rate αn. This parallels the observation that certain brain injuries (e.g., radiation-induced) can lead to an often fatal “infective” necrosis that presumably destroys healthy tissue in its path (9).
Figure 1.
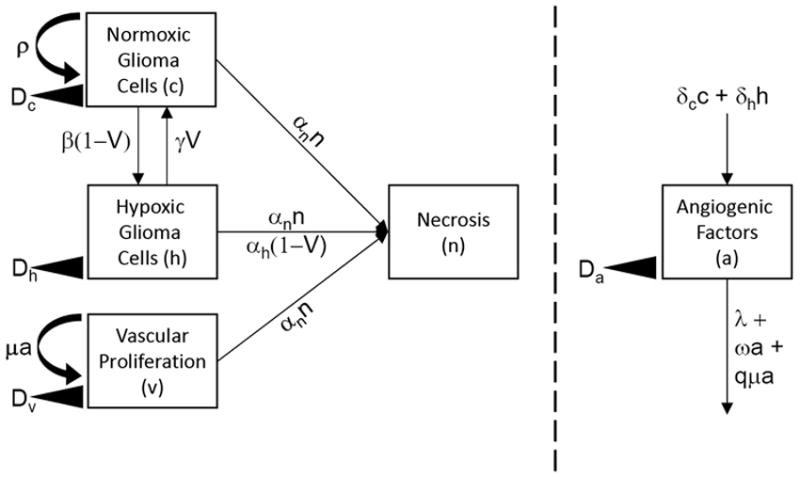
Outline of the PIHNA mathematical model illustrating the interaction of glioma cells (normoxic and hypoxic) with the available vasculature which can be affected by the production of angiogenic factors.
Both normoxic and hypoxic glioma cells produce tumor angiogenic factors (TAF). Normoxic cells produce these factors (e.g., EGF(10)) at a rate δc and hypoxic cells (e.g., VEGF (11)) at a rate δh, where it is known that δh is much greater than δc to reflect both the virulent production of VEGF by hypoxic cells, as well as the strong pro-angiogenic effect of the VEGF. Once produced by cells, angiogenic factors diffuse in the tissue at a rate Da (12) and decay at a rate λ (13). Endothelial cells (ECs) are also assumed to move randomly at a rate Dv (14). The migration and proliferation (at a maximal rate μ (15)) of these ECs results in the growth of new glioma vasculature that has to compete along with all the other cells for space in the brain. We have used the Monod model (16) for proliferation to describe the interaction between TAF and ECs.
Materials and Methods
Parameter Estimates
One of the major limiting factors in the usefulness of mathematical models lies within the ability to parameterize them. Multiple parameters are often difficult to obtain and may only be guesstimates at best; however, the strength of models is that one can examine a wide range of parameter space that incorporates the extremes of real world values, thus allowing a full understanding of the possible model outcomes. Our previous work has shown a marked quantitative heterogeneity in the net rates of proliferation and invasion, even within each grade of gliomas – e.g. GBMs (4). We can use this previous work to provide reasonable bounds for exploration for D and ρ. The rest of the model parameters are estimated from literature sources or calculated to approximate physiological conditions (Supplementary Table 1).
In Silico Grade Maps and Virtual Immunohistchemistry
Because of the diffuse nature of this disease, the in silico grade of each simulated tumor will vary spatially (as is the case in vivo). To obtain an appropriate summarized grade, analogous to the WHO grading scheme for astrocytomas (22), for each case (D, ρ and time combination), we sample virtual biopsies at random locations within the tumor region visible on imaging (i.e., the simulated T2-weighted tumor region). Following our previous work (4, 17), the simulated T2 region corresponds to a simulated total cell density greater than the T2 threshold of detection (i.e., cell density greater than a threshold, approximated to be 16% of the maximal tissue carrying capacity K). Each virtual biopsy is the size of a typical microscopic high-power field (40X HPF equals about 0.067 mm2). We consider the in silico pathological specimen to consist of 10 random HPFs to capture the typical clinical pathological review protocol. For example, based on the WHO criteria, if within those there is a region of hypercellularity (i.e. cell density greater than a threshold) and more than 2 mitoses encountered (but not a significant amount of neo-vasculature or necrosis), then the lesion would be considered a grade III anaplastic glioma.
Results
PIHNA simulations exhibit key characteristics of all grades of invasive gliomas
Figure 2 illustrates simulations of the PIHNA model varying only the net proliferation rate (ρ) revealing stereotypical differences between glioma grades. Simulations are represented as plots of the density or concentration of the model variables (normoxic, hypoxic, and necrotic tissue, vasculature and VEGF) with respect to the distance from the center of the in silico tumor. Note the increasing cellularity, vasculature, hypoxia and necrosis with increasing grade consistent with the currently used histopathological grading scheme. The simulated pattern showing hypoxia as a wave at the leading edge of the hypercellular tumor was recently observed by Swanson and colleagues when comparing MRI observations of human glioblastoma with imaging of hypoxia with [18F]-fluoromisonidazole PET (18).
Figure 2.
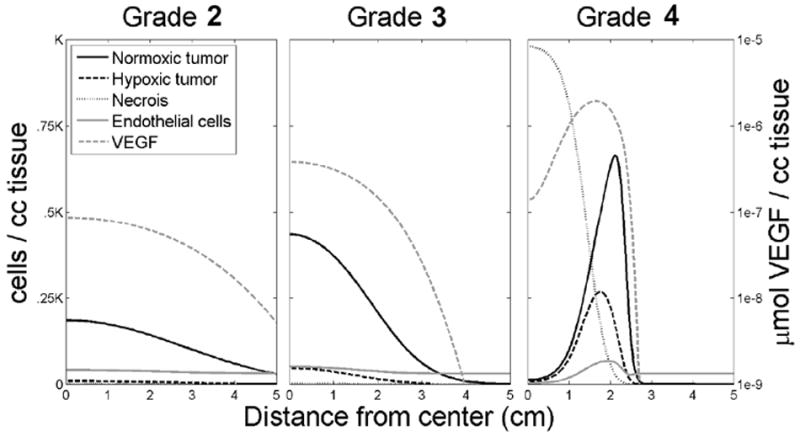
Simulations illustrating each grade of glioma represented as a plot of density or concentration of the model variables (normoxic, hypoxic, and necrotic tissue, vasculature and VEGF) with respect to the distance from the center of the in silico tumor. Note the increasing cellularity, vasculature, hypoxia and necrosis with increasing grade.
In silico PIHNA model-predicted survival time is dependent on glioma proliferation and invasion kinetics
To explore the prognostic possibilities of knowing D and ρ, we created 225 virtual patients representing 15 × 15 combinations of D and ρ. The range of D and ρ was chosen to cover those observed in our experience with glioblastoma patients, for whom we have estimated these parameters from serial MRIs (4, 19, 20). We anticipate a wider range (specifically lower D and ρ values) to encompass a wider spectrum of low-grade gliomas. On this log-log scaled plot of ρ (y-axis) and D (x-axis), lines of constant velocity (for velocities of a form similar to the Fisher velocity of ) would be parallel diagonal lines. Assuming a fatal tumor burden (FTB) – (20) equivalent to a T2 radius of 4 cm, we calculated the in silico survival time, beginning at (probably) the smallest detectable size (T2 radius = 1 cm), as shown in Figure 3. The pattern clearly is a diagonal one, related to the velocity of radial expansion, with short survival to be expected with high D/high ρ and long survival with low D/low ρ. Note, however, that as D gets large, the tumor is dominated by its diffuse extent so that it takes longer for the detection threshold to be met; but once it is met, the in silico tumor immediately fills the virtual brain, albeit at a low cell density – similar to gliomatosis cerebri. Such a constant size for the FTB may represent the average but does not seem realistic in the view of the great variability of glioma size at autopsy (21). If we assume a FTB equivalent to a total number of glioma cells (our current best estimate is about 1.1×1011 cells – (20)), the pattern of survival is quite different (results not shown), more vertical, with short survival expected with high ρ and long survival with low ρ but again, biased by the invisible invasive cells included distantly due to high D.
Figure 3.
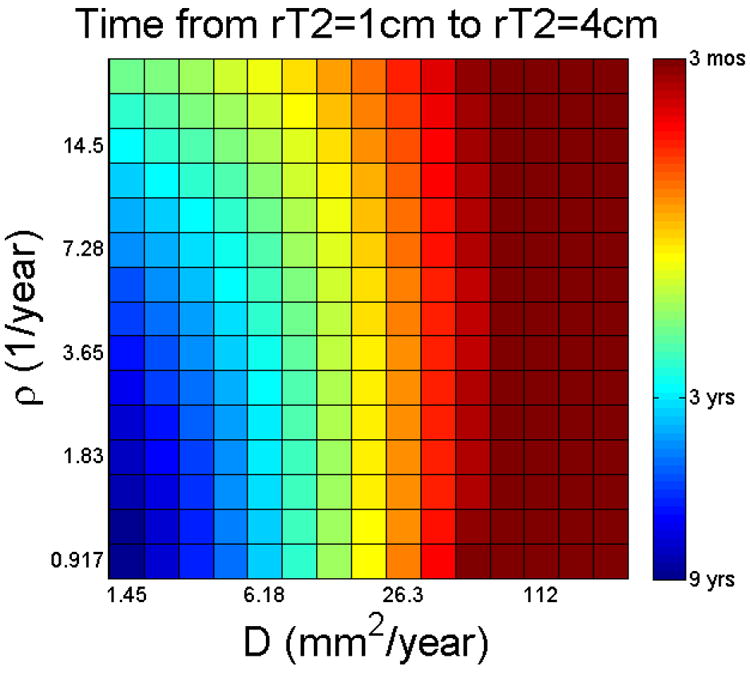
In silico (survival) time from a typical diagnostic size of 1cm on T2 MRI to a typically fatal size of 4cm, as a function of D and ρ.
Tumor grade increases with tumor size and time
Motivated by the clinical paradigm, we considered the effect of both tumor size and time on the evolution of grade. For each pair of ρ and D (and tumor size or time point), as shown in Figure 3 for mapping predicted survival times for a spectrum of in silico patients, we apply the in silico grading scheme (see Methods) to each simulated tumor to create Figure 4a. Because of the diffuse nature of this disease, the in silico grade of each simulated tumor will vary spatially. We therefore, as before, took virtual biopsies at random locations within the tumor region to obtain an appropriate summarized grade (expressed as a probability of observing a given grade on virtual biopsy) for each case (D, ρ, and time/size combination) generating Figure 4a:
As a function of tumor size, the progression wave of the in silico grading map appears to descend obliquely (diagonally) from the top left to the bottom right. Thus, for small tumors to attain the histological features of GBM, there must be both low D and high ρ, however, for larger tumors, there is a wider range of D and ρ that will generate the features of GBM.
As a function of time, the progression wave of the in silico grading map quickly spreads obliquely (diagonally) from high ρ low D (upper left corner) but then more vertically, such that practically all are grade IV. Again, this is evidence of the biasing effect of high D and the invisible low densities of invading glioma cells.
Figure 4.
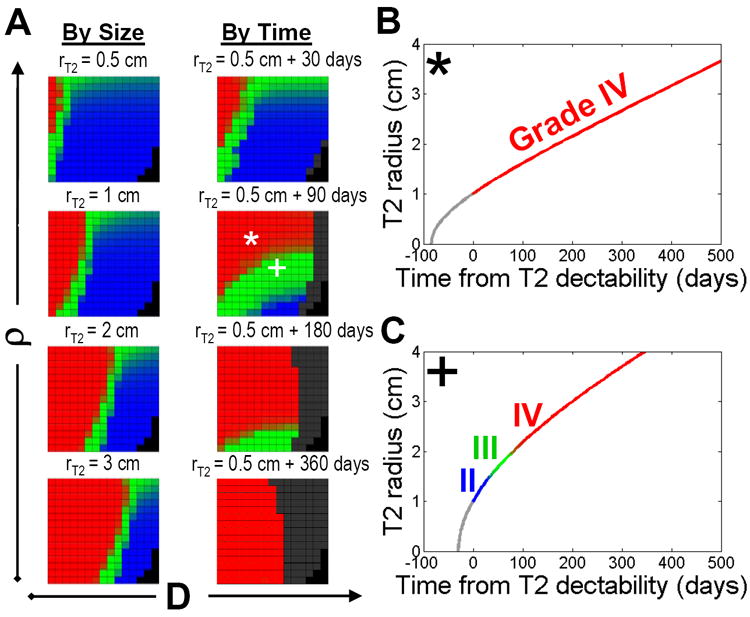
a) Maps of tumor grade as a function of tumor size (on T2 MRI) or of time. The blackened boxes indicate that the T2-visible portion of the simulated lesion has grown to a size sufficient to fill the whole brain. b) The * combination (high ρ, low D) produces a “primary” GBM (i.e., GBM from its first detectability), while comparatively c) the + combination (low ρ, high D – relative to *) produces a “secondary” GBM (i.e., “progressing” from lower grade).
The color gradient between each grade is a reflection of the probability that a given grade is observed on virtual biopsy. Movies comparing the progression of these 2 simulated patients (* and +) are available in supplementary material.
Malignant progression separates primary and secondary GBMs according to proliferation and invasion kinetics
Figure 4 shows the time course of the radial expansion of the simulated abnormality seen on T2 MRI for two different choices of the model parameters – a high ρ and low D case, *, (Figure 4B) and a lower ρ and higher D case, + (Figure 4C). These two simulated cases differ in their time course for appearance on imaging, as well as their in silico grade at the time of first detection (assumed here to be 1 cm in radius on T2 MRI). In fact, these two cases represent in silico “primary” and “secondary” glioblastomas (pGBM and sGBM, respectively) in the sense that from the time of detectability on T2 MRI (assumed to be 1 cm in radius), the * case maintained a Grade IV in silico grade while the + case was considered a lower-grade until it was over 2 cm in radius on T2 MRI (22). To further explore this, we illustrate the portion of parameter space in which pGBMs and sGBMs live in Figure 5, denoting the location of the * and + examples.
Figure 5.
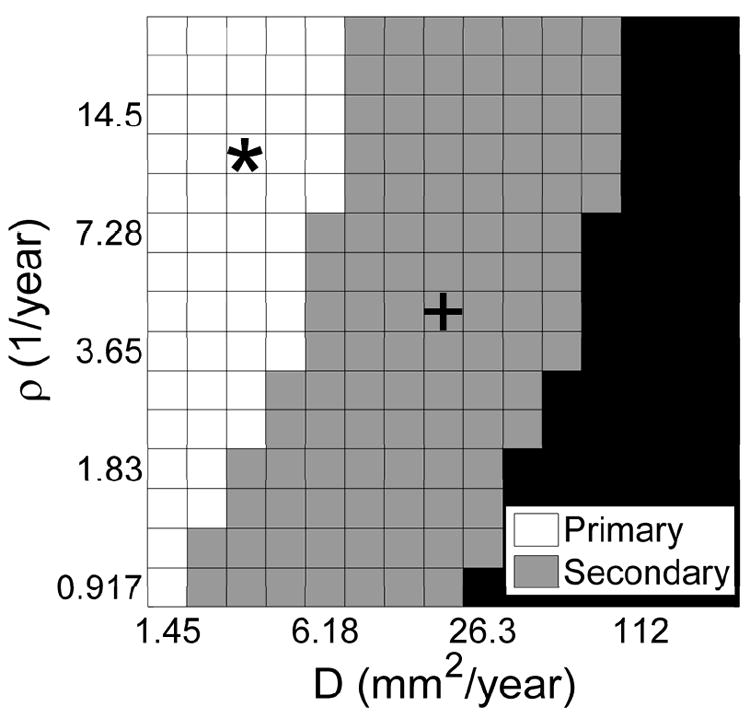
A map of simulated “primary” (white) and “secondary” (grey) glioblastomas and gliomas that become fatal is size before malignant progression (black).
Despite malignant progression, in silico growth dynamics reveal a constant velocity of imageable growth
As reviewed by Harpold et al (4), our studies with the PI model have culminated in a characterization of pre-treatment glioma kinetics with a net proliferation rate (ρ) and a net invasion rate (D). An important consequence of the prior proliferation-invasion (PI) model is an asymptotically constant rate of radial expansion visible on imaging (23). This constant (untreated) radial growth has been confirmed in both low-grade (24) and high-grade (19) gliomas. Further, this rate of radial expansion, which is mathematically related to the model parameters as , is prognostically significant in gliomas (8, 17, 20, 25, 26). We explored this in the simulations of the expanded PIHNA model (c.f., Figures 4B and 4C) and found a similar pattern of approaching a constant speed of radial expansion (dependent predominantly upon the D and ρ).
In silico patterns of growth factor expression and other tissue markers are similar to those observed in human gliomas
By randomly sampling from the regions of D-ρ space containing in silico pGBMs and sGBMs, we noticed several similarities to published observations. Specifically, we confirmed the in vivo observation that pGBMs tend to have increased expression of VEGF when compared to Grade 3 and sGBMs (22, 27), as shown in Figure 6a. For the simulations resulting in Grade 3, sGBM, and pGBM with rT2=3cm were identified. Then from each category, 10 simulations were selected at random, and the ratio of total VEGF to total tumor cells (obtained by integrating concentrations over the whole domain) was calculated. The error bars represent the full range of data in each category, while the gray boxes indicate the 25th to 75th percentile. Similarly, with increasing grade, an increasing number of these virtual biopsy cells express VEGF or HIF1α consistent with the in vivo observations of Korkolopoulou et al. (28) and Flynn et al (29) – Figure 6b.
Figure 6.
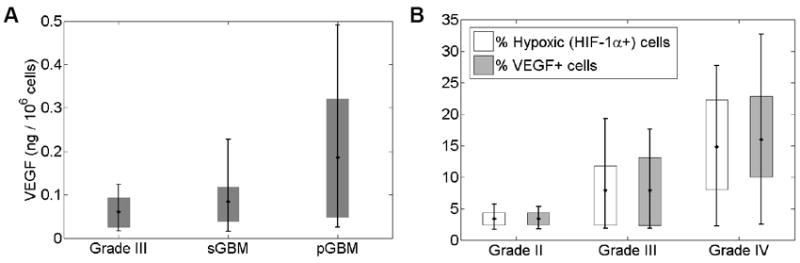
a) The total VEGF (integral amount of model TAF) normalized to the local glioma cell density for in silico Grade 3, sGBM and pGBM tumors showing increasing VEGF concentrations qualitatively consistent with the observations of (27). b) Increasing in silico expression of VEGF and hypoxia marker, HIF-1α, with increasing histological grade qualitatively consistent with the observations of (28, 29).
PIHNA model accurately predicts hypoxia, necrosis and histologic grade in patients
Using patient-specific estimates of net proliferation rate (ρ) and net invasion rate (D) (8, 25, 30, 31), it is possible to assess the validity of patient-specific PIHNA simulations bridging the elements interpretable through the model such as histology, immunohistochemistry (e.g., as in Figure 6) and other imaging features. Specifically, we considered 3 patients who had a similar size on T2 MRI of approximately 2 cm but who had different hypoxic burdens (% of the cells on histological specimens expressing HIF1α), different necrotic core sizes seen on T1Gd MRI and different D-ρ combinations. Figure 7 shows a) a map of histological grade, b) a map of the maximum of the hypoxic cell population (h) and c) the size of the necrotic core (n, assuming a cutoff of 5% of the max cell density defining the edge of the necrotic core) all at simulated time point for which the simulated T2 radius is 2 cm. Figure 7 shows a direct mapping of parameters of net rates of proliferation (ρ) and invasion (D) for 3 glioblastoma patients (stars) allowing a direct correlation between model-predicted and actual observations of histologic grade (Figure 7a) and hypoxia measured histologically by HIF1-α expression (Figure 7b). Moreover, those same model simulations also accurately predict the volume of necrosis seen in these patients as assessed by regions of hypo-intensity on contrast-enhanced T1-weighted MRI (Figure 7c). All patients were accurately predicted to be of histological grade IV (red in Figure 7a). Patient 1 with the lowest ρ was accurately predicted as having a smaller hypoxic fraction (30%) and smaller necrotic radius (8mm) than others. Patients 2 and 3 were predicted to have similar levels of hypoxia (red/orange on Figure 7b) which was observed on histology (40% HIF1a+ in both cases). Patient 2 had a 16mm necrotic radius on T1Gd MRI while Patient 3 had a slightly larger 18mm necrotic core which is consistent with the model predictions seen in Figure 7c. In summary, these patient-specific simulations are compared with patient-specific information from imaging and histology and we find that the simulations provide consistent patterns between these 5 distinct data points: overall T1Gd radius, T2 radius, tumor grade, central necrosis visible T1Gd MRI and HIF1-α immunohistochemistry. Further, Gu et al (32) recently reported using the PIHNA model to generate patient-specific simulated [18F]-fluoromisonidazole PET images of hypoxia.
Figure 7.
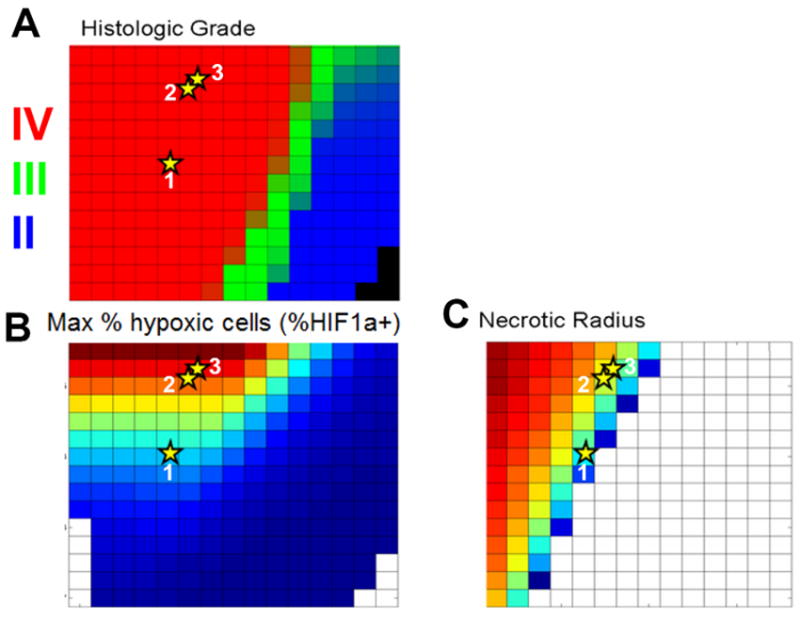
Connecting tissue level and imaging level model PIHNA model predictions of a) histological grade, b) fraction of cells that are hypoxic, and c) the radial size of central necrosis visible on T1-Gd MRI. Each grid cell represents a single virtual tumor with a specified combination of D and ρ, with in silico histologic grade, hypoxia and necrosis assessed within the virtual tumor and graphed separately. The PIHNA model accurately predicts relative amounts of hypoxia and necrosis as well as histologic grade for the 3 individual glioblastoma patients (stars). Each patient was approximately 2 cm in radius on T2 MRI with variable sizes on T1-Gd corresponding to the different D, ρ combinations graphed (quantified as in (25, 31)). Each patient was assessed for hypoxia quantified as the maximum HIF1α on immunohistochemistry (Patient 1: 30%, 2: 40%, 3: 40%) and a necrotic core radius on T1Gd MRI (Patient 1: 8mm, 2: 16mm, 3: 18mm).
Discussion
“Histological grading is a means of predicting the biological behavior of a neoplasm” (22). This definition consists of a challenge, namely, to explain what is assumed to be true, that histology has some innate ability to explain why it can, if it indeed does, predict the future. For the past 15 years, we have been exploiting a bio-mathematical model of gliomas (23) (the PI model) based on only two key cellular characteristics, net rates of proliferation (ρ) and of diffusion/invasion (D) of glioma cells, revealing predictive insights related to surgical resection (5), chemotherapy (6, 7), and radiation therapy.
By directly extending the PI model we produced the PIHNA model which encompass the angiogenic cascade and allowed us to define tumor grade as a combination of both cellular and microenvironmental model specific variables. Critically, this allowed us to summarize intuitively a complete parameter study using a graphical representation of changing grade. This highlighted a disparity between the current in vivo grading schemes and observed tumor behavior, and suggests that malignant “progression” may be less related to acquisition of new genetic abnormalities than a natural consequence of a relatively small set of initial key molecular-genetic changes that “turn on” the glioma cells to proliferate and invade. That is the net rates of proliferation (ρ) and diffusion (D) of any particular glioma may ultimately be what drive the in silico glioma’s behavior (including the whole spectrum from slow to fast and from localized to diffuse) and histologic appearances, (including increasing degrees of cellularity, mitoses, hypoxia-driven neo-angiogenesis, and necrosis) provided the microenvironment responds appropriately.
The goal of a good grading scheme is to capture prognosis implied by cellular behavior; however, the in silico grade maps (Figure 4) suggest that the WHO grading scheme, independent of size and time, is preferentially biased towards assessing proliferative capacity, although Figure 4 suggests that invasiveness significantly impacts on survival as well. A key difference between the grade maps in terms of size and time is in the ability to capture the impact of invasion rates on overall tumor growth kinetics. One would presume that rapid growth (as a result of a combination of invasion and proliferation) would be associated with shorter survival times. To illustrate this, we map the time from a typical diagnostic size (say 1cm in radius on T2) to a typical fatal size (say 4 cm in radius on T2) to approximate a survival map. A comparison of this survival map with the grade map (for the same initial size of a T2 radius of 1 cm) shows that the shortest survival times are for those in silico tumors with both high D and high ρ whereas the WHO grading scheme applied to the simulated tumors would suggest only high ρ is important. Further, these model results suggest exploration of invasion-specific markers (e.g. MMP-2 (33), vimentin (34)) may enhance the prognostic accuracy of the current grading scheme. Admittedly, assessing invasion from fixed histological sections of dead tissue is a true challenge. However, this challenge may be mediated by dynamic modeling tools like that presented here.
Obviously, histopathological grading is based on a static snapshot of the tumor tissue at a certain size (and time in the tumor’s natural history). Applying our in silico WHO grading scheme, a virtual patient presenting with a 1 cm tumor with a relatively high D and low ρ (+ in Figures 4), would appear histologically as a grade 2. However, according to our model, following this patient in time would show a rapid progression to GBM, whereas a patient with a relatively high ρ and low D (*) would have an overall slower growth rate but would have histological characteristics of a GBM from diagnosis. This is consistent with clinical observation showing wide variability in survival and growth patterns even amongst histologically similar tumors and suggests that morphological based grading is naturally limited (22). Such cases of malignant progression as illustrated in Figure 4c (+) is common amongst low-grade (grade II or III) gliomas but is currently considered a relatively unpredictable event associated with a dramatic decrease in median survival time (22).
Figure 6 provides preliminary confirmation that the PIHNA model maps directly to, and connects with, clinical metrics of malignant progression, medical imaging and histology in individual patients. These data suggest that patient-specific simulations of the PIHNA model may be able to recapitulate and, in fact, predict characteristics of each patients’ tumor onto an orthogonal data set that is not obtainable directly from the original patient imaging used as input to the model. Although a similar analysis on a broader patient set is needed to confirm these preliminary findings, these data support an integrative role of the PIHNA model in bridging imaging, histology and immunohistochemistry in individual patients.
Traditional molecular-genetic characterization of gliomas shows a marked heterogeneity within and across grades (22). We propose that our model parameters may be simply consequences (downstream) of these molecular signatures. Our model simulation results suggest that one can think of each glioma as having an implicit invasive and proliferative potential. That potential is manifest quantitatively in terms of our model parameters D and ρ. As the cells grow and invade according to their pre-programmed D, ρ combination, the local microenvironment may become harsh (e.g. hypoxic) which may lead to the production of angiogenic factors that induce vascular growth (via HIF1α and similar pathways). Other than those cells within the hypoxic tumor becoming less proliferative and the vascular endothelial cells beginning to proliferate, the kinetics of the cell population (c) driving the growth of the tumor overall have not changed – i.e. D and ρ remain the same while depending on the size and time of observation, the morphological features (seen on histological specimen) can be strikingly different. These results are consistent with recent findings (35) suggesting that microenvironment and/or tumor-stromal interactions are the key to malignant progression, and changes in cell kinetics may not be necessary to see these pathological changes.
It is important to note that this conclusion does not preclude changes in cellular behavior as a result of gene-expression changes (caused by any number of mechanisms including environmental influences as well as mutational events). Nor does it exclude the possibility that early in glioma development (and perhaps post treatment) the tumor consisted of a more heterogeneous population with a range of D and ρ. Rather, these results simply illustrate that changes in the dominant cellular kinetics (e.g., D, ρ) are not necessary to obtain the range of morphological patterns seen in vivo within individual patients. Meaning, the wildly different morphological changes on imaging and histopathology observed in some patients through their disease course may not actually be a result of an underlying change in the dominant tumor cellular phenotype (D, ρ) but simply a manifestation of the evolution of that cellular phenotype interacting with its microenvironment.
We realize, of course, that we are proposing a simplified model for the dynamic interactions between the tumor cells and the microenvironment and therefore misses several key characteristics which can be readily explored in future models. Overall, our proposed minimal PIHNA model for glioma-microenvironment interactions, which explicitly incorporates the interactions of normoxic glioma cells, hypoxic glioma cells, vascular endothelial cells, diffusible angiogenic factors and the formation of necrosis is sufficient to describe the spectrum of in vivo dynamics of gliomas visualized with medical imaging and may be an invaluable research analysis and clinical assessment tool. An important area of future exploration for this model is to understand the changing tumor-microenvironment in the context of treatment. This is particularly relevant to increasing our understanding of the effects of antiangiogenics on disease progression.
Supplementary Material
Major Findings.
Although commonly attributed to molecular-genetic factors such as the accumulation of genetic mutation, this study combines mathematical modeling with experimental and clinical data for human gliomas to suggest that changes in cell kinetics are not necessary to generate the imaging and histological features of malignant progression seen in vivo.
Acknowledgments
KRS gratefully acknowledges the generous and timely support of the McDonnell Foundation, the Dana Foundation, the Academic Pathology Fund and the NIH/NCI Moffitt-UW Physical Sciences Oncology Center U54 CA143970.
References
- 1.Fischer I, Gagner JP, Law M, Newcomb EW, Zagzag D. Angiogenesis in gliomas: biology and molecular pathophysiology. Brain Pathol. 2005;15:297–310. doi: 10.1111/j.1750-3639.2005.tb00115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brem S. The role of vascular proliferation in the growth of brain tumors. Clin Neurosurg. 1976;23:440–53. doi: 10.1093/neurosurgery/23.cn_suppl_1.440. [DOI] [PubMed] [Google Scholar]
- 3.Plate KH, Breier G, Weich HA, Mennel HD, Risau W. Vascular Endothelial Growth-Factor and Glioma Angiogenesis - Coordinate Induction of Vegf Receptors, Distribution of Vegf Protein and Possible in-Vivo Regulatory Mechanisms. Int J Cancer. 1994;59:520–9. doi: 10.1002/ijc.2910590415. [DOI] [PubMed] [Google Scholar]
- 4.Harpold HL, Alvord EC, Jr, Swanson KR. The evolution of mathematical modeling of glioma proliferation and invasion. J Neuropathol Exp Neurol. 2007;66:1–9. doi: 10.1097/nen.0b013e31802d9000. [DOI] [PubMed] [Google Scholar]
- 5.Swanson KR, Alvord EC, Jr, Murray JD. Virtual resection of gliomas: effects of location and extent of resection on recurrence. Mathematical and Computer Modeling. 2003;37:1177–90. [Google Scholar]
- 6.Swanson KR, Bridge C, Murray JD, Alvord EC., Jr Virtual and real brain tumors: using mathematical modeling to quantify glioma growth and invasion. J Neurol Sci. 2003;216:1–10. doi: 10.1016/j.jns.2003.06.001. [DOI] [PubMed] [Google Scholar]
- 7.Swanson KR, Alvord EC, Jr, Murray JD. Quantifying efficacy of chemotherapy of brain tumors with homogeneous and heterogeneous drug delivery. Acta Biotheoretica. 2002;50:223–37. doi: 10.1023/a:1022644031905. [DOI] [PubMed] [Google Scholar]
- 8.Rockne R, Rockhill JK, Mrugala M, Spence AM, Kalet I, Hendrickson K, et al. Predicting the efficacy of radiotherapy in individual glioblastoma patients in vivo: a mathematical modeling approach. Phys Med Biol. 2010;55:3271–85. doi: 10.1088/0031-9155/55/12/001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oshiro S, Tsugu H, Komatsu F, Ohnishi H, Ueno Y, Sakamoto S, et al. Evaluation of intratumoral administration of tumor necrosis factor-alpha in patients with malignant glioma. Anticancer Res. 2006;26:4027–32. [PubMed] [Google Scholar]
- 10.Dunn IF, Heese O, Black PM. Growth factors in glioma angiogenesis: FGFs, PDGF, EGF, and TGFs. J Neurooncol. 2000;50:121–37. doi: 10.1023/a:1006436624862. [DOI] [PubMed] [Google Scholar]
- 11.Johansson M, Brannstrom T, Bergenheim AT, Henriksson R. Spatial expression of VEGF-A in human glioma. J Neurooncol. 2002;59:1–6. doi: 10.1023/a:1016317407043. [DOI] [PubMed] [Google Scholar]
- 12.Levine HA, Pamuk S, Sleeman BD, Nilsen-Hamilton M. Mathematical modeling of capillary formation and development in tumor angiogenesis: penetration into the stroma. Bull Math Biol. 2001;63:801–63. doi: 10.1006/bulm.2001.0240. [DOI] [PubMed] [Google Scholar]
- 13.Serini G, Ambrosi D, Giraudo E, Gamba A, Preziosi L, Bussolino F. Modeling the early stages of vascular network assembly. Embo J. 2003;22:1771–9. doi: 10.1093/emboj/cdg176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sherratt JA, Murray JD. Models of epidermal wound healing. Proc Biol Sci. 1990;241:29–36. doi: 10.1098/rspb.1990.0061. [DOI] [PubMed] [Google Scholar]
- 15.Xiu M, Turner SM, Busch R, Gee TA, Hellerstein MK. Measurement of endothelial cell proliferation rate in vivo using 2H2O labeling: a kinetics biomarker of angiogenesis. FASEB J. 2006;20:A718-a. [Google Scholar]
- 16.Murray JD. Mathematical biology I: an introduction. 3. New York: Springer-Verlag; 2002. [Google Scholar]
- 17.Swanson KR, Rostomily RC, Alvord EC., Jr A mathematical modelling tool for predicting survival of individual patients following resection of glioblastoma: a proof of principle. Brit J Cancer. 2008;98:113–9. doi: 10.1038/sj.bjc.6604125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swanson KR, Chakraborty G, Wang CH, Rockne R, Harpold HL, Muzi M, et al. Complementary but distinct roles for MRI and 18F-fluoromisonidazole PET in the assessment of human glioblastomas. J Nucl Med. 2009;50:36–44. doi: 10.2967/jnumed.108.055467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swanson KR, Alvord EC., Jr Serial imaging observations and postmortem examination of an untreated glioblastoma: A traveling wave of glioma growth and invasion. Neuro-Oncol. 2002;4:340. [Google Scholar]
- 20.Swanson KR, Harpold HL, Peacock DL, Rockne R, Pennington C, Kilbride L, et al. Velocity of radial expansion of contrast-enhancing gliomas and the effectiveness of radiotherapy in individual patients: a Proof of Principle. Clin Oncol (R Coll Radiol) 2008;20:301–8. doi: 10.1016/j.clon.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 21.Concannon JP, Kramer S, Berry R. The extent of intracranial gliomata at autopsy and its relationship to techniques used in radiation therapy of brain tumors. Am J Roentgenol Radium Ther Nucl Med. 1960;84:99–107. [PubMed] [Google Scholar]
- 22.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. WHO Classification of Tumours of the Central Nervous System. 4. Geneva, Switzerland: Renouf Publishing Co Ltd; 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swanson KR. Mathematical Modeling of the Growth and Control of Tumors [PhD] Seattle, WA: University of Washington; 1999. [Google Scholar]
- 24.Mandonnet E, Delattre JY, Tanguy ML, Swanson KR, Carpentier AF, Duffau H, et al. Continuous growth of mean tumor diameter in a subset of grade II gliomas. Ann Neurol. 2003;53:524–8. doi: 10.1002/ana.10528. [DOI] [PubMed] [Google Scholar]
- 25.Wang CH, Rockhill JK, Mrugala M, Peacock DL, Lai A, Jusenius K, et al. Prognostic significance of growth kinetics in newly diagnosed glioblastomas revealed by combining serial imaging with a novel biomathematical model. Cancer Res. 2009;69:9133–40. doi: 10.1158/0008-5472.CAN-08-3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pallud J, Mandonnet E, Duffau H, Kujas M, Guillevin R, Galanaud D, et al. Prognostic value of initial magnetic resonance imaging growth rates for World Health Organization grade II gliomas. Ann Neurol. 2006;60:380–3. doi: 10.1002/ana.20946. [DOI] [PubMed] [Google Scholar]
- 27.Karcher S, Steiner HH, Ahmadi R, Zoubaa S, Vasvari G, Bauer H, et al. Different angiogenic phenotypes in primary and secondary glioblastomas. Int J Cancer. 2006;118:2182–9. doi: 10.1002/ijc.21648. [DOI] [PubMed] [Google Scholar]
- 28.Korkolopoulou P, Patsouris E, Konstantinidou AE, Pavlopoulos PM, Kavantzas N, Boviatsis E, et al. Hypoxia-inducible factor 1alpha/vascular endothelial growth factor axis in astrocytomas. Associations with microvessel morphometry, proliferation and prognosis. Neuropathol Appl Neurobiol. 2004;30:267–78. doi: 10.1111/j.1365-2990.2003.00535.x. [DOI] [PubMed] [Google Scholar]
- 29.Flynn JR, Wang L, Gillespie DL, Stoddard GJ, Reid JK, Owens J, et al. Hypoxia-regulated protein expression, patient characteristics, and preoperative imaging as predictors of survival in adults with glioblastoma multiforme. Cancer. 2008;113:1032–42. doi: 10.1002/cncr.23678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Swanson KR, Alvord EC, Jr, Murray JD. Virtual brain tumours (gliomas) enhance the reality of medical imaging and highlight inadequacies of current therapy. Brit J Cancer. 2002;86:14–8. doi: 10.1038/sj.bjc.6600021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szeto MD, Chakraborty G, Hadley J, Rockne R, Muzi M, Alvord EC, Jr, et al. Quantitative metrics of net proliferation and invasion link biological aggressiveness assessed by MRI with hypoxia assessed by FMISO-PET in newly diagnosed glioblastomas. Cancer Res. 2009;69:4502–9. doi: 10.1158/0008-5472.CAN-08-3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gu S, Chakraborty G, Champley K, Alessio A, Claridge J, Rockne R, et al. Applying A Patient-Specific Bio-Mathematical Model of Glioma Growth to Develop Virtual [18F]-FMISO PET Images. Mathematical Medicine and Biology. 2011 doi: 10.1093/imammb/dqr002. IN PRESS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jaalinoja J, Herva R, Korpela M, Hoyhtya M, Turpeenniemi-Hujanen T. Matrix metalloproteinase 2 (MMP-2) immunoreactive protein is associated with poor grade and survival in brain neoplasms. J Neurooncol. 2000;46:81–90. doi: 10.1023/a:1006421112839. [DOI] [PubMed] [Google Scholar]
- 34.Hilbig A, Barbosa-Coutinho LM, Toscani N, Ribeiro Mde C, da Cunha BS. Expression of nestin and vimentin in gliomatosis cerebri. Arq Neuropsiquiatr. 2006;64:781–6. doi: 10.1590/s0004-282x2006000500015. [DOI] [PubMed] [Google Scholar]
- 35.Basanta D, Strand DW, Lukner RB, Franco OE, Cliffel DE, Ayala GE, et al. The role of transforming growth factor-beta-mediated tumor-stroma interactions in prostate cancer progression: an integrative approach. Cancer Res. 2009;69:7111–20. doi: 10.1158/0008-5472.CAN-08-3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.