

Abstract
In this Letter, we describe the first total synthesis of cremastrine, a pyrrolizidine alkaloid from Cremastra appendiculata, with anticholinergic activity as well as an unnatural analogue. The streamlined synthesis proceeds in 9 steps, 7 steps longest linear sequence, in 25.2% overall yield, and features novel methodology to construct the pyrrolizidine core. Biological evaluation of cremastrine and the unnatural analogue indicated that both are pan-mAChR functional antagonists.
Keywords: pyrrolizidine, cremastrine, anticholinergic, sulfinimine, Mitsunobu
Recently, Ikeda and co-workers reported on the isolation and characterization of cremastarine (1), a pyrrolizidine alkaloid from the Japanese tuber Cremastra appendiculata, a species widely used in traditional medicine in Asia for a number of therapeutic indications.1 Preliminary biological investigation, employing a [3H]-NMS binding assay, indicates that 1 is a moderately selective muscarinic acetylcholine receptor subtype 3 (M3) ligand (M1 Ki = 505 nM, M2 Ki >5,000 nM M3 Ki = 126 nM, M4 Ki = 498 nM, M5 Ki = 1,220 nM) an interesting pharmacological profile if the binding profile translated into functional antagonism.1,2 As such, 1 would have the potential to treat irritable bowel syndrome, chronic obstructive pulmonary disease and asthma.2 Based on our lab’s interest in muscarinic drug discovery3 and our recently developed methodology to rapidly construct enantiopure idolizines, pyrrolo[1,2-a]azepines and pyrrolo[1,2-a]azocines,4 application of this methodology to the synthesis of the related pyrrolizidine core seemed warranted. In this letter, we report the first total synthesis of cremastarine (1) employing a new synthetic strategy, distinct from previous pyrrolizidine syntheses,5–8 (Scheme 1), that afforded 1 in 7 total synthetic steps and an overall yield of 25.2% that enabled biological evaluation.
Scheme 1.

Retrosynthetic analysis of cremastrine (1).
Efforts first focused on the synthesis of the α-hydroxy carboxylic acid 3. Conversion of D-isoleucine 6 to α-hydroxy carboxylic acid 3 was accomplished following the Shin procedure wherein the α-amino acid is converted to the α-hydroxy acid with retention of stereochemistry.9,10 Treatment of 6 with aqueous nitrous acid leads to diazotization of the α-amine stereocenter followed by hydrolysis to furnish the enantiopure α-hydroxy acid 3.9,10 Treatment of acid 3 with excess tert-butyldimethylsilyl chloride, followed by basic hydrolysis, provides coupling partner 7 in 65% yield over 3 steps, without epimerization and in agreement with literature precedent (Scheme 2).9
Scheme 2.

Synthesis of α-hydroxy acid 3 and protected congener 7.
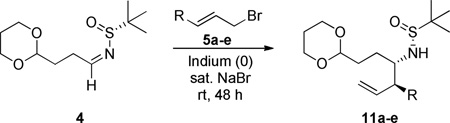
With 7 in hand, we then targeted the synthesis of the pyrrolizidine core of 1 employing our new methodology for enantiopure azacine construction employing Ellman sulfinimine technology via 4.4,11 Requisite aldehyde 10 is commercially available, but we also developed two complimentary routes to access the key aldehyde precursor 10 to sulfinamide 4 (Scheme 3). 4-Pentenal 8 was protected as the 1,3-dioxane, followed by oxonolysis to provide 10 in 78% yield. Alternatively, bromide 9 was converted to the nitrile congener, and reduced with DIBALH to deliver 10 in 84% yield. Condensation of commercially available (R)-tert-butanesulfinamide and aldehyde 10 gives chiral aldimine 4 in 93% yield.
Scheme 3.

Two approaches for the synthesis of sulfinamide 4.
From aldimine 4, we next sought to install two stereocenters through an indium mediated allylation using an appropriately functionalized allyl bromides (Table 1).12 Interestingly, esters and free hydroxyl moieties afforded poor conversion (30–55%), but good dr (3:1 to >6:1). Ultimately, good yield and moderate diastereoselectivity was observed when TBS-ether 5 was used providing 11 (85% conversion, >4:1 dr), and the stereochemistry assigned based on the established literature precedent of a six-membered ring chelation control model.13,14 For large scale preparations, we found that stirring and sonication of the indium metal and bromide before adding the aldimine was crucial to deliver homoallylic sulfinamide 11c with yields comparable to small-scale reactions.
Table 1.
In(0) allylations of sulfinamide (4) with diverse allyl bromides.
 | |||||
|---|---|---|---|---|---|
| entry | R | bromide | sulfinamide | yield (%)a | drb |
| 1 | CO2Me | 5a | 11a | 55 | >6:1 |
| 2 | CO2Et | 5b | 11b | 50 | >6:1 |
| 3 | CH2OTBS | 5c | 11c | 85 | 4:1 |
| 4 | CH2OPMB | 5d | 11d | 0 | - |
| 5 | CH2OH | 5e | 11e | 31 | 3:1 |
Hydroboration-oxidation of the terminal olefin of 11c furnished primary alcohol 12 in excellent yield. Initial attempts to form the substituted pyrrolidine ring system involved mesylation of the alcohol and displacement in a 5-exo-tet cyclization fashion. This was unfortunately unsuccessful, but under Mitsunobu conditions, we successfully obtained substituted pyrrolidine 13. NOESY-NMR data confirms the syn-stereochemical relationship of the two chiral centers on the pyrrolidine ring. From here, TBAF deprotection of the tert-butyldimethylsilyl ether gave primary alcohol 14. Deprotection of the acetal and the sulfinyl exposed the aldehyde and amine, respectively. This was theorized to undergo an intramolecular condensation to form the imine, upon which an addition of a reducing agent would produce 2,11–13 the pyrrolizidine core structure. Both classical conditions,11–13 and a variety of acids (TFA, HCl, HOAc) and reducing agents (Et3SiH, NaBH(OAc)3, NaBH3CN) failed to provide the desired product 2 (Scheme 4).
Scheme 4.

Attempted synthesis of pyrrolidine core 2.
Due to the failure of this key transformation, we presumed that the presence of the free hydroxyl in the protic environment led to undesired side reactions leading to very polar, complex mixtures. Therefore, we modified our approach to cyclize after formation of the ester bond. As shown in Scheme 5, an EDCI/DMAP mediated coupling between acid 7 and alcohol 14 delivered ester 15 in excellent yield. Application of the classical deprotection/reductive amination conditions (TFA:H2O (95:5) followed by triethyl silane as the reducing agent) afforded both global deprotection of 15 as well as intramolecular condensation/reductive amination to furnish the natural product cremastrine (1) in 80% yield from ester 15. Our synthetic 1 was in agreement with the data reported for natural 1;15 however, only tabular NMR data is available and authentic samples could not be acquired. Thus, the first total synthesis of 1 has been completed in seven steps (five steps longest linear sequence) with an overall yield of 25.2% from commercial starting materials. This expedited route also allowed for the preparation of quantities of 1 to support biological studies.
Scheme 5.

Completion of the synthesis of cremastrine (1).
Following the route in Scheme 2, we also prepared a benzyl protected congener 16 of 7, to explore strucutre-activity relationships (SAR) with a non-hydrolyzable protecting group in place of the free hydroxyl of 1. As shown in Scheme 6, unnatural analog 17 was readily prepared and once again in high overall yield (26.1%).16 Unnatural analogue 17 also provided confidence for the synthesis of 1 (since we were unable to acquire NMRs or an authentic sample), as spectral data for 17 agreed well with synthetic and natural 1.
Scheme 6.

Synthesis of unnatural analog 17.
As cremastrine (1) was reported to display modest selectivity for binding to the M3 mAChR (2- to 20-fold),1 we evaluated both our synthetic 1 and unnatural analog 17 against all five human mAChRs (M1–M5) in a functional assay to determine if the modestly selective M3 binding would translate into selective M3 functional antagonist activity.17 Interestingly, we found that 1 was a pan-mAChR functional antagonist (Fig. 1), moderately inhibiting M1–M5 (M1 IC50 = 2.8 µM, M2 IC50 > 10 µM, M3 IC50 > 10 µM, M4 IC50 > 10 µM, M5 IC50 = 4.0 µM), with a preference for M1 and M5, and very weak acitivty at M3. Thus, 1 is not a selective M3 antagonist. Unnatural analog 17 was more potent and selective than 1 (M1 IC50 = 1.9 µM, M2 IC50 > 10 µM, M3 IC50 = 5.0 µM, M4 IC50 = 5.2 µM, M5 IC50 = 2.0 µM), but still categorized as a pan-mAChR antagonist. These data suggest that the azabicyclo[3.3.0]octane ester chemotype is comparable to the pan-mAChR antagonist tropane ester chemotypes, such as atropine.15 However, this data is still intriguing and the synthesis and evaluation of additional unnatural analogs would be of scientific value.
Figure 1.

Human mAChR (M1–M5) functional assay with cremastrine 1 and unnatural analog 17, indicating that both are functional antagonists of all five mAChRs.
In summary, we have successfully extended our methodology to rapidly construct enantiopure idolizidines, pyrrolo[1,2-a]azepines and pyrrolo[1,2-a]azocines,4 to the synthesis of pyrrolizidine alkaloids. Here, we completed the first total synthesis of the pyrrolizidine alkaolid cremastrine (1) in seven steps from commercial materials and in 25.2% overall yield. The methodology also allowed for the synthesis of unnatural analog such as 17. Moreover, we demonstrated in mAChR functional assays, that both 1 and unnatural 17 are not selective M3 antagonists, as previous binding data suggested, but rather pan-mAChR antagonists. This concise synthesis will allow for the synthesis of additional unnatural analogs of 1 for biological evaluation as well as for the synthesis of related pyrrolizidine-based alkaloid natural products. These efforts are in progress and will be reported in due course.
Acknowledgments
The authors gratefully acknowledge funding from the Department of Pharmacology, Vanderbilt University Medical Center and the NIH/Molecular Libraries Production Center Network (MLPCN) (U54MH084659).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Ikeda Y, Nonaka H, Furumai T, Igarashi I. J. Nat. Prod. 2005;68:572–573. doi: 10.1021/np049650x. [DOI] [PubMed] [Google Scholar]
- 2.Peretto I, Petrillo P, Imbimbo BP. Med. Res, Rev. 2009;29:867–902. doi: 10.1002/med.20158. [DOI] [PubMed] [Google Scholar]
- 3.Conn PJ, Jones C, Lindsley CW. Trends in Pharm. Sci. 2009;30:148–156. doi: 10.1016/j.tips.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fadeyi OO, Senter TJ, Hahn KN, Lindsley CW. Chem. Eur. J. doi: 10.1002/chem.201200629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vanecko JA, West FG. Org. Lett. 2005;7:2949–2952. doi: 10.1021/ol050915o. [DOI] [PubMed] [Google Scholar]
- 6.El-Naggar M, Piggott AM, Capon RJ. Org. Lett. 2008;10:4247–4250. doi: 10.1021/ol8016512. [DOI] [PubMed] [Google Scholar]
- 7.Fürstner A, Kortte A. Chem. Asian J. 2008;3:310–318. doi: 10.1002/asia.200700288. [DOI] [PubMed] [Google Scholar]
- 8.Ishibashi H, Sasaki M, Taniguchi T. Tetrahedron. 2008;64:7771–7773. [Google Scholar]
- 9.Shin MR, Lee J, Lee M, Jung W, Lee J, Yoon J. Org. Chem. 2000;65:7667–7675. doi: 10.1021/jo0006573. [DOI] [PubMed] [Google Scholar]
- 10.Sugiura M, Mori C, Kobayashi S. J. Am. Chem. Soc. 2006;128:11038–11039. doi: 10.1021/ja064106r. [DOI] [PubMed] [Google Scholar]
- 11.Brinner KM, Ellman JA. Org. Biomol. Chem. 2005;3:2109–2113. doi: 10.1039/b502080h. [DOI] [PubMed] [Google Scholar]
- 12.Sun X-W, Liu M, Xu M-H, Lin G-Q. Org. Lett. 2008;10:1259–1262. doi: 10.1021/ol8001514. [DOI] [PubMed] [Google Scholar]
- 13.Schulte ML, Lindsley CW. Org. Lett. 2011;13:5684–5687. doi: 10.1021/ol202415j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robak MT, Herbage MA, Ellman JA. Chem. Rev. 2010;110:3600–3740. doi: 10.1021/cr900382t. [DOI] [PubMed] [Google Scholar]
- 15.A solution of ester 15 (50 mg, 0.0914 mmol) in 2 mL of 95:5 trifluoroacetic acid:H2O was stirred for 1 hr at room temperature then 0.19 mL of triethyl silane was added. This solution was stirred overnight and concentrated in vacuo to give a crude, yellow oil. This was dissolved in 1 mL DMSO and purified by reverse phase chromatography (2% to 45 H2O(0.1%TFA):AcCN) to give (19 mg, 80% yield) as a colorless oil. [α]D20 = −15.2 (c = 0.97 CHCl3; [α]D20 = −27.2 (c = 1.0, EtOH); 1H NMR (400.1 MHz, CDCl3) δ (ppm) = 6.87 (m, 1H), 4.49 (d, J = 5.2, 1H), 4.33 (m, 1H), 4.20 (m, 1H), 4.18 (m, 1H), 3.55 (m, 1H), 3.49 (t, J = 7 Hz 1H), 2.88 (m, 1H), 2.8 (m, 1H), 2.65 (dt, J = 8.1, 1H), 1.99 (m, 1H), 1.90 – 1.80 (brm, 4H), 1.72 (m, 2H), 1.59 (m, 1H), 1.42 (m, 1H), 1.1 (d, J = 4 Hz, 3H), 0.99 (t, J = 7 Hz, 3H). 13C NMR (100.6 MHz, pyr-d5) δ (ppm): 174.6, 72.6, 68.1, 63.1, 55.38, 53.27, 39.9, 39.46, 26.64, 26.2, 25.93, 24.78, 15.81, 11.75; HRMS (TOF, ES+) C14H26NO3 [M+H]+ calc’d 256.1913, found 256. 1911.
- 16.A solution of ester (50 mg, 0.0914 mmol) in 2 mL of 95:5 trifluoroacetic acid:H2O was stirred for 1 hr at room temperature then 0.19 mL of triethyl silane was added. This solution was stirred overnight and concentrated in vacuo to give a crude, yellow oil. This was dissolved in 1 mL DMSO and purified by reverse phase chromatography (2% to 45% H2O(0.1%TFA):AcCN) to give (19 mg, 80% yield) as a colorless oil. [α]D20 = (c = 1.12, CHCl3); 1H NMR (400.1 MHz, CDCl3) δ (ppm) = 7.33 (m, 5H), 4.66 (d, J = 11.49, 1H), 4.38 (d, J = 11.50, 1H) 4.15 (dt, J = 11.26, 4.89 Hz, 2H), 3.74 (d, J = 5.65 Hz, 1H), 3.67 (m, 1H), 3.46 (m, 1H), 3.11 – 2.92 (m, 3H), 2.41 (sextet, J = 6.77 Hz, 1H), 2.01 (m, 1H), 1.88 – 1.78 (m, 3H), 1.58 (m, 2H), 1.25 (m, 3H), 0.91 (d, J =6.77 Hz, 3H), 0.87 (t, J = 7.91, 3H); 13C NMR (100.6 CDCl3) δ (ppm): 172.44, 137.48, 128.37, 127.86, 82.65, 72.53, 64.69, 62.72, 61.18, 44.29, 41.49, 38.00, 31.30, 29.68, 28.75, 28.05, 24.71, 15.17, 11.28; HRMS (TOF, ES+) 346.2382, found 346.2381.
- 16.For full details of the mAChR functional assay see: Lebois EP, Bridges TM, Dawson ES, Kennedy Jp, Xiang Z, Jadhav SB, Yin H, Meiler J, Jones CK, Conn PJ, Weaver CD, Lindsley CW. ACS Chemical Neurosci. 2010;1:104–121. doi: 10.1021/cn900003h.
- 17.Schulte ML, Lindsley CW. In: Bioactive Heterocyclic Compound Classes: Pharmaceuticals and Agrochemicals. Lamberth N, Dinges J, editors. Wiley-VCH Verlag GmbH & Co; 2012. pp. 23–36. [Google Scholar]