

Abstract
Obesity and patient morbidity has become a health concern worldwide. Obesity is associated with over activity of the endocannabinoid system, which is involved in the regulation of appetite, lipogenesis and insulin resistance. Hypothalamic cannabinoid-1 receptor (CB1R) inverse agonists reduce body weight and improve cardiometabolic abnormalities in experimental and human obesity but displayed neuropsychiatric side effects. Hence, there is a need to develop therapeutics which employs blocking peripheral CB1 receptors and still achieve substantial weight loss. In view of the same, adipose tissue CB1 receptors are employed for this study since it is more specific in reducing visceral fat. Computer aided structure based virtual screening finds application to screen novel inhibitors and develop highly selective and potential drug. The rational drug design requires crystal structure for the CB1 receptor. However, the structure for the CB1 receptor is not available in its native form. Thus, we modelled the crystal structure using a lipid G-Protein coupled receptor (PDB: 3V2W, chain A) as template. Furthermore, we have screened a herbal ligand Quercetin [- 2- (3, 4-dihydroxyphenyl) - 3, 5, 7-trihydroxychromen-4-one] a flavonol present in Mimosa pudica based on its better pharmacokinetics and bioavailability profile. This ligand was selected as an ideal lead molecule. The docking of quercetin with CB1 receptor showed a binding energy of -6.56 Kcal/mol with 4 hydrogen bonds, in comparison to the known drug Rimonabant. This data finds application in proposing antagonism of CB1 receptor with Quercetin, for controlling obesity.
Keywords: Obesity, Cannabinoid receptor 1, CB1 inverse agonists, Quercetin, Rimonabant
Background
Peripheral CB1 receptors are located in the gastrointestinal (GI) tract, liver and in adipose tissue. CB1 receptor is a 7- transmembrane G-protein coupled receptor associated with the endocannabinoid signalling system. It inhibits adenyl cyclase and activates mitogen-activated protein (MAP) kinase. In addition, CB1 receptors inhibit presynaptic N- and P/Q-type calcium channels and activate inwardly rectifying potassium channels [1]. As seen earlier many potent drugs have been utilized in blocking the CB1 receptor but still pose a lot of neuropsychiatric effects which emphasizes the need of a novel drug which targets the peripheral CB1 receptor [2]. The drug Rimonabant which is a highly selective and potent inverse agonist for the hypothalamic CB1 receptor has been banned due to neuropsychiatric effects [3]. Recently, LH-21 a peripherally acting cannabinoid receptor neutral antagonist and possessing lower brain penetration profile was used for the treatment of obesity. It showed potentially reduced side effects indicating that peripheral blockage provides a safer means to alleviate obesity [4]. It has been reported that flavonoids have various biological activities such as antioxidant, antibacterial and anticancer effect and play roles in prevention of obesity [5]. Hence, flavonoids may act as novel drug candidates. For the present study, Quercetin a flavonol from Mimosa pudica leaves was employed. Quercetin is an extensively used flavonoid in research, which possesses good pharmacophore and bioavailability properties [6–9]. Secondly, it is an herbal adaptogen having drug like properties and obesity related studies have been carried out. Blocking of adipose tissue CB1 receptor with Quercetin is a novel approach since it is much more specific and potent approach in reducing the visceral fat. The major key role in obesity is the excess endocannabinoid production by adipocytes driving CB1 in a feed-forward dysfunction. Several health consequences attributable to obesity include coronary heart disease, diabetes, sleep apnea, digestive disorders and cancer [10]. Endocannabinoids (eicosanoids) act at the CB1 receptors to increase hunger and promote feeding and it is speculated that they decrease intestinal peristalsis and gastric emptying. Thus, antagonism at these receptors can inverse these effects [11]. CB1 antagonists produce inverse cannabimimetic effects that are opposite in direction from those produced by agonists for these receptors [12]. Antagonism of CB1 receptors increases insulin sensitivity in peripheral tissues and oxidation of fatty acids in muscles and the liver [13]. Structure prediction enables to explain the mechanisms of interaction between G-protein coupled receptors and variety of ligands, enzymes, ion channels and current drugs which underlies the basic concept of drug designing [14]. The major focus of the study is prediction of putative adipose tissue CB1 receptor – Quercetin binding conformations. Based on extensive literature studies we propose antagonism by computational docking thereby discovering novel herbal adaptogen for controlling obesity.
Methodology
Selection of ligand:
Endocannibinoids stimulate appetite and also regulate fat metabolism. This is evident from the fact that CB1 -/- mice are resistant to diet induced obesity even though their calorie intake is similar to that of wild type [15]. Thus, CB1 antagonists showing promise in treatment of obesity. It is possible to find out the druggish properties of herbal compounds by computer aided screening and the data will be useful to screen best lead molecules. The 3D structure of plant based ligand Quercetin is available in drug data base and the structure was retrieved from PubChem [16]. The required file format corresponding to the analysis such as modelling, docking, validation and ADME studies was generated by Open Babel [17].
ADMET studies:
The absorption, distribution, metabolism, excretion and toxicity (ADMET) are the most important part of pharmacological studies of the concerned molecule required for drug based discovery. Pre-ADMET is the tool that provides drug-likeliness, ADME profile and toxicity analysis for the ligand. Quercetin identified from the selected plant Mimosa pudica was tested by Pre-ADMET to study the pharmacological properties. It uses Caco2-cell (heterogeneous human epithelial colorectal adenocarcinoma cell lines) and MDCK (Madin-Darby Canine Kidney) cell models for oral drug absorption prediction and skin permeability, and human intestinal absorption model for oral and trans-dermal drug absorption prediction. Distribution is predicted using BBB (blood brain barrier) penetration and plasma protein binding. Pre-ADMET predicts toxicity based on the mutagenicity of Ames test and rodent carcinogenicity assays [18–19].
Receptor characterization:
The amino acid sequence of human CB1 [Accession: NP_001153731] was retrieved from GenBank [20] and the FASTA sequence is used for our studies. The secondary structure conformation of CB1 protein sequence was predicted by GORIV [21]. The domains and motifs, and transmembrane regions were predicted by TMHMM [22]. Information related to the CB1 signaling was given by KEGG [23] and concise genomic related information on human CB1 gene was retrieved from Gene Cards [24]. The availability of good template dictates quality of the model, so it is important to identify the best template structure. The template was selected based on percentage of identity, similarity, expectation value and alignment scores using BLASTP [25] algorithm across Protein Data Bank (PDB).
Homology modelling:
Previous studies have reported that blocking CB1 receptor circumvents obesity condition [26, 27]. Computer aided drug design provides the platform to understand the receptor and ligand interactions. A three dimensional structure of the protein from its amino acid sequence was generated by Modeller. Easy Modeller, based on programming language PerlTk uses a very simple and easy interface to implement the features of Modeller v9.7 [28]. The crystal structure of CB1 was modelled using Easy Modeller v2.0 and Swiss PDB Viewer [29] with 3V2W, chain A used as the template from Protein Data Bank [30].
Model validation:
The X-ray diffraction structure of template (PDB ID: 3V2W, Chain A) was identified as the best template. The initial model building and structural alignment was performed and the modelled protein was visualized using PyMol [31]. Parameters like covalent bond distances and angles, stereochemical validation and atom nomenclature were validated using PROCHECK [32]. The overall quality factor of non-bonded interactions between different atoms types were calculated by ERRAT program [33]. DaliLite [34] is used to calculate root mean square deviation (RMSD) between the set of targets and template protein to check deviation of modelled protein from the template protein structure.
Docking studies:
The selected herbal ligand, Quercetin was used to dock the target protein using AutoDock 4.2 [35] by Lamarckian genetic algorithm. The program uses a Monte Carlo simulated annealing for configurational exploration using grid based molecular affinity potentials and provides bioactive conformation by energy minimization. The catalytic and binding site of the target has been identified by Auto Grid. The grid box was set in such a way that includes only the selected active site residues. The rigidity parameters were set for the receptor, keeping the ligand flexible. The structure and chemical properties of the active site allow the recognition and binding of the ligand. The best conformations were screened in terms of lowest binding energy among several bioactive conformations generated by various iterations [36].
Discussion
The three dimensional coordinates of Quercetin was retrieved from PubChem. The molecular properties were deduced from Molinspiration [37] which is tabulated Table 1 (see supplementary material). The PreADMET tool provided information about the absorption, distribution and toxicity details about the herbal ligand and was found to be in acceptable limits, Table 2 (see supplementary material). The seconadary structure of CB1 receptor was predicted and which contains 100 alpha helices, 135 extended strands and 237 random coils. It was found that coils were predominant in the modelled protein and helices were present in small proportion. CB1 receptor has 7 transmembrane helices in which residues 118-140, 153-175, 190-212, 233-255, 275-297, 345-367 and 377-399 span the membrane. CB1 receptor [Gene name: CNR1] is a GPCR and mediates through MAP kinase pathway.
Homology searches in PDB were performed using the program BLASTP, by comparing and aligning the target sequence with each sequence in PDB. The sequence identity between the template (PDB ID: 3V2W chain A; crystal structure of a lipid G Protein-coupled receptor) and cannabinoid receptor 1 isoform a (Homo sapiens) is 40%. Further, structure-based sequence alignment of CB1 of Homo sapiens against the selected template was performed. The 3D homology model of CB1 receptor in Homo sapiens (Accession NP_001153731) was predicted using Easy Modeller v2.0, which used Modeller automodel to build homology models. High quality models demands an accurate sequence alignment between the model and the template protein. The annotated model structure of the receptor was predicted, which was “CB1 receptor. B99990001”. It was further mutated with same residues and torsion angles were adjusted by SPDBV to generate the best model “CB1 receptor.pdb” for further docking studies (Figure 1A). The energy value of the model was calculated before and after the minimization. It has noted that after minimization the energy of the model reaches its energy minima which showed more stable structure. Structural evaluations and validation of protein model were performed by the programs called ProCheck and Verify 3D. The evaluation of CB1 receptor model revealed that stereochemical and geometrical parameter implemented in ProCheck was satisfied in this model. The path with the best RMSD is subject to dynamic programming to achieve an optimal alignment. The z-score can be used to filter less significant results or alternatively look for weak similarities. In spite of that, the relationship between backbone RMSD in Å and structure quality for X-ray structure ensemble for protein structure comparison showed that they were barely acceptable and have a very closely related to each other [19, 36]. Therefore, the model predicted was acceptable. The RMSD value of modelled protein was found to be 1.0 Å and z-score of 40.7. Stereochemical validation shows out of 377 residues, 85.1% residues are in allowed region, 11.9% in additional allowed regions, 1.5% in generously allowed regions and 1.5% in disallowed regions of Ramachandran plot (Figure 1B). Further, validation was done by various empirical force fields. Overall quality factor of the model identified to be 81.30 and error values of individual residues are negligible. The docking was performed effectively with the selected ligand. Active site was deduced from Q-site finder [38], a ligand binding site prediction tool. It works by binding hydrophobic (CH3) probes to the protein, and finding clusters of probes with the most favourable binding energy. These clusters are placed in rank order of the likelihood of being a binding site according to the sum total binding energies for each cluster. The residues selected for the grid were Met 103, Asp 104, Glu 106, Cys 107, Val 110, Leu 111, Asn 112, Pro 113, Ser 114, Gln 115, Gln 116, Leu 117, Asp 176, Phe 177, His 178, Val 179, His 181, Arg 182, Phe 189, Lys 192, Leu 193, Asp 266, Ile 267, Phe 268 and Pro 269. The main interacting residues of protein with Quercetin are Ser 114, His 181, Asp 266, His 178, Ile 267, Asn 112, Asp 104 and Lys 192. A total of 10 conformations were generated. Information about the number of hydrogen bonds and binding energy is provided Table 3 (see supplementary material). The best docked conformation was the 4th one with -6.56 kcal/mol binding energy and 4 hydrogen bonds (Figure 2A).
Figure 1.
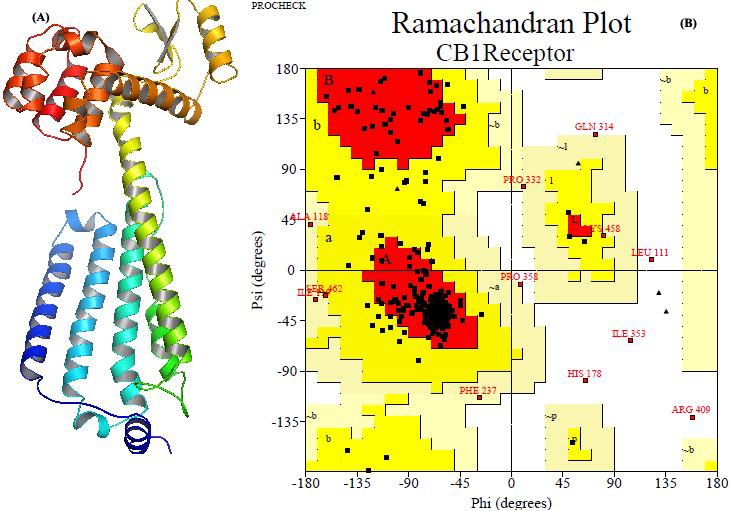
(A) The generated 3D model of protein (CB1 receptor) consists of stable secondary structure (helices and coils) which gives the catalytic sites for drug interaction is visualized; (B) Ramachandran plot of the model depict that 85.1% residues are in allowed region, 11.9% in additional allowed regions, 1.5% in generously allowed regions and 1.5% in disallowed regions.
Figure 2.
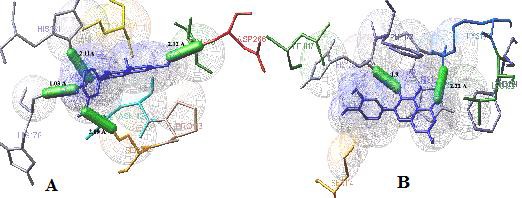
Docked structures of Quercetin with modelled CB1 receptor (A& B) by AutoDock. (A) The ligand – receptor interaction in the 4th conformation was stabilized by four H bonds (represented as green colored stick). The bond length was 1.03 Ao with His178, 2.11 Å with His181, 2.09 Å with Ser114 and 2.12 Å with Asp266 respectively, indicating a stronger and stable interaction The binding energy of docked complex was found to be -6.56 kcal/mol implies more stable docking. (B) Since, inverse agonism on CB1 receptor is based on binding to Lys192, Quercetin was docked to observe for the same. The interaction was stabilized by two hydrogen bonds and bond length was 1.9 Å with His178 and 2.22 Å with Lys192 respectively. This interaction indicated that Quercetin can be implicitly used to block the receptor with better binding affinities as compared to known drug Rimonabant.
Though the use of drugs has been utilized to curb obesity, it still shows prominent side effects making it not viable for long term use. Herbal compounds are better therapeutic substances with fewer side effects. Based on the above studies we have identified the phytoligand Quercetin as a novel inhibitor against the receptor. Previous studies have revealed that blockage of CB1 receptor is mainly mediated through transmembrane helix (TMH) 3−4−5−6 region which includes an aromatic microdomain comprised of residues F3.25, F3.36, W4.64, Y5.39, W5.43 and W6.48. Earlier reports devised that aromaticity at position 5.39 in CB1 is crucial for proper function of CB1. Modelling studies reported that in the inactive state of CB1, the binding site of the CB1 inverse agonist/antagonist SR141716A (rimonabant) is within the TMH 3−4−5−6 aromatic microdomain and involves direct aromatic stacking interactions with F3.36, Y5.39 and W5.43, as well as hydrogen bonding with K3.28 [39–42]. CB1 receptor displays a high level of constitutive activity and displays two state-model of receptor activation in which receptors are in equilibrium between two states, active and inactive. An agonist will stabilize the active state leading to activation, a neutral antagonist binds equally to active and inactive states, whereas an inverse agonist will preferentially stabilize the inactive state. Rimonabant has been reported in many cases to behave as an inverse agonist rather than as a neutral antagonist and it is likely that it binds preferentially to the inactive state of the CB1, thereby decreasing the activation of the signalling pathway. The key binding interaction is a hydrogen bond formed between the carbonyl group of Rimonabant and the Lys192 residue of the CB1 receptor. From our study, it has been deduced that Quercetin not only shows binding efficiency to the Lys192 (Table 3) &(Figure 2B) indicating it will be suitable to block the receptor but also has efficient binding capacity due to its 4 hydrogen bonding with Ser114, His 181 and Asp 266. Thus, a potential inhibitor against obesity by blocking CB1 receptor has been proposed.
Conclusion
Computer aided drug discovery is an imminent and effective platform for identification of novel therapeutic substances. Naturally available herbal compound and its antagonistic effectiveness against the CB1 receptor are tested by molecular docking. Quercetin was identified to be an effective phytoligand for CB1 receptor with better binding and inhibitory properties. Hence, it is a novel approach against obesity with better efficacy in comparison to known drug Rimonabant. Present study will boost the use of herbal adaptogens as a remedy with potentially less side effects for treating obesity and its related disorders. Henceforth, drugs will be designed and derived from plant origin and discovery and development of flavonoids as ideal drug candidates through computer aided screening will be a major contribution to pharmaceutical sectors.
Supplementary material
Acknowledgments
The authors thankfully acknowledge the R & D Centre of Life Sciences and Engg, Dayananda Sagar Institutions for providing the necessary facilities and also grateful to Dr. P.S Rao, Vice President, R & D in Life Sciences for his constant support and encouragement throughout the study.
Footnotes
Citation:Shrinivasan et al, Bioinformation 8(11): 523-528 (2012)
References
- 1.K Mackie. Annu Rev Pharmacol Toxicol. 2006;46:101. doi: 10.1146/annurev.pharmtox.46.120604.141254. [DOI] [PubMed] [Google Scholar]
- 2.J Tam, et al. J Clin Invest. 2010;8:2953. [Google Scholar]
- 3.J Steurer. Praxis (Bern 1994) 2010;99:1439. doi: 10.1024/1661-8157/a000314. [DOI] [PubMed] [Google Scholar]
- 4.M Alonso, et al. Br J Pharmacol. 2012;165:2274. doi: 10.1111/j.1476-5381.2011.01698.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.H Kamisoyama, et al. Biosci Biotechnol Biochem. 2008;72:3225. doi: 10.1271/bbb.80469. [DOI] [PubMed] [Google Scholar]
- 6.S Enomoto, et al. Circulation. 2008;118:S1124. [Google Scholar]
- 7.J Ahn, et al. Biochem Biophys Res Commun. 2008;373:545. doi: 10.1016/j.bbrc.2008.06.077. [DOI] [PubMed] [Google Scholar]
- 8.EU Graefe, et al. J Clin Pharmacol. 2001;41:492. doi: 10.1177/00912700122010366. [DOI] [PubMed] [Google Scholar]
- 9.PC Hollman, et al. FEBS Lett. 1997;418:152. doi: 10.1016/s0014-5793(97)01367-7. [DOI] [PubMed] [Google Scholar]
- 10.GA Bray, et al. J Clin Endocrinol Metab. 2004;6:2583. doi: 10.1210/jc.2004-0535. [DOI] [PubMed] [Google Scholar]
- 11.KA Bronander, MJ Bloch. Vasc Health Risk Manag. 2007;3:181. doi: 10.2147/vhrm.2007.3.2.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.V Di Marzo. Drug Discov Today. 2008;13:1026. doi: 10.1016/j.drudis.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 13.PN Patel, R Pathak. Am J Health Syst Pharm. 2007;64:481. doi: 10.2146/060258. [DOI] [PubMed] [Google Scholar]
- 14.AL Hopkins, CR Groom. Nat Rev Drug Discov. 2002;1:727. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 15.D Osei-Hyiaman, et al. J Clin Invest. 2005;5:1298. doi: 10.1172/JCI23057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Y Wang, et al. Nucleic Acids Res. 2010;38:D255. doi: 10.1093/nar/gkp965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.NM O'Boyle, et al. J Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.A Seal, et al. Bioinformation. 2011;5:430. [Google Scholar]
- 19.S Skariyachan, et al. Bioinformation. 2011;7:222. [Google Scholar]
- 20.M Manich, et al. PLOS One. 2008;3:e3764. doi: 10.1371/journal.pone.0003764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.F Xia, et al. BMC Bioinformatics. 2011;12:S5. doi: 10.1186/1471-2105-12-S12-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.A Krogh, et al. J Mol Biol. 2001;3:567. [Google Scholar]
- 23.M Kanehisa, et al. Nucleic Acids Res. 2010;38:D355. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.M Rebhan, et al. Bioinformatics. 1998;14:656. [Google Scholar]
- 25.M Johnson, et al. Nucleic Acids Res. 2008;36:W5. [Google Scholar]
- 26.AN Verty, et al. Int J Obes (Lond) 2012;3:10. [Google Scholar]
- 27.HN Ginsberg, SC Woods. Obesity (Silver Spring) 2009;17:1821. [Google Scholar]
- 28.J Tian, et al. BMC Bioinformatics. 2010;11:370. doi: 10.1186/1471-2105-11-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.B Knapp, et al. Protein Sci. 2008;6:977. [Google Scholar]
- 30.HM Berman, et al. Nucleic Acids Res. 2000;28:235. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.D Seeliger, BL de Groot. J Comput Aided Mol Des. 2010;24:417. doi: 10.1007/s10822-010-9352-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.A Sharma, A Nigam. Bioinformation. 2010;5:136. [Google Scholar]
- 33.C Colovos, TO Yeates. Protein Sci. 1993;2:1511. [Google Scholar]
- 34.L Holm, J Park. Bioinformatics. 2000;16:566. doi: 10.1093/bioinformatics/16.6.566. [DOI] [PubMed] [Google Scholar]
- 35.AP Norgan, et al. J Cheminform. 2011;3:12. [Google Scholar]
- 36.S Skariyachan, et al. Bioinformation. 2011;6:375. [Google Scholar]
- 37.J Jaganatharaja, R Gowthaman. Bioinformation. 2006;1:112. doi: 10.6026/97320630001112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.AT Laurie, RM Jackson. Bioinformatics. 2005;21:1908. [Google Scholar]
- 39.GG Muccioli, DM Lambert. Curr Med Chem. 2005;12:1361. doi: 10.2174/0929867054020891. [DOI] [PubMed] [Google Scholar]
- 40.S Ortega-Gutiérrez, ML López-Rodríguez. Mini Rev Med Chem. 2005;5:651. doi: 10.2174/1389557054368754. [DOI] [PubMed] [Google Scholar]
- 41.JH Lange, CG Kruse. Drug Discov Today. 2005;10:693. doi: 10.1016/S1359-6446(05)03427-6. [DOI] [PubMed] [Google Scholar]
- 42.SD McAllister, et al. J Med Chem. 2003;46:5139. doi: 10.1021/jm0302647. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.