

Abstract
Hexokinases (HKs) are the enzymes that catalyses the ATP dependent phosphorylation of Hexose sugars to Hexose-6-Phosphate (Hex-6-P). There exist four different forms of HKs namely HK-I, HK-II, HK-III and HK-IV and all of them share a common ATP binding site core surrounded by more variable sequence that determine substrate affinities. Although they share a common binding site but they differ in their kinetic functions, hence the present study is aimed to analyze the binding mode of ATP. The analysis revealed that the four ATP binding domains are showing 13 identical, 7 similar and 6 dissimilar residues with similar structural conformation. Molecular docking of ATP into the kinase domains using Molecular Operating Environment (MOE) soft ware tool clearly showed the variation in the binding mode of ATP with variable docking scores. This probably explains the variable phosphorylation rates among hexokinases family.
Keywords: Hexokinase, Molecular Docking, MOE
Background
HK catalyzes the phosphorylation process by transferring the γ- phosphoryl group from ATP to the OH group of sixth carbon of Hexose sugars to give Hex-6-P. There are four isozymes of mammalian HKs namely HK-I, HK-II, HK-III and HK-IV, which are tissue specific and are located in different organs of the body [1, 2]. Liver contains all four types of HKs while kidney and intestine lacks HK-IV. HK-I and HK-II are found in epididymal fat pad, skeletal muscle, brain and heart. However, HK-I is predominantly present in brain and kidney and HK-II is predominant in skeletal muscle and epididymal fat pad [2]. The formation of Hex-6-P by HKs commits hexose sugars to alternative metabolic pathways: the formation of glycogen and short-term carbohydrate storage in liver, immediate use in energy production by glycolysis and the formation of pentose phosphates in the anabolic reactions [3] (Figure 1). Up regulation and down regulation of metabolic pathways can be linked to the different organs in the body and these differences may be attributed to the structure, affinity for substrates, inhibitors and sub cellular location of the isozymes [3]. HK-I and HK-II have a tail on the N-terminus that is important to bind with mitochondria whereas, HK-III and HK-IV lacks such structures and hence they are unable to bind to mitochondria. Thus, these isozymes may be associated with metabolic pathways other than glycolysis. All HKs share a common ATP binding site core surrounded by more variable sequence that determines substrate affinities. Although they share a common ATP binding site, the difference in their kinetic functions was observed [4]. This may be probably due to the variation in the active site residues and conformations which will finally affect the phosphorylation machinery. In order to ascertain these variations we carried out an insight structural analysis of all HKs concentrating on the kinase domain conformations. These different conformations may results in variable binding of ATP among HKs and hence there may be variation in the phosphorylation mechanism. In the present study we have carried out molecular docking study to predict the catalytic interactions between ATP and kinase domains of all HKs.
Figure 1.
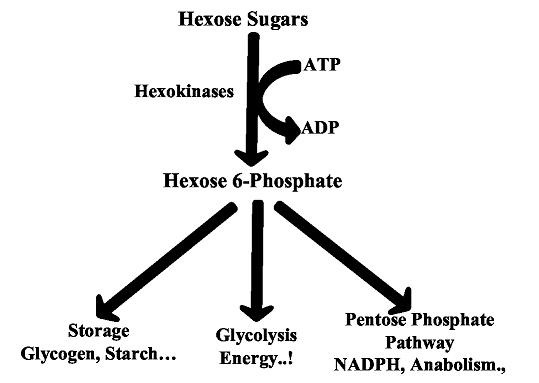
Fate of Hexose Sugars by Hexokinases
Methodology
Hexokinase Structures:
The three dimensional structures of HK-I (1HKC), HK-II (2NZT) and HK-IV (1V4S) were obtained from Protein Data Bank (PDB) [5]. As the structure of HK-III is not available so far in the PDB we have constructed its 3D model by homology modeling method.
Homology modeling of HK-III:
The three dimensional model of the HK-III was constructed by using Modeller 9v8 tool [6]. The HK-III protein sequence was retrieved from NCBI [7] (AC No: NP_002106.2) and it was subjected to BLASTp [8] against PDB and the crystal structure of human Hexokinase-II (PDB ID: 2NZT) was chosen as template for modeling which is having a maximum identity of 56%. The protein sequence and 3D structure of the template were retrieved. A sequence alignment file was generated in PIR format for Query and template sequences using ClustalX tool [9], a Python script was written and 20 models were generated. Among 20, the model with the lowest DOPE score was selected for further analysis.
Validation of HK-III Model:
The stereo chemical quality of energy minimized HK-III model was assessed and validated by PROCHECK validation server [10]. The recognition of errors in this theoretical protein model is also a critical point employed in protein structure validation. Hence the overall quality of the structure was calculated by ProSA web server [11]. It reads the atomic coordinates of the model and generates the Z-Score that is a determinant of the quality of the model.
Identification and alignment of kinase domains of Hexokinases:
The kinase domains of all HKs were identified by scanning their protein sequences against PROSITE data base that consists of documentation entries describing protein domains, families and functional sites as well as associated patterns and profiles [12]. The identified domains were aligned by multiple sequence alignment process using ClustalX tool to find out the similarities and dissimilarities among the domains.
Superimposition of kinase domains:
The kinase domains of all HK structures were superimposed to find out the conformational variations using multiple 3D alignment module of MATRAS tool [13].
Molecular Docking:
Molecular docking was carried using MOE docking soft ware tool (MOE 2011.10). The 3D structure of ATP was retrieved from PubChem [14] and its geometry was optimized in MOE working environment. All the HK structures were protonated and energy minimized individually in the MMFF94x force field at a gradient value of 0.05 by enabling all bonded and non bonded interactions. Individual dockings were performed between the kinase domains and ATP to find out the binding modes and affinity variations. The Proxy triangle methodology was applied where the conformers are pre-superposed prior to be placed in the kinase domains. The docked conformers are ranked by alpha HB scoring function, which is a linear combination of two terms, i.e. the geometric fit of the ligand to the binding site and the hydrogen bonding effects and finally both terms are summed over all ligand atoms. The conformations were refined and rescored in the same force filed to remove the duplicate conformations. At the end of docking process the conformation with least docking score was chosen in each docking to study the binding orientations of ATP among all kinase domains.
Results
Homology modeling of the HK-III structure using Modeller 9v8 generated 20 best models and among them, the lowest DOPE score of -111452.515 was found with the 18th model and this structure was chosen to validate the stereo chemical quality by PROCHECK and ProsaWeb servers. The Ramachandran plot generated by PROCHECK showed that 93.4% of residues in allowed regions, 6.1% in additional allowed regions, 0.5% in generously allowed regions and no residues were found in the disallowed regions (Figure 2). The ProsaWeb analysis showed a quality Z-Score of –10.56, which falls in the range of native XRay crystallographic, structures (Figure 3). Both of these results indicate that the generated HK-III model is valid with good stereo chemical quality. The kinase domains of HKs identified from PROSITE database showed that they are all having an identical length of 26 residues. The sequence alignment of these domains showed that they share 13 identical, 7 similar and 6 dissimilar residues (Figure 4). To find out the conformational variations, these domain structures were superimposed and it was found that they share a common structural conformation (Figure 5) with two anti parallel β-sheets and their RMSD values were found to be nearby zero, which indicated the close structural identity among those domains Table 1 (see supplementary material), but no significant structural variation was observed in the conformations during alignment.
Figure 2.
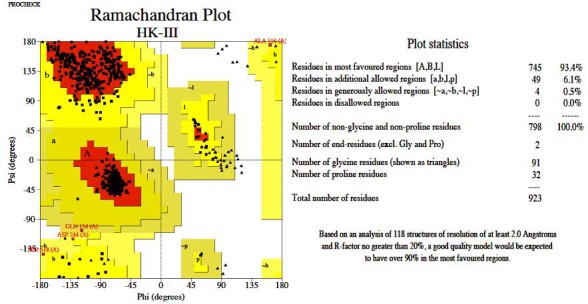
Ramachandran plot showing the stereo chemical quality of homology model of HK-III generated by PROCHECK validation server.
Figure 3.
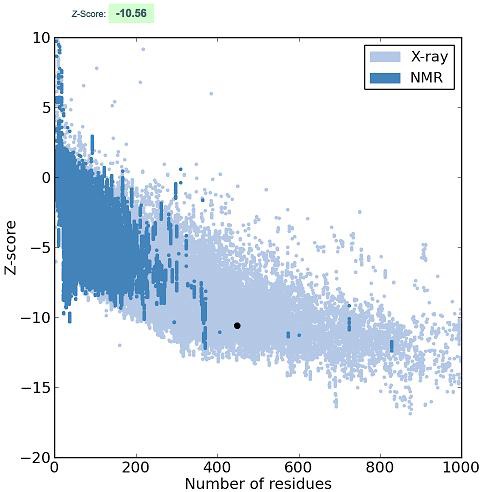
ProsaWeb quality plot of HK-III model showing a Zscore of -10.56 that is observed in the range of native X-Ray crystallographic structures.
Figure 4.

Sequence alignment of kinase domains of HKs. * indicates the identical residues: indicates similar residues and the remaining residues are dissimilar residues.
Figure 5.

The super imposed kinase domain structures of HK-I (Yellow), HK-II (Green), HK-III (Pink) and HK-IV (Blue).
The molecular docking of ATP into these kinase domains revealed that the binding orientations are completely different among all HKs (Figure 6). ATP is found to be interacting with G170, K621, S682 and E742 residues of HK-I with six hydrogen bonds and among S682 is showing predominant role to bind with ATP with three hydrogen bonds. HK-II is showing interaction with ATP with H504, Q608 and N609 residues forming three hydrogen bonds. HK-III is found to be interacting with ATP by T626, R520, L623 and N624 residues with five hydrogen bonds and among N262 showing two hydrogen bonds with ATP. Finally HK-IV is showing interaction using H156 and E157 residues with a single hydrogen bond each. The interaction of phosphate of ATP was observed with all kinase domains except HK-IV, where the interaction of ATP was found with its 2'-OH group of ribose sugar and H156 residue and amino group of Adenine ring with E157 residue of the kinase domain. In HK-I the three phosphates of ATP (α, β and γ) were found to be interacting with the kinase domain residues.
Figure 6.
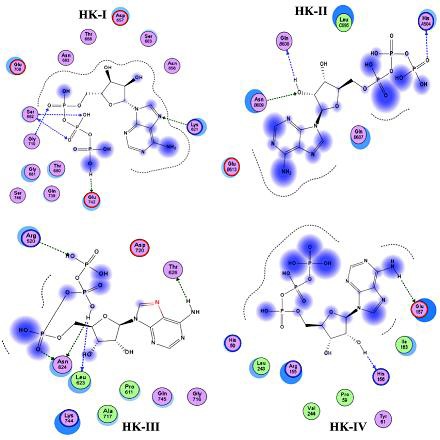
Molecular docking of ATP showing binding mode variations in the kinase domains of HKs. Arrow marks indicate hydrogen bonding between ATP and kinase domain residues and blue shaded region indicates the solvent contacts made by ATP.
In HK-II only the γ−phosphate of ATP is found to be interacting with kinase domain along with its 2'-OH group of ribose sugar. The three phosphate groups and amino group of Adenine ring of ATP were found to be interacting with HK-III kinase domain (Figure 6). The docking scores for the four HKs indicated that HK-IV is showing best lowest docking score of -13.1834 and the remaining three HKs are showing almost similar docking scores of -12.417, -12.2306, -12.6445 for HK-I, HK-II and HK-III respectively Table 2 (see supplementary material). These values indicate the ATP-kinase domains complex strengths where the lowest value indicates the greater stability.
Discussion
HKs work continuously to provide energy and thereby keeping the cells alive although they are tissue specific and confined to specific organs of the body. Their activity differs based on the requirement of energy sources at different stages [15]. Among the four HKs, the first three shows a low Km and HK-IV shows high Km and hence it is called as a predominant HK among HK family i.e. Glucokinase. This Km values determines the binding ability of the enzyme with substrate or ATP. This variation in the Km will result in the variable catalytic mechanism among their kinase domains [16]. This Km variation may be due to many factors and here in the present study we aimed to explain an important reason with respect to the binding affinities and orientation of ATP molecule in the binding site. Glucokinase which is HK-IV shows high affinity for glucose than other HKs and is the major enzyme associated with glucose phosphorylation in the body to process them to successive metabolic processes. Such a high phosphorylation rates may be due to high affinity towards substrate and ATP by its kinase domain [17]. Although the similar domains are shown by other HKs they are not showing high affinity as to that of HK-IV. The kinase domains of all HKs showed the similar conformation with two anti parallel β-sheets that providing the stability of the domains during catalysis. The structural alignment of kinase domains cleared that there is no conformational variation among them and the catalytic variation may not be due to the conformational variation of kinase domains (Figure 5). The residual composition of the domains is also found to be similar to major extent (Figure 4). The molecular docking study revealed the variation in the interaction of ATP which may explain the reasons for variable phosphorylation rates among four kinases. Interestingly it was found that the phosphate moiety of the ATP was in free form without interacting with the kinase domain residues in the HK-IV and hence it may be readily available for the phosphorylation of the sugars. But, in HK-I and HK-III, the α, β,and γ phosphates of the ATP were found to be in bound condition with their kinase domain residues, where as in HK-II, only γ phosphate is found to be in bound condition to its kinase domain (Figure 6). In all these hexokinases, during catalytic process only γ phosphate that is at free end will be donated to the OH group of the sixth carbon of hexose sugar [1]. In this study it was found that among first three HKs there is no availability of the free γ phosphate where as it is available in HK-IV so that facilitating its transfer to hexose sugars. This might be an important evidence for the high Km and rapid phosphorylation rates by HK-IV than remaining HKs. This is more strengthened by their docking score variations where HK-IV showed a lowest docking score among all indicating the highest affinity for ATP than other HKs Table 2 (see supplementary material). Finally this study gave an insight into the variation of binding modes of ATP in the respective kinase domains. These orientations along with their variable docking scores may explain the variation and provide a theoretical basis for the variable phosphorylation rates among hexokinase family.
Conclusion
The molecular docking of ATP into the kinase domains of four HKs revealed the binding mode variations and the phosphate moiety of ATP is found to be available only in HK-IV but not in remaining HKs. This might be responsible for their variable catalytic rates of phosphorylation.
Supplementary material
Acknowledgments
We are highly thankful for DST INSPIRE Division, Department of Science and Technology (DST), Government of India, for supporting with INSPIRE Fellowship.
Footnotes
Citation:Kumar et al, Bioinformation 8(12): 543-547 (2012)
References
- 1.C Gonzalez, et al. Biochem Biophys Res Commun. 1964;16:347. doi: 10.1016/0006-291x(64)90038-5. [DOI] [PubMed] [Google Scholar]
- 2.HM Katzen, RT Schimke. Proc Natl Acad Sci U S A. 1965;54:1218. [Google Scholar]
- 3.JE Wilson. J Exp Biol. 2003;206:2049. doi: 10.1242/jeb.00241. [DOI] [PubMed] [Google Scholar]
- 4. http://www.proteopedia.org/wiki/index.php/Hexokinase.
- 5.HM Berman, et al. Nucleic Acids Res. 2000;28:235. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.N Eswar, et al. Curr Protoc Bioinformatics. 2006;5:5. [Google Scholar]
- 7. http://www.ncbi.nlm.nih.gov/
- 8.SF Altschul, et al. Nucleic Acids Res. 1997;25:3389. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.JD Thompson, et al. Nucleic Acids Res. 1997;25:4876. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.RA Laskowski, et al. J. Appl. Cryst. 1993;26:283. [Google Scholar]
- 11.M Wiederstein, MJ Sippl. Nucleic Acids Res. 2007;35:W407. [Google Scholar]
- 12.E De Castro, et al. Nucleic Acids Res. 2006;34:W362. [Google Scholar]
- 13.T Kawabata, et al. Nucleic Acids Res. 2003;31:3367. doi: 10.1093/nar/gkg581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Y Wang, et al. Nucleic Acids Res. 2009;37:623. [Google Scholar]
- 15.H Kanno. Baillieres Best Pract Res Clin Haematol. 2000;13:83. doi: 10.1053/beha.1999.0058. [DOI] [PubMed] [Google Scholar]
- 16.L Grossbard, et al. J Biol Chem. 1966;241:3546. [PubMed] [Google Scholar]
- 17.S Kawai, et al. J Biosci Bioeng. 2005;99:320. doi: 10.1263/jbb.99.320. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.