

Abstract
Oxidized phospholipid species are important, biologically relevant, lipid signaling molecules that usually exist in low abundance in biological tissues. Along with their inherent stability issues, these oxidized lipids present themselves as a challenge in their detection and identification. Often times, oxidized lipid species can co-chromatograph with non-oxidized species making the detection of the former extremely difficult, even with the use of mass spectrometry. In this study, a normal-phase and reverse-phase two dimensional HPLC-mass spectrometric system was applied to separate oxidized phospholipids from their non-oxidized counterparts, allowing unambiguous detection in a total lipid extract. We have utilized bovine heart cardiolipin as well as commercially available tetralinoleoyl cardiolipin oxidized with cytochrome c (cyt c) and hydrogen peroxide as well as with lipoxygenase to test the separation power of the system. Our findings indicate that oxidized species of not only cardiolipin, but other phospholipid species, can be effectively separated from their non-oxidized counterparts in this two dimensional system. We utilized three types of biological tissues and oxidative insults, namely rotenone treatment of lymphocytes to induce mitochondrial damage and cell death, pulmonary inhalation exposure to single walled carbon nanotubes, as well as total body irradiation, in order to identify cardiolipin oxidation products, critical to the cell damage/cell death pathways in these tissues following cellular stress/injury. Our results indicate that selective cardiolipin (CL) oxidation is a result of a non-random free radical process. In addition, we assessed the ability of the system to identify CL oxidation products in the brain, a tissue known for its extreme complexity and diversity of CL species. The ability of the two-dimensional HPLC-mass spectrometric system to detect and characterize oxidized lipid products will allow new studies to be formulated to probe the answers to biologically important questions with regard to oxidative lipidomics and cellular insult.
Keywords: lipids, mass spectrometry, oxidative lipidomics, cardiolipin, apoptosis, high performance liquid chromatography
1.1 Introduction
Oxidation of phospholipids and accumulation of their oxygenated intermediates have long been considered as participants of two possibly interrelated processes: cell/tissue damage and danger signaling [1]. However, direct experimental evidence supporting these views is scarce, mostly due to technological difficulties in quantitative assessments of peroxidized phospholipids. The traditional point of view is that oxidative stress causes – through yet to identified mechanisms – random free radical peroxidation of phospholipids whereby the rule of preferable oxidation of most polyunsaturated fatty acids dominates over the types of phospholipids based on the specificity of their polar head-groups. In contrast, peroxidation pathways catalyzed by specific catalysts – such as cytochrome c in apoptotic cells – should non-randomly oxidize those classes of phospholipids that are localized in the immediate proximity to the catalytic sites. Among those of particular interest in the context of peroxidation reactions are cardiolipins (CLs), a critical phospholipid component of cellular membranes from bacteria to mammals. In bacteria, CL is localized in the plasma membrane while in mammals, CL is exclusively confined to the inner membrane of mitochondria [2]. While bacterial CL species are mostly saturated or monounsaturated [2], mammalian CLs contain polyunsaturated fatty acid residues, hence are highly prone to oxidation [3-5]. The importance of CL peroxidation in mammals is also emphasized by its critical role in the function of numerous mitochondrial enzymes involved in electron transport chain function and other proteins involved in energetic metabolism which include cytochrome c (cyt c) oxidase [6], creatine kinase [7], ATP synthase [8] and the mitochondrial ADP carrier [9] among others.
Our laboratory has pioneered the work describing the specific oxidation of CL which is catalyzed by cyt c that acts as a CL-specific peroxidase in mitochondria during the early part of the apoptotic program. Specifically, we have demonstrated that CL oxidation is required for the release of cyt c and other pro-apoptotic factors into the cytosol and it is an important step in the mitochondrial stage of apoptosis [10]. Since CL is sequestered primarily in mitochondria of mammalian cells, it has also been viewed as a potent “danger signal” to the immune system when tissue damage occurs as evidenced by the presence of anti-CL antibodies in plasma of patients with a variety of diseases [11,12,13,14,15] and the report of CL binding to the CD1 protein and stimulating a host T-cell response to bacterial lipids and potentially host CL [16]. These results suggest that the CL-responsive T cells may play a role in immune surveillance during infection and tissue injury. The fact that CL is a normal component of plasma lipoproteins [17], suggests that the anti-phospholipid antibodies present in various disease states, could recognize CL or oxidized CL in conjunction with lipoproteins and/or plasma proteins. In addition, CL has been implicated as a modulator of proteins like reactive protein c, acting as a cofactor thereby promoting its anticoagulant activity [18].
As mentioned above, a high percentage of CL species that are found in mitochondria are enriched in polyunsaturated fatty acids, making them excellent candidates for oxidation in general, and to cytochorme c-catalyzed peroxidation during apoptosis. A high degree of structural variability of CL species exists depending upon the tissue in question, ranging from a few major species in heart to over two hundred plus species in brain. Oxidized forms of CL are now pursued with high interest due to their role in apoptosis via the activation of cyt c peroxidase activity and eventual loss of mitochondrial permeability and asymmetry [19,20] and as potential signaling markers that are released during cellular damage.
Analysis of oxidized lipids, especially peroxides, has become a daunting task in mass spectrometry due to their low abundance, chemical and thermal instability and the potential for further propagation of lipid oxidation and eventual degradation. Peroxidized lipids are very prone to oxidation and degradation by heat and this characteristic was indeed an early obstacle for their measurement which was overcome by the use of derivatization protocols. Lately, liquid chromatography-mass spectrometry (LC-MS) has become an indispensable tool for the analysis of lipids. Separation of lipids can be performed by normal and reverse phase chromatography and numerous methods have been described. Normal phase separation is based in adsorption and polarity of the lipids to the stationary surface whereas in reverse-phase, hydrophobicity plays the major role. Both techniques have their advantages and disadvantages. Normal phase chromatography allows separation of lipid classes although both oxidized and non-oxidized species may overlap and the signals for the low abundant (oxidized) species may be suppressed. While reverse-phase separation allows for the separation of individual molecular species, multiple lipid classes may overlap. In the case of CL and its oxidized products, additional difficulties arise due to their inherent structural characteristics: the presence of two phosphate groups in the head group and four fatty acyl moieties that give rise to multiple structural combinations. Therefore, only the most abundant oxidized species are visible when samples of CL are run under normal phase conditions.
The combination of both normal phase and reverse phase chromatography for the separation of individual lipid species for each group was originally described by Pulfer [21] and this approach has been utilized by several groups [22,23] although the literature in the area of chromatographic separation and analysis of lipids and their oxidation products is extensive [24-31]. Minkler and collaborators [32] recently developed a solvent system that resolved individual CL species on a reverse-phase column. This system was also shown to be applicable not only for the separation of intact forms of CL but also their oxidized species [5].
In the present work, we show the ability to separate not only oxidized species of CL but other classes of oxidized phospholipids first by separating phospholipids in a normal phase system in the first dimension (1D) followed by a reverse-phase ion-pair based system for separation in the second dimension (2D). We tested the power of this system to separate oxidized and non-oxidized species of lipids in three different tissues; in human lymphocytes treated with rotenone, in murine lung tissue after inhalation of single walled carbon nanotubes (SWCNTs) and in gut (small intestine) tissue after total body irradiation. In addition, we applied the 1D/2D LC-MS analysis on samples from rat brain, to further test the system with a tissue containing extensive and diverse species of CL. The use of this chromatographic protocol allowed us to characterize individual species for each phospholipid and when present, their oxidized species, with a special emphasis on CL, in lipid extracts from cells and tissues subjected to various insults. While the principle of non-random peroxidation, specifically with regard to the preferential oxidation of CL and PS, has been propagated in our previous work, the current work allows for better resolution of the oxidized products and confirms that CL is one of the major substrates for the oxidative process induced by a variety of cellular stresses, particularly those leading to apoptotic cell death.
2.1 Materials and Methods
2.1.1 Reagents
Chloroform (HPLC grade), bovine heart cardiolipin (BHC) and cyt c were purchased from Sigma-Aldrich (St. Louis, MO, USA). Methanol, triethylamine, acetic acid and water were all LC-MS grade from Fisher, Scientific (Pittsburgh, PA, USA). Diethylenetriaminepentacetic acid (DTPA), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), deoxycholate and soybean lipoxygenase (LOX) were also purchased from Fisher Scientific. Phospholipid standards including tetralinoleoyl cardiolipin (TLCL) were purchased from Avanti Polar Lipids (Alabaster, AL, USA).
2.1.2 Oxidation of CL and other phospholipids by cyt c and lipoxygenase as a model system
Cyt c was used to oxidize TLCL in the following buffer: HEPES (20mM) pH 7.4, (DTPA (100μM), TLCL (50μM) pH 7.4. Oxidation was started after addition of cyt c (5μM) and H2O2 (100μM) and the reaction mixture was incubated for 1 hour at room temperature. LOX was used for a more robust and efficient oxidation of CL in HEPES buffer (100mM) pH 8.0, containing DTPA (0.5mM), deoxycholate (0.2%), and BHC (50μM). A similar concentration of other lipids (phosphatidylcholine (PC), phosphatidylethanolamine (PE) and phosphatidylserine (PS)) was utilized for their oxidations. LOX (15U/ml) was added to the reaction system and incubated for 1 hour at room temperature. Samples were vortexed every 15 min to allow oxygenation of the solution to occur.
2.1.3 Lipid extraction
Lipid extraction was performed according to the Folch procedure described previously [33]. Briefly, choloroform:methanol (2:1) was added to the sample that was resuspended in KCl (0.75%) in a ratio 5/1. The sample was kept on ice for 1 hour with vortexing every 15 min. The organic layer was removed and filtered with Whatman number 4 filter paper (Whatman, Inc., Piscataway, NJ) and dried over nitrogen. Samples were resuspended in the appropriate 1D or 2D solvent system.
2.1.4 1D separation of phospholipid species and LC-MS/MS analysis
Separation of individual phospholipid classes was accomplished by normal phase chromatography using a silica column (Luna, 3micron, 15 cm × 2mm i.d. or a Luna 5 micron, 25cm × 2 mm i.d., Phenomenex, Inc., Torrance, CA). Phospholipids were eluted from the column using the following solvent system: Solvent A: chloroform:methanol:triethylamine:acetic acid, (80:19:0.5:0.5); Solvent B: chloroform:methanol:water:triethylamine:acetic acid, (60:33.5:5.5:1:0.065). Flow was maintained at 0.2 ml/min and a linear gradient of 0-100% B in 30 min was run (15 cm column). Alternatively, the following gradient system was utilized: 0-6 min 100% A; 6-40 min 100% B; 40-50 min 100% B; 50-55min 100% A; 55-70 min 100% A (25 cm column, 0.3ml/min). Separation was performed at room temperature.
Fifty to eighty percent of the flow was diverted to a fraction collector while the remainder was utilized for MS analysis. Chromatographic systems included either a Hewlett Packard 1050 liquid chromatograph (Hewlett Packard, Inc., Palo Alto, CA) or a Shimadzu Prominence HPLC system (Shimadzu, Inc., Kyoto, Japan). Spectra were analyzed either on a T7000 triple quadrupole mass spectrometer (ThermoElectron, Inc. San Jose, CA ) or on a Waters Q-TOF mass spectrometer (Waters, Inc., Milford, MA). Parameters for the T7000 were as follows: spray voltage, 5.0 kV, negative mode; capillary, 250°C; sheath gas, 20 psi. Spectra were collected in a data dependent mode. Tuning and collision energies were optimized for individual phospholipid classes. Parameters for the Q-TOF were as follows: capillary voltage, 2.85 kV, negative mode; source temperature, 100°C; desolvation gas, 400 L/h; sampling cone, 60V; extraction cone: 4.5V; ion guide, 3.0V. Tuning was optimized for all lipids across the scan range.
2.1.5 2D separation of phospholipid species and LC-MS/MS analysis
Fractions corresponding to individual phospholipid classes were collected from the 1D separation, combined and dried over nitrogen. The lipids were dissolved in the appropriate 2D solvent (see below) and were subsequently analyzed in a second dimension system. For CL, either a C8 column (5 micron, 4.6mm × 15 cm, Phenomenex, Inc.) or a C18 column (5 micron, 4.6mm × 15 cm, Phenomenex, Inc.) was used with either an isocratic solvent system consisting of 2-propanol: water: triethylamine: acetic acid (45:5:0.25:0.25) or with a gradient solvent system consisting of acetonitrile: water: triethylamine: acetic acid (45:5:0.25:0.25, solvent A) and 2-propanol: water: triethylamine: acetic acid (45:5:0.25:0.25, solvent B). The gradient program was as follows: 0-5 min 50% B; 5-10 min 65% B; 10-25 min 75% B; 25-45 min 95% B; 45-50 min 95% B; 50-55 min 50% B and 55-60 min 50% B for re-equilbration. Flow was maintained at 0.4 ml/min and analysis was performed on a Waters Premier Q-TOF mass spectrometer. The remaining phospholipids were analyzed on an ion trap mass spectrometer (LCQ-Duo, ThermoElectron, Inc.) using a C18 column (Luna, 5 micron, 4.6mm × 15 cm, Phenomenex, Inc.) with a gradient that was modified from a previously described solvent system [5] and was as follows: 0-5 min 50% B, 5-15 min 80% B, 15-30 min 100% B, 30-45 min 100% B, 45-50 min 50% B, 50-55 min 50% B for re-equilibration of the column using a Spectra 6000 solvent delivery system (Thermo-Electron, Inc.). Flow was maintained at 0.4 ml/min. In some experiments, data was collected in a data dependent mode in which both MS and MS/MS acquisitions were performed sequentially on the most abundant mass ions. Instrument conditions for the Waters Premier Q-TOF mass spectrometer were as follows: capillary voltage, 2.85 kV, negative mode; source temperature, 100°C; desolvation gas, 400 L/h; sampling cone, 60V; extraction cone: 4.5V; ion guide, 3.0V. Tuning was optimized for all lipids across the scan range. Instrument conditions for the ion trap analysis were as follows: capillary temperature, 250°C; spray voltage, 4.5 kV; sheath gas, 30 psi. All other tuning parameters were optimized for each individual lipid class. All of the above separations were performed at room temperature. Alternatively, using a smaller dimension C18 column (1.0 mm × 15 cm, 3 micron Luna 2 reverse phase, Phenomenex, Inc.), a modified solvent system was used which consisted of solvent A: acetonitrile: water: triethylamine:acetic acid (45:5:0.25:0.25) containing 0.01% formic acid and 5mM ammonium acetate and solvent B: 2-propanol:water:triethylamine:acetic acid (45:5:0.25:0.25) containing 0.01% formic acid and 5mM ammonium acetate. Column temperature was 30°C and flow was maintained at 55 μl/min on a Dionex Ulimate 3000 HPLC system (Thermo, Inc.). Spectra were acquired on a Themro LXQ ion trap mass spectrometer (Thermo, Inc.) using a spray voltage of 5.0 kV and a capillary temperature of 150°C. Scans were collected in a data dependent mode with an isolation width of 1.5 Da and a normalized collision energy of 30 V. The HPLC gradient was as follows: 0-5 min 50% B; 5-10 min 75% B; 10-25 min 85% B; 25-40 min 95% B; 40-50 min 95% B; 50-60 min 50% B and 60-70 min 50% B.
2.1.6 Treatment of lymphocytes with rotenone
Human peripheral blood lymphocytes were isolated from whole blood with the use of Histopaque (Sigma-Aldrich, St. Louis, MO, USA) according to the manufacturer's instructions. Monocytes were removed by their characteristic adherence to plastic (1h, 37°C). The non-adherent cells (human lymphocytes) were washed with PBS and incubated with rotenone (250 μM) for 18h at 37°C. Cells were harvested and lipids were extracted according to the protocol described above.
2.1.7 Particles and exposure of mice to SWCNTs
SWCNTs were purchased from Carbon Nanotechnology (CNI, Houston, TX). The nanotubes were manufactured using the high-pressure CO disproportionation process (HiPco) and were used in the inhalation studies. The supplied SWCNTs contained nanometer-scale Fe catalyst particles inherent in the HiPco process. Analysis indicates that these SWCNTs contained elemental carbon (82% wt), Fe (17.7%). Trace elements present included Cu (0.16%), Cr (0.049%), and Ni (0.046%). C57BL/6 mice were exposed to SWCNTs via inhalation (5mg/m3, whole body inhalation for 4 consecutive days, 5 h/day) [34]. All procedures were pre-approved and performed according to the protocols established by the NIOSH Institutional Animal Care and Use Committee and the Institutional Animal Care and Use Committee of the University of Pittsburgh.
2.1.8 Isolation of crude mitochondrial/synaptosomal (P2) fraction from rat brain tissue
Rats were perfused transcardially with ice-cold saline and then decapitated. Brains (minus cerebellum) were rapidly removed and ipsilateral pericontusional cortex was isolated and placed in ten volumes of ice-cold 0.32M sucrose in 1mM Tris buffer (pH 7.4). The tissue was homogenized in a Teflon/glass homogenizer by ten gentle strokes. The homogenate was spun at 1,000g for 10 minutes to remove nuclei and cell debris. The resulting supernatant was centrifuged at 10,000g for 20 minutes to obtain the crude mitochondrial pellet. The pellet was washed and centrifuged (4 minutes, 10,000g at 4°C).
2.1.9 Total body irradiation
Groups of C57BL/6NHsd female mice were irradiated to a dose of 9.5 Gy of total body irradiation using a Shepherd Mark 1 Model 68 cesium irradiator at a dose rate of 80 cGy/min as previously described [35]. The mice were sacrificed at 5 days post irradiation by CO2 inhalation. A 5-cm piece of small intestine was removed, cut open, washed in PBS to remove fecal material, blotted dry and frozen in liquid nitrogen. All procedures were pre-approved and performed according to protocols established by the Institutional Animal Care and Use Committee of the University of Pittsburgh.
3.1 Results
3.1.1 First dimension separation of phospholipids
A total lipid extract from murine brain was separated by LC-MS using a normal phase column in the first dimension. Excellent separation of phospholipid classes resulted as shown in Figure 1. Ceramides and glycolipids eluted within the first four minutes of the gradient. This was followed by phosphatidylglycerol (PG), CL, phosphatidylinositol (PI), PE, PS, PC and sphingomyelin (SM). In this particular profile, a 1:1 split of the column effluent was performed, diverting half of the flow to a fraction collector and the remaining half to the mass spectrometer. Fractions containing individual phospholipid classes were pooled, dried over nitrogen and dissolved in appropriate running solvent for chromatography in the second dimension.
Figure 1. First-dimension (1D) separation of rat brain phospholipids by normal-phase HPLC-MS/MS.
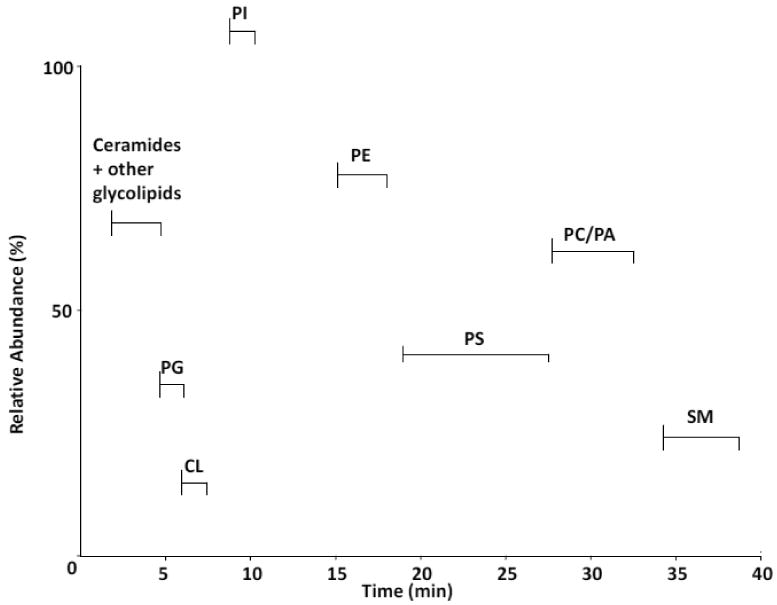
Lipids were extracted from rat brain tissue (non-mitochondrial enriched) and were subjected to 1D LC-MS analysis on a normal phase column. Individual lipids classes were identified based on retention times and MS/MS analyses. Major phospholipid and glycolipid fractions eluted in the following order; ceramide/glycolipids, PG, CL, PI, PE, PS, PC/PA, SPH as indicated. A base peak intensity plot is shown.
3.1.2 Second dimension separation of phospholipids
Individual classes of phospholipids from the first dimension chromatography were re-chromatographed on a reversed-phase C18 column for the second dimension analysis. Figure 2A displays the power of the second dimension system where PI, PG, SM and CL, which eluted as one peak in the first dimension, were resolved into multiple peaks in the second dimension. The dominant peak for PI (m/z 885, Fig. 2B) eluting at 19.1 min had a fatty acyl chain composition of 18:0/20:4 upon MS/MS analysis. The dominant peak for PG eluting at 20.6 min was identified as a 16:0/18:1 PG (m/z 747, Fig. 2B). The dominant SM species in brain (m/z 789, Fig. 2B) was identified as a d18:0/18:1 SM which eluted as an acetate adduct. In contrast, while some degree of species separation was obtained with brain CL (Figure 2A), multiple overlapping species still existed within each of the baseline resolved peaks. A summed spectrum over all of these peaks (Fig. 2B) still displays extreme complexity of this phospholipid in brain tissue.
Figure 2. Second-dimension (2D) separation of rat brain phospholipids by reverse-phase HPLC-MS/MS.
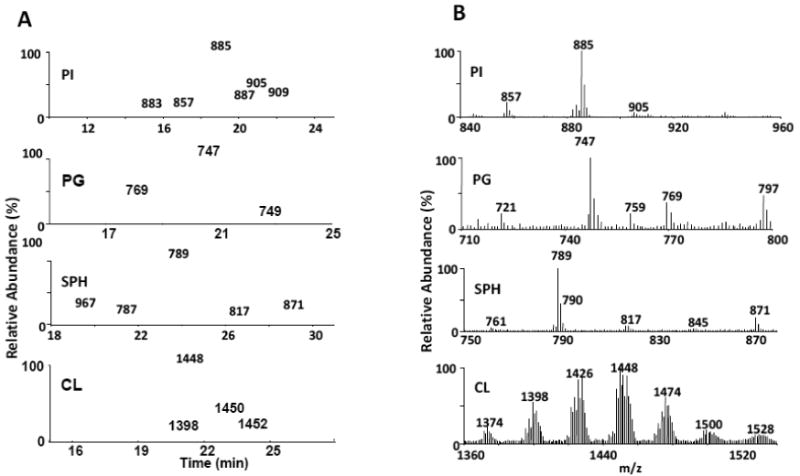
Individual fractions corresponding to PI, PG, SPH and CL were from the 1D analysis were pooled based on phospholipid class and re-chromatographed in the 2D C18 reverse-phase system. Lipids were identified based on retention time and MS/MS analysis. The following rat brain phospholipid classes which eluted as essentially single peaks in the 1D system, were resolved into the following number of peaks in the second dimension: (PI, 6), (PG, 3), (SPH, 5) and (CL, 4), 2A. Chromatograms for PI, PG and SPH. The chromatogram for CL was collected in full MS mode. Summed spectra across all peaks for each phospholipid class are shown in 2B.
3.1.3 Oxidized phospholipids elute at earlier retention times in the second dimension system
One of the goals of this work was to develop a system that would separate oxidized phospholipid species from non-oxidized phospholipid species. During a 1D elution of lipids on a normal phase column, oxidized species can co-elute with non-oxidized species. Since oxidized species usually exist in lower abundance, these biologically important lipids may go undetected. While this partly depends upon the column chemistry chosen and the solvent system utilized, our experience indicates that this is generally the case. In order to determine whether or not the 1D/2D system would meet these requirements, we tested the system by pooling individual phospholipid species of PC, PE and PS from the one dimensional run and oxidizing them in the presence of LOX as indicated in the Materials and Methods section. As shown in Figure 3A, oxidized species of PC, PE and PS eluted earlier (within the first 20 min retention time window) than their non-oxidized counterparts eluting after 20 min. These results are in agreement with an increased hydrophilic character of the oxidized species which should decrease their retention time in a reverse-phase system. Figure 3B indicates the major non-oxidized masses of brain PC, PE and PS. Major species of PC included 16:0/16:0 PC, acetate adduct (m/z 792) and 16:0/18:1 PC, acetate adduct (m/z 818). Additional major species of PE and PS are also noted. Some of the major oxidized species of PE, PC and PS (Figure 3C) that were summed over the first 20 min retention time window and eluted earlier than their non-oxidized species included m/z 822 (PE 18:0/22:6 +OO), m/z 900 (PC 20:5 +O/22:6 +OO) and m/z 866 (PS 18:0/22:6 +OO), respectively. Oxidations were confirmed by MS/MS analysis. One example is shown in Figure 3D where MS/MS analysis of PS (m/z 866) indicated a fatty acyl composition of 18:0/22:6 +OO, indicating a +32 Da increase in the 22:6 fatty acyl chain (m/z 359) which normally exhibits a mass at 327 Da. A fragmentation scheme is shown for the main fragments indicating the loss of stearic acid (m/z 283), the loss of the serine from the head group (m/z 779) as well as the loss of the oxidized fatty acid chain (m/z 359). The fragment at m/z 418 is a result of the loss of both the serine residue from the head group and the oxidized fatty acid chain. The species at m/z 761 indicates a loss of water.
Figure 3. Second dimension (2D) analysis of brain PC, PE and PS oxidized in the presence of lipoxygenase.
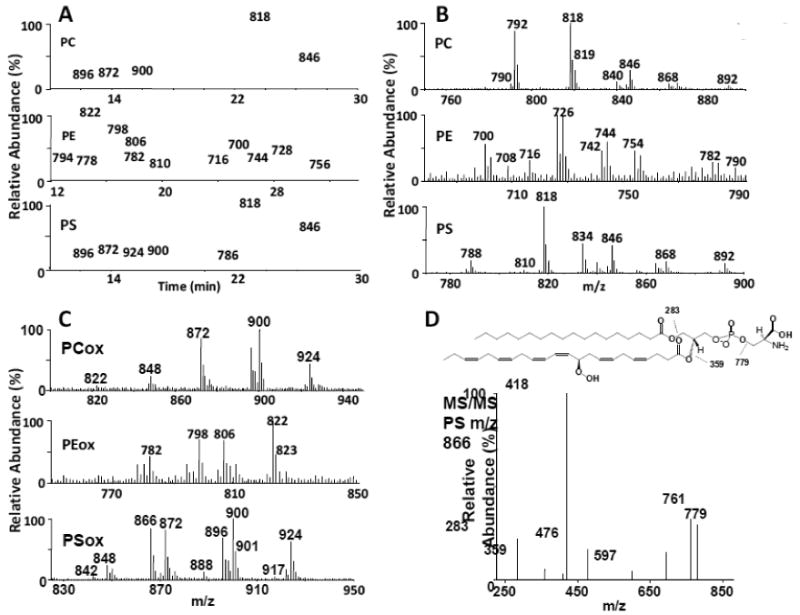
A total rat brain lipid extract was chromatographed in the 1st dimension. Fractions corresponding to individual phospholipid classes were pooled, oxidized with lipoxygenase and chromatographed in the second dimension system using a C18 colum. Lipids were identified based on retention time and MS/MS analysis. In all cases, oxidized species eluted at earlier retention times as compared to their non-oxidized counterparts. 3A: Chromatograms for PC, PE and PS. 3B: Spectra from the individual 2D chromatograms for non-oxidized PC, PE and PS were summed over the retention times associated with the non-oxidized species and are as follows; PC (22-28 min), PE (22-33 min) and PS (20-32 min). 3C: Spectra from the individual 2D chromatograms for oxidized PC, PE and PS were summed over the retention times associated with the oxidized products and are as follows: PC (10-18 min), PE (6-20 min) and PS (6-18 min). 3D: MS/MS analysis was performed on the oxidized PS species (m/z 866). Fatty acyl chain analysis is shown with an oxidation of a 22:6 fatty acid chain (m/z 359) indicating the addition of two oxygens. A fragmentation scheme is shown for the main fragments indicating the loss of stearic acid (m/z 283), the loss of the serine residue (m/z 779) as well as the loss of the oxidized fatty acid chain (m/z 359). The fragment at m/z 418 is a result of the loss of both the serine residue from the head group and the oxidized fatty acid chain.
3.1.4 Retention time of oxidized species of bovine heart cardiolipin (BHC) and TLCL in a 1D/2D separation system is dependent on the degree of oxidation
To further test the 1D/2D separation system, we oxidized TLCL as indicated in the Materials and Methods section. As in the case with other phospholipid species tested, oxidized species of TLCL exhibited earlier retention times (Figure 4A), this being dependent on the degree of oxidation. Commercially available TLCL contains tetralinoleoyl CL (m/z 1448) as its dominant CL species, with a minor sodiated species at m/z 1470 (Figure 4B). Oxidation can be induced in the presence of cyt c and H2O2, as well as in the presence of LOX (Figure 4B, C). The main oxidized products when cyt c was used consisted of hydroperoxy (m/z 1480) TLCL. A small amount of monohydroxy TLCL (m/z 1464, Fig. 4B) existed as well as two minor species consisting of hydroxy/hydroperoxy (m/z 1496) and di-hydroperoxy (m/z 1512) TLCL species (Figure 4B). All of the oxidized species eluted between 3 and 11 min, well separated from the non-oxidized TLCL species eluting at 16 min on a C18 reversed-phase column using an isocratic solvent system of 100% solvent B. On the other hand, oxidation with LOX, a more robust oxidizing system, produced a shift in the chromatogram (Figure 4C) to a more highly oxidized, earlier eluting species with a m/z of 1576 (Figure 4B) which consisted of TLCL with a combination of hydroperoxy and hydroxy modifications (+ 128 Da increase).
Figure 4. Treatment of TLCL and BHC with cyt c/H2O2 and LOX induces products with varying degrees of oxidation.
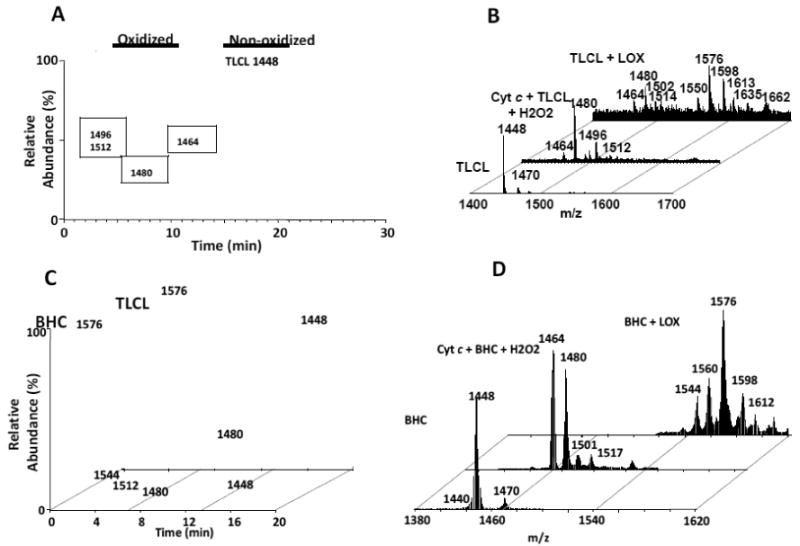
4A: TLCL was oxidized with cyt c/H2O2 as described in Materials and Methods and chromatographed on the 2D system on a C18 column (isocratic, 100% sovent B). Increased oxidation correlated with decreased retention time. 4B: TLCL was oxidized either with cyt c/H2O2 or with LOX as described in Materials and Methods. Oxidation with cyt c was biased toward di-hydroxy and/or monoperoxy (m/z 1480) species. Monohydroxy (m/z 1464), monoperoxy/hydroxy (m/z 1496) and dihydroperoxy (m/z 1512) species represented minor modifications. Oxidation with LOX produced numerous species of highly oxidized TLCL, one of which contained a combination of peroxy/hydroxy modifications (m/z 1576). 4C: 2D chromatograms of BHC and TLCL treated with LOX. Oxidized products of BHC and TLCL eluted at earlier retention times (3- 11 min) than their non-oxidized counterparts eluting at 16-19 min. 4D: Mass spectra of BHC and its oxidation products after treatment with cyt c/H2O2 or LOX. Major oxidized products resulting from treatment with cyt c/H2O2 (m/z 1464 and 1480) or LOX (m/z 1576) are noted.
Similar results were obtained when BHC was used as a substrate. Second dimension separation of oxidized from non-oxidized CL species indicated oxidized products eluting earlier (Figure 4C), which consisted of mono-hydroxy and mono-hydroperoxy BHC (m/z 1464 and 1480, respectively, Fig. 4D) as the major species when cyt c was used (Figure 4D). Oxidation with LOX produced a chromatogram consisting of highly oxidized species (Figure 4C), with the major peak eluting at 3.5 min. This peak contained a main species at m/z 1576 (Figure 4D) which consisted of BHC with a mixture of peroxy and hydroxy modifications. These results indicated that the 1D/2D system is capable of separating oxidized species of CL from non-oxidized species. In addition, these results also indicated that the 1D/2D system could then make the detection of low abundant oxidized CL species possible.
3.1.5 Treatment of lymphocytes with rotenone induces oxidation of cardiolipin that is detectable after 1D/2D LC-MS analysis
We next tested the 1D/2D system for its ability to detect oxidized species of CL after treatment of lymphocytes with the pesticide, rotenone. Rotenone inhibits electron transport chain function by interfering with the transfer of electrons from the iron-sulfur centers in complex I to ubiquinone producing an increase in reactive oxygen species (ROS) production. This mimics the mitochondrial death pathway via mitochondrial dysfunction seen in many disease states. For these studies, human lymphocytes were treated with 250 μM of rotenone for 18h. Lipids were extracted and separated in a 1D system as described in Materials and Methods. A total of 91 lipid species were identified between the first and second dimension analyses. The CL fractions from the first dimension were isolated, pooled and re-chromatographed in the second dimension utilizing, in this case, a C8 column using a isocratic solvent system of 100% solvent B. We hypothesized that the oxidized species would be in low abundance in this biological system. We reasoned that utilization of a C8 reverse-phase column would allow better concentration of oxidized species thereby producing a higher signal to noise ratio. This results in the elution of CL-ox species in the 4-6 min retention time window and the CL species in the 6-8 min retention time window. The CL species from lymphocytes eluted at 6.4 min in the second dimension (Figure 5A). Our results indicated that human lymphocytes contained TLCL (m/z 1448) as their major CL species (Figure 5B), although 11 species were identified. Treatment with rotenone induced oxidation of CL, with two major species appearing at m/z 1464 (mono-hydroxy) and m/z 1480 (mono-hydroperoxy/or di-hydroxy, Fig. 5C). These species, although in low abundance, eluted between 4-5.5 min in the 2D system. Minor species indicating higher degrees of oxidation were also detected. As expected, these species were undetectable in the first dimension (not shown), due to the ion suppression produced by the more highly abundant non-oxidized CL species. These results indicated that the 1D/2D system was capable of detecting oxidized CL species from a biological model system. Interestingly, the effect of rotenone on lipid oxidation appeared to be specific for CL as no other oxidized lipids were detected in the 1D/2D system. This most likely reflects the specificity of rotenone as a mitochondrial complex I inhibitor.
Figure 5. Treatment of human lymphocytes with rotenone induces monohydroxy and monoperoxy modifications of mitochondrial CL.
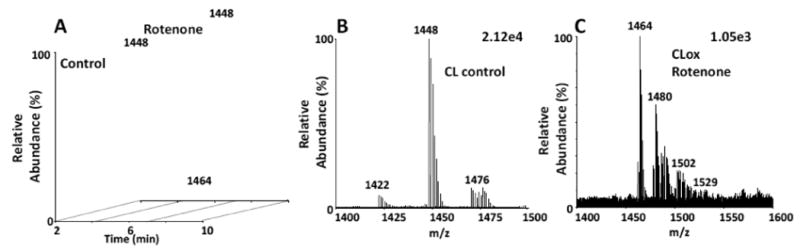
5A: Human lymphocyte contained tetralinoleoyl CL (m/z 1448) as its major CL species which eluted at 6.45 min in the second dimension system (C8 column, 100% sovent B). Lymphocytes treated with 250 μM rotenone (Ro) for 18h also displayed oxidized species eluting between 4-6 min. 5B: Non-oxidized CL spectrum from lymphocytes. 5C: Spectrum of CL-ox from lymphocytes treated with rotenone. Monoperoxy (m/z 1464) and hydroperoxy (m/z 1480) were the dominant oxidized CL species. Elemental compositions for CL masses are as follows: 1421.9443, C79H139O17P2; 1447.9654, C81H141O17P2; 1464.9632, C81H141O18P2; 1479.9638, C81H141O19P2.
3.1.6 Exposure to SWCNTs induces oxidation of cardiolipin in murine lung tissue as detected by the 1D/2D LC/MS system
Mice exposed to SWCNTs via inhalation were sacrificed on day 7. Lung tissue was harvested and extracted for lipids. CL was isolated in the first dimension and analyzed in the second dimension using the protocol described in this study. Using this protocol, we were able to resolve non-oxidized from oxidized species of CL that differed significantly in their retention times. The non-oxidized CL profile from lung is shown in Figure 6A. Species at m/z 1448, 1470 and 1476 have been previously identified as CLs with the following fatty acyl composition: C18:2/C18:2/C18:2/C18:2, C18:2/C18:2/C18:2/C18:2 + sodium and C18:2/C18:1/C18:1/C20:4 , respectively [35]. Both control and SWCNT samples displayed similar chromatographic profiles with non-oxidized CL eluting at 6.5 min and oxidized CL species eluting within the 4-6 min retention time window on the C8 column, again using an isocratic solvent system of 100% solvent B (not shown). Summation of spectra within this oxidized region indicated the presence of characteristic species of oxidized CL at m/z values of 1480, 1502 and 1508 at approximately a 3-fold higher intensity for the SWCNT exposed sample (SWCNT treatment, day 7) as compared to control (control, Figure 6B). Controls displayed only minor amounts of m/z species 1480 and 1502. These oxidized species at m/z 1480, 1502 and 1508 were generated from the non-oxidized CL species at m/z 1448, 1470 and 1476 with +32 Da increases reflecting mono-peroxy or di-hydroxy additions.
Figure 6. Inhalation of SWCNT's induces CL oxidation.
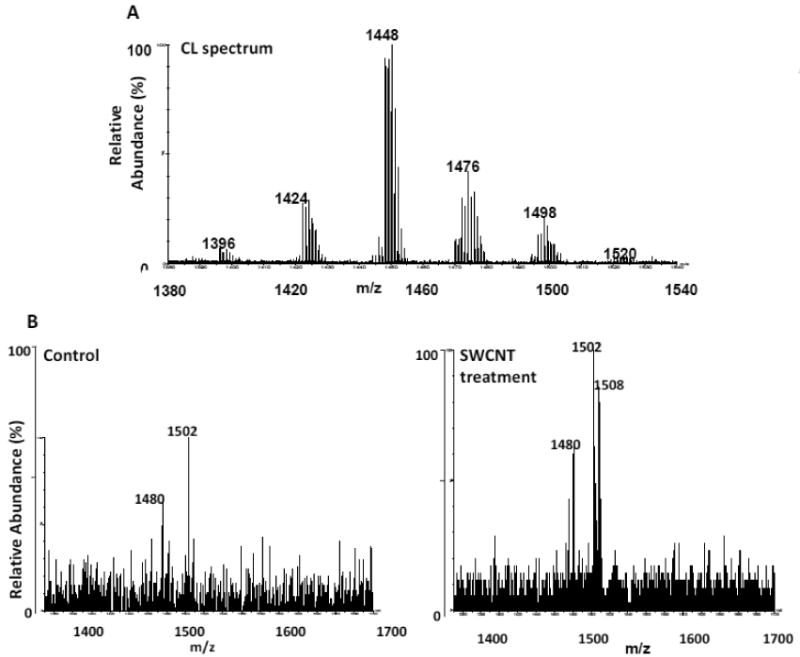
Murine lung tissue was harvested on day 7 after four days of SWCNT exposure (5h/day). Lung tissue was extracted for lipids and analyzed on the 1D/2D LC-MS system (C8 column, 100% solvent B). 6A: non-oxidized CL spectrum.6B: Increases in CL-ox species (m/z 1480, 1502 and 1508, which eluted in the CL-ox retention time window (4-6 min), were noted. Elemental composition for CL masses are as follows: 1395.9331, C77H137O17P2; 1423.9635, C79H141O17P2; 1447.9660, C81H141O17P2; 1475.9931, C83H145O17P2; 1497.9871, C85H143O17P2; 1519.9675, C87H141O17P2.
3.1.7 Whole body irradiation induces oxidation of CL in murine gut tissue as detected by the 1D/2D LC-MS system
Mice receiving 9.5 Gy of whole body irradiation were sacrificed 5 days post-irradiation. Small intestine tissue was harvested as described in Materials and Methods and processed for lipid extraction. CL was isolated in the first dimension (not shown) and analyzed in the second dimension utilizing the modified HPLC solvent system containing formic acid and ammonium acetate on the 1.0 mm C18 column described in Materials and Methods. The method utilizing this particular column and solvent system utilized a flow rate of 55 μl/min, thus adding to the extended retention times for CL and CL-ox products. The small intestine non-oxidized CL for the irradiated sample eluted in a series of peaks in the reverse-phase system over a retention time of 30-40 min and consisted of 5-6 main CL clusters (Figure 7A). Summation of spectra in the CL-ox region (20-30 min retention time) indicated two mass ions at m/z 1464 (25.7 min) and 1480 (23.3 min), which correlated with their base peak chromatograms (Figure 7B). We compared these results from irradiated tissue to a model system where CL was oxidized with cyt c and H2O2 as described in Materials and Methods. Indeed, oxidized CL eluted within the 20-30 min retention time window with corresponding mass ions at m/z 1464 and 1480 (not shown). When we compared fragmentation patterns of mass ions with m/z 1480 and 1464 from our model system to the same mass ions seen in the irradiated small intestine sample, similar fragmentation patterns were seen. The m/z 1480 ion from the irradiated tissue displayed linoleic acid as one of the fatty acyl chains (m/z 279) as well as fragment ions at m/z 695 and 727, all which are seen in the TLCL sample oxidized with cyt c/H2O2 (Fig. 7C,a), indicating that oxidation had occurred as determined by the +32 Da mass increase. A fragmentation scheme is shown above for the main fragments indicating the loss of the oxidized fatty acyl chain (m/z 311), the loss of one oxidized phosphatidic acid (PA) structure (m/z 727) and one non-oxidized PA structure (m/z 695) as well as the loss of linoleic acid (m/z 279). In addition, the daughter ion spectrum of the m/z 1464 mass ion from the irradiated tissue also displayed the same fragments at m/z 695 and 711 indicating a +16 Da mass increase, analogous to the TLCL sample oxidized with cyt c/H2O 2 containing one linoleic (m/z 279) and one hydroxy linoleic fatty acyl chain (m/z 295, Fig. 7C,b). A fragmentation scheme is also shown above for the main fragments indicating the loss of hydroxy-linoleic acid (m/z 295), linoleic acid (m/z 279) as well as one oxidized and one nonoxidized PA structure (m/z 711 and 695, respectively). The fragment at m/z 415 indicates m/z 695 minus linoleic acid. Thus, it was concluded that the CL species at m/z 1464 and 1480 in the irradiated tissue was oxidized as determined from their respective retention times and fragmentation patterns.
Figure 7. Second dimension (2D) analysis of CL from irradiated murine small intestine.
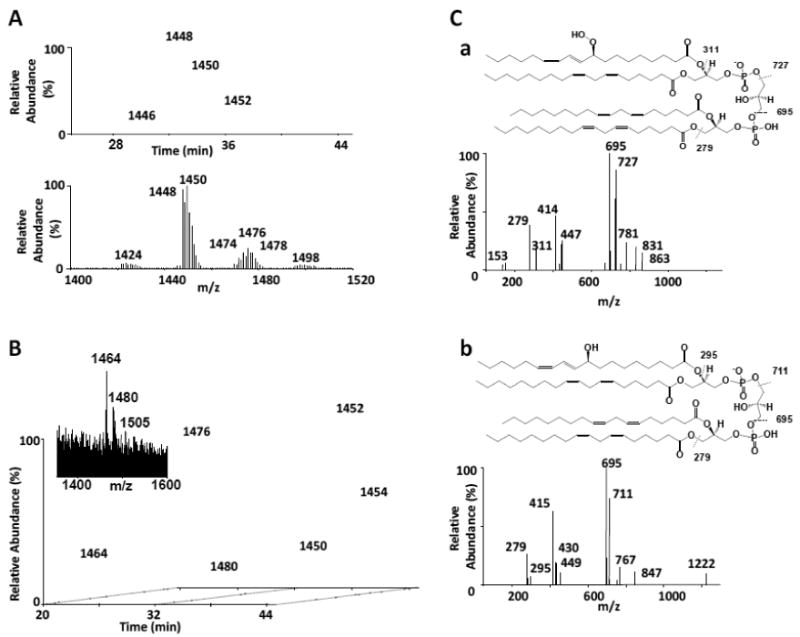
Mice were exposed to whole body irradiation and lipids were extracted from murine small intestine tissue and were subjected to 1D LC-MS analysis on a normal phase column. The CL fraction from the 1D run was re-chromatographed in the 2D on a 1.0mm C18 column (full gradient). 7A: The base peak chromatogram indicated a separation of non-oxidized CL species which eluted within the 30-40 min retention time window. A summed spectrum is also shown for the non-oxidized CL. 7B: 2D analysis of CL from irradiated small intestine. Base peak chromatograms for m/z 1464 and m/z 1480 are shown along with a summed spectrum from the oxidized region (22-27 min retention time window, inset). 7C: LC-MS/MS analysis of m/z 1480 and m/z 1464 species from TLCL oxidized with cyt c/H2O2. (7C,a and 7C,b, respectively). A fragmentation scheme is shown above for the main fragments of m/z 1480 (7C, a) indicating the loss of the oxidized fatty acyl chain (m/z 311), the loss of one oxidized phosphatidic acid (PA) structure (m/z 727), one non-oxidized PA structure (m/z 695) as well as the loss of linoleic acid (m/z 279). A fragmentation scheme is also shown above for the main fragments of m/z 1464 (7C, b) indicating the loss of hydroxy-linoleic acid (m/z 295), linoleic acid (m/z 279) as well as one oxidized and one nonoxidized PA structure (m/z 711 and 695, respectively). The fragment at m/z 415 indicates m/z 695 minus linoleic acid.
3.1.8 Evidence for CL oxidation in normal rat brain tissue
We next determined whether the 1D/2D LC-MS system could be applied to normal brain tissue, where the complexity and diversity of CL species is apparent. Crude mitochondrial fractions were isolated from rat brain tissue and lipids were extracted. CL was isolated in the first dimension and re-chromatographed in the second dimension system. Figure 8 (top panel) displays the 2D LC-MS chromatogram in which the non-oxidized CL eluted within the 6-8 min retention time window using a C8 column with 100% solvent B. The spectrum for the non-oxidized CL species (Fig. 8, bottom right panel) displays the extreme diversity of brain CL with 8 clusters of CL signals within the m/z 1340 to 1540 range. Also seen in the normal brain was a small amount of CL-ox species, which eluted within the 4-6 min retention time window (Fig. 8 bottom left panel). Interestingly, these species fell within a m/z range of 1540 to 1680. While these species were in too low abundance for structural identification, preliminary experiments with an in vitro model system (not shown) indicated that one of the major species (m/z 1552) suggested the presence of two linoleic chains with hydroxy/peroxy modifications (+ 48 Da per chain). It should be noted here that tissue homogenization can, in some cases, be a potential causative agent for oxidation. However, this is usually associated with excessive handling/homogenization of tissue which was not the case in these studies. Here, a glass Dounce homogenizer was used to deliver only ten gentle strokes for tissue disruption. Oxidation induced by homogenization would have undoubtedly resulted in the accumulation of peroxidation products in most of the polyunsaturated lipids, which was not the case in these studies. These results suggest that CL oxidation products can be assessed in tissues such as brain, containing a high degree of CL complexity and diversity. In addition, significantly elevated levels of CL-ox have been observed after traumatic brain injury (Jing, et al, submitted), [36], lending support to the non-random and specific, rather than nonspecific, character of the oxidative process.
Figure 8. Assessment of oxidized CL species in normal rat brain tissue.
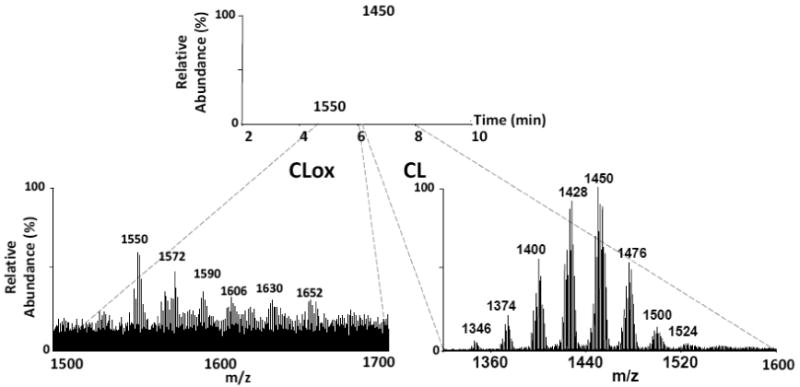
Lipids were extracted from rat brain tissue and fractionated on the 1D LC-MS system. The CL fraction was collected and re-chromatographed in the 2D system (C8 column, 100% solvent B). Fig. 8, top panel: The 2D CL base peak chromatogram indicated a separation CL and CLox species. Fig. 8, bottom right panel: Representative spectrum for non-oxidized brain CL. Fig. 8, bottom left panel: Representative spectrum for oxidized brain CL. Elemental composition for CL masses are as follows: 1345.9111, C73H135O17P2; 1373.9478, C75H139O17P2; 1399.9681, C77H141O17P2; 1427.9907, C79H145O17P2; 1449.9856, C81H143O17P2; 1475.9945, C83H145O17P2; 1449.9953, C85H145O17P2; 1523.9994, C87H145O17P2.
4.1 Discussion
Peroxidation of lipids is believed to occur through two major pathways: 1) a non-enzymatic free radical process and 2) an enzymatic reaction process where radical intermediates are “caged” within the regulated protein environment [37]. The two most prominent features of non-enzymatic peroxidation are 1) its random character with regards to the major classes of phospholipids whereby specificity is driven by the degree of unsaturation of the substrates [38,39], and 2) a plethora of different oxidation products from hydroperoxides to carbonyl compounds and truncated species formed as a result of beta-scission [37]. By contrast, enzymatic peroxidation is specific with regards to both oxidation substrates as well as reaction products. Until recently, a detailed characterization of these most important characteristics of peroxidation, with a notable exception of oxygenated fatty acids, represented an extremely challenging and in many cases, an almost impossible task. With the advent of mild ionization techniques and contemporary MS protocols, these technological issues slowly find their solutions and characterization of lipid peroxidation products in complex biological mixtures becomes a more realistic goal.
Here, we have utilized a normal phase/reverse-phase two dimensional HPLC MS/MS method for the analysis of oxidized lipid species, especially as it applies to CL. We first tested the 2D separation system with CL oxidized in the presence of cyt c/H2O2 as well as with LOX. Oxidized CL species were clearly separated from non-oxidized species in the second dimension due to their increased hydrophilicity. In addition, elution profiles correlated with the degree of oxidation, with the more highly oxidized species eluting first. The 1D/2D system was also tested on PC, PE and PS species isolated in the first dimension from a total brain lipid extract. These isolated phospholipid classes were then treated with LOX to induce oxidation. The products of the reaction were then re-chromatographed in the second dimension. As seen in the case of CL and oxidized CL, all oxidized species of PC, PE and PS were clearly separated from the non-oxidized species and eluted at earlier time points on the reverse-phase system, indicating that this system could be utilized for all phospholipid classes. Oxidation of TLCL or BHC with cyt c/H2O2 produced oxidized species mainly consisting of hydroxy and hydroperoxy species. A more robust oxidation was seen when LOX was used which generated higher degrees of oxidation across all fatty acyl chains as evidenced by the increased mass of the oxidized products.
We further tested the system with four biologically relevant model systems in four different tissue settings. The first model system involved the treatment of human lymphocytes with the pesticide rotenone, an agent known to induce mitochondrial damage via complex I and increased ROS. Our results indicated that oxidized CL species eluted earlier and were clearly separated from non-oxidized CL species. Two major CL oxidations occurred with rotenone treatment and included hydroxylations and peroxidations, mainly associated with the oxidizable linoleic fatty acyl chains. Oxidations in other lipids were not detected most likely due to the direct effect of rotenone on mitochondrial complex I activity, resulting in an increase in ROS, an oxidative event which is biased toward CL. This strongly suggests that in the rotenone/lymphocyte system, oxidation of CL is a non-random event, occurring within two CL species, all containing two or more linoleic fatty acyl chains.
Our second system involved a biologically different but relevant SWCNT inhalation model. Here, CNT inhalation caused an enhancement of two oxidized CL species at m/z 1480, 1502 (sodium adduct) and 1508. These represented +32 Da increases from their respective parent masses indicating di-hydroxy and/or monoperoxy modifications. Here again, oxidation appeared to be specific (biased mostly toward oxidation of linoleic species) strongly suggesting a non-random pattern of oxidation.
Our third system involved the effect of radiation on gut (small intestine) tissue. Here, we identified two oxidized species of CL (m/z 1464 and 1480) five days after radiation exposure. Fragmentation of these species indicated that hydroxy (m/z 1464) and di-hydroxy/monoperoxy (m/z 1480) modifications were present. Linoleic fatty acyl chains were once again identified as the target of oxidation. The presence of oxidized species of CL was also confirmed by their decreased and essentially identical retention times in the 2D reverse-phase system to that of CL oxidized in the presence of cyt c/H2O2.
Finally, we were able to assess oxidized CL within normal rat brain tissue. Oxidized CL species were detected in this tissue despite their low abundance. Unlike lymphocyte, lung and small intestine, oxidized CL species in brain appear at much higher m/z values. While this is most likely due to the extreme diversity in CL species in this tissue, the results still converge on linoleic fatty acyl chains as one of the main targets of oxidative attack. A comparison between oxidized CL species generated in vitro in a model system with cyt c and LOX versus those species found in rotenone-treated lymphocytes, SWCNT inhalation, whole body irradiation and normal brain tissue indicates a preference or bias toward the involvement of enzymatic pathways of oxidation over non-enzymatic pathways. The specificity and substrate preference found in all four biological samples strongly implies that non-random oxidation is the driving force behind these processes. In all of these cases it should be explained that the non-random oxidation process that is referred to here are those species of CL and PS that appear to be specifically susceptible to oxidation despite the fact that other oxidizable phospholipids are plentiful, containing multiple double bonds in their fatty acyl chains. Still, CL and PS with lower levels of polyunsaturated fatty acids present themselves as the main phospholipid species that are oxidized. With respect to CL, the number of oxidation products is directly related by the abundance and most likely the particular species of CLs that interact with cyt c.
Structural analysis indicated that oxidations of CL containing linoleic fatty acyl chains were most common, which included hydroxy and monoperoxy/di-hydroxy modifications. While other polyunsaturated fatty acyl chains exist, which are all susceptible to oxidation, it is interesting to speculate as to why the linoleic fatty acyl chain is favored in CL. One obvious reason would be because of its higher abundance. However, location of the particular CL species as well as the substrate preference of the oxidative enzyme system may also be partially responsible. It is interesting to note that other mitochondrial lipids, such as PG, containing linoleic fatty acyl chains, were not oxidized as determined by this system. This suggests a specific oxidative preference toward CL. However, the potential of rapid remodeling for other mitochondrial lipid species containing oxidized fatty acyl chains may also exist.
In this study, CL oxidation was noted in three different biological systems and in four representative tissues. A plausible explanation for these results may be that oxidized CL functions as both a signaling molecule and as a danger signal for damaged tissue with the degree of oxidation and the particular species oxidized playing a key role in this regard.
While the current study represents an improvement in the analysis and characterization of oxidized lipid species, progress in the area of quantitation of these species is recognized and is still needed. Much of the difficulty in this area stems from the lack of proper oxidized phospholipid standards. The number of different phospholipid oxidation products can be enormous such that the number of oxidized standards that are needed become almost impractical to obtain. Commercial standards for various oxidized phospholipids are extremely scarce and are severely limited in scope. While in vitro oxidation of phospholipid classes can be considered as potential sources of standards, controlling the type and extent of oxidation is difficult and there is no guarantee that these in vitro oxidations will directly mimic those seen in a particular biological system. The authors recognize this shortcoming with regard to quantitation of oxidized phospholipid species and as a result, we are developing alternative biochemical/enzymatic approaches that could potentially circumvent the problem of quantitation of oxidized lipids species. The major intent of this approach is to convert myriads of possible phospholipid peroxidation products into a manageable number of different oxygenated PUFA. In this case, lipid extracts could be treated with a combination of phospholipases to remove oxidized fatty acid chains (lipoprotein phospholipase A2, secreted) and non-oxidized fatty acid chains (secreted/cytosolic phospholipase A2). With the more readily available oxidized fatty acid standards, the analytical/quantitative power of the system can be improved remarkably by employment of this approach with a total lipid extract to quantitate the total amount of oxidation sites. The enzymatic approach could then be applied to individual phospholipid classes after 1D separation for a more selective distinction of oxidation products. Additionally, comparison of the levels of isoprostanes and lipid peroxidation products would allow for the discrimination between random, non-enzymatic pathways and that of specific enzymatic pathways in damaged tissue. This would allow for the development of more efficient therapies against oxidative stress related diseases.
With the application of the 1D/2D HPLC-MS system, a variety of questions can now be formulated to probe the answers to a variety of biologically important questions with regard to oxidative lipidomics.
Highlights.
Oxidized lipids have been separated from non-oxidized lipids using a 2D HPLC-MS system
Oxidized CL species have been detected in four different tissue settings
Oxidation of CL is a non-random process
Similar oxidized CL species appear with different biological insults
Acknowledgments
Supported by NIOSH OH008282; NIH U19 AI068021, HL70755, HL094488, ES020693, ES021068, NS061817. AKSA is a recipient of a research fellowship from La Junta de Extremadura y el Fondo Social Europeo (2010063090).
Abbreviations
- HPLC
high performance liquid chromatography
- LC-MS
liquid chromatography-mass spectrometry
- Q-TOF
quadrupole-time of flight
- ADP
adenosine dinucleotide diphosphate
- NDPK
nucleoside diphosphate kinase
- CCI
control cortical impact
- CL
cardiolipin
- BHC
bovine heart cardiolipin
- TLCL
1,1′,2,2′-tetralinoleoylcardiolipin
- PG
phosphatidylglycerol
- PI
phosphatidylinositol
- PE
phosphatidylethanolamine
- PS
phosphatidylserine
- PC
phosphatidylcholine
- SM
sphingomyelin
- cyt c
cytochrome c
- DTPA
diethylenetriaminepentaacetic acid
- H2O2
hydrogen peroxide
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- PBS
phosphate buffered saline
- TBI
traumatic brain injury
- 1D
1st dimension
- 2D
2nd dimension
- ROS
reactive oxygen species
- SWCNTs
single-walled carbon nanotubes
Footnotes
Disclaimer: The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the National Institute for Occupational Safety and Health.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bochkov VN, Oskolkova OV, Birukov KG, Levonen AL, Binder CJ, Stockl J. Generation and biological activities of oxidized phospholipids. Antioxidants & redox signaling. 2010;12:1009–1059. doi: 10.1089/ars.2009.2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoch FL. Cardiolipins and biomembrane function. Biochimica et biophysica acta. 1992;1113:71–133. doi: 10.1016/0304-4157(92)90035-9. [DOI] [PubMed] [Google Scholar]
- 3.Ardail D, Privat JP, Egret-Charlier M, Levrat C, Lerme F, Louisot P. Mitochondrial contact sites. Lipid composition and dynamics. The Journal of biological chemistry. 1990;265:18797–18802. [PubMed] [Google Scholar]
- 4.Kagan VE, Bayir HA, Belikova NA, Kapralov O, Tyurina YY, Tyurin VA, Jiang J, Stoyanovsky DA, Wipf P, Kochanek PM, Greenberger JS, Pitt B, Shvedova AA, Borisenko G. Cytochrome c/cardiolipin relations in mitochondria: a kiss of death. Free radic2al biology & medicine. 2009;46:1439–1453. doi: 10.1016/j.freeradbiomed.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim J, Minkler PE, Salomon RG, Anderson VE, Hoppel CL. Cardiolipin: characterization of distinct oxidized molecular species. Journal of lipid research. 2011;52:125–135. doi: 10.1194/jlr.M010520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robinson NC. Specificity and binding affinity of phospholipids to the high-affinity cardiolipin sites of beef heart cytochrome c oxidase. Biochemistry. 1982;21:184–188. doi: 10.1021/bi00530a031. [DOI] [PubMed] [Google Scholar]
- 7.Cheneval D, Carafoli E. Identification and primary structure of the cardiolipin-binding domain of mitochondrial creatine kinase. European journal of biochemistry / FEBS. 1988;171:1–9. doi: 10.1111/j.1432-1033.1988.tb13750.x. [DOI] [PubMed] [Google Scholar]
- 8.Zhou M, Morgner N, Barrera NP, Politis A, Isaacson SC, Matak-Vinkovic D, Murata T, Bernal RA, Stock D, Robinson CV. Mass spectrometry of intact V-type ATPases reveals bound lipids and the effects of nucleotide binding. Science. 2011;334:380–385. doi: 10.1126/science.1210148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoffmann B, Stockl A, Schlame M, Beyer K, Klingenberg M. The reconstituted ADP/ATP carrier activity has an absolute requirement for cardiolipin as shown in cysteine mutants. The Journal of biological chemistry. 1994;269:1940–1944. [PubMed] [Google Scholar]
- 10.Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, Vlasova II, Zhao Q, Zou M, Di P, Svistunenko DA, Kurnikov IV, Borisenko GG. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nature chemical biology. 2005;1:223–232. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- 11.Colaco CB, Scadding G, Newsom-Davis J. Anti-cardiolipin antibodies in neurological diseases. Lancet. 1984;1:164. doi: 10.1016/s0140-6736(84)90093-x. [DOI] [PubMed] [Google Scholar]
- 12.Bamberga P, Asero R, Vismara A, Brucato A, Riboldi P, Meroni PL. Anti-cardiolipin antibodies in progressive systemic sclerosis (PSS) Clinical and experimental rheumatology. 1987;5:387–388. [PubMed] [Google Scholar]
- 13.Klemp P, Cooper RC, Strauss FJ, Jordaan ER, Przybojewski JZ, Nel N. Anti-cardiolipin antibodies in ischaemic heart disease. Clinical and experimental immunology. 1988;74:254–257. [PMC free article] [PubMed] [Google Scholar]
- 14.Paggi A, Caccavo D, Ferri GM, Di Prima MA, Amoroso A, Vaccaro F, Bonomo L, Afeltra A. Anti-cardiolipin antibodies in autoimmune thyroid diseases. Clinical endocrinology. 1994;40:329–333. doi: 10.1111/j.1365-2265.1994.tb03927.x. [DOI] [PubMed] [Google Scholar]
- 15.Bernard C, Exquis B, Reber G, de Moerloose P. Determination of anti-cardiolipin and other antibodies in HIV-1-infected patients. Journal of acquired immune deficiency syndromes. 1990;3:536–539. [PubMed] [Google Scholar]
- 16.Dieude M, Striegl H, Tyznik AJ, Wang J, Behar SM, Piccirillo CA, Levine JS, Zajonc DM, Rauch J. Cardiolipin binds to CD1d and stimulates CD1d-restricted gammadelta T cells in the normal murine repertoire. Journal of immunology. 2011;186:4771–4781. doi: 10.4049/jimmunol.1000921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deguchi H, Fernandez JA, Hackeng TM, Banka CL, Griffin JH. Cardiolipin is a normal component of human plasma lipoproteins. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:1743–1748. doi: 10.1073/pnas.97.4.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fernandez JA, Kojima K, Petaja J, Hackeng TM, Griffin JH. Cardiolipin enhances protein C pathway anticoagulant activity. Blood cells, molecules & diseases. 2000;26:115–123. doi: 10.1006/bcmd.2000.0285. [DOI] [PubMed] [Google Scholar]
- 19.Petrosillo G, Casanova G, Matera M, Ruggiero FM, Paradies G. Interaction of peroxidized cardiolipin with rat-heart mitochondrial membranes: induction of permeability transition and cytochrome c release. FEBS letters. 2006;580:6311–6316. doi: 10.1016/j.febslet.2006.10.036. [DOI] [PubMed] [Google Scholar]
- 20.Petit JM, Huet O, Gallet PF, Maftah A, Ratinaud MH, Julien R. Direct analysis and significance of cardiolipin transverse distribution in mitochondrial inner membranes. European journal of biochemistry / FEBS. 1994;220:871–879. doi: 10.1111/j.1432-1033.1994.tb18690.x. [DOI] [PubMed] [Google Scholar]
- 21.Pulfer M, Murphy RC. Electrospray mass spectrometry of phospholipids. Mass spectrometry reviews. 2003;22:332–364. doi: 10.1002/mas.10061. [DOI] [PubMed] [Google Scholar]
- 22.Lesnefsky EJ, Stoll MS, Minkler PE, Hoppel CL. Separation and quantitation of phospholipids and lysophospholipids by high-performance liquid chromatography. Analytical biochemistry. 2000;285:246–254. doi: 10.1006/abio.2000.4783. [DOI] [PubMed] [Google Scholar]
- 23.Houjou T, Yamatani K, Imagawa M, Shimizu T, Taguchi R. A shotgun tandem mass spectrometric analysis of phospholipids with normal-phase and/or reverse-phase liquid chromatography/electrospray ionization mass spectrometry. Rapid communications in mass spectrometry : RCM. 2005;19:654–666. doi: 10.1002/rcm.1836. [DOI] [PubMed] [Google Scholar]
- 24.Bird SS, Marur VR, Sniatynski MJ, Greenberg HK, Kristal BS. Lipidomics profiling by high-resolution LC-MS and high-energy collisional dissociation fragmentation: Focus on characterization of mitochondrial cardiolipins and monolysocardiolipins. Anal Chem. 2011;83:940–949. doi: 10.1021/ac102598u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu W, Porter NA, Schneider C, Brash AR, Yin H. Formation of 4-hydroxynonenal from cardiolipin oxidation: Intramolecular peroxyl radical addition and decomposition. Free Rad Biol Med. 2011;50:166–178. doi: 10.1016/j.freeradbiomed.2010.10.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu C, Dane A, Spijksma G, Wang M, van der Greef J, Hankemeier T, Vreeken RJ. An efficient hydrophilic interaction liquid chromatography separation of 7 phospholipid classes based on a diol column. J Chromatogr A. 2012;1220:26–34. doi: 10.1016/j.chroma.2011.11.034. [DOI] [PubMed] [Google Scholar]
- 27.McLaren DG, Miller PL, Lassman ME, Castro-Perez JM, Hubbard BK, Roddy TP. An ultraperformance liquid chromatography method for the normal-phase separation of lipids. Anal Biochem. 2011;414:266–72. doi: 10.1016/j.ab.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 28.Pang LQ, Liang QL, Wang YM, Ping L, Luo GA. Simultaneous determination and quantification of seven major phospholipid classes in human blood using normal-phase liquid chromatography coupled with electrospray mass spectrometry and the application in diabetes nephropathy. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;869:118–25. doi: 10.1016/j.jchromb.2008.05.027. [DOI] [PubMed] [Google Scholar]
- 29.Brouwers JF. Liquid chromatographic-mass spectrometric analysis of phospholipids. Chromatography, ionization and quantification. Biochim Biophys Acta. 2011;1811:763–75. doi: 10.1016/j.bbalip.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 30.Spickett CM, Wiswedel I, Siems W, Zarkovic K, Zarkovic N. Advances in methods for the determination of biologically relvant lipid peroxidation products. Free Radic Res. 2010;44:1172–1202. doi: 10.3109/10715762.2010.498476. [DOI] [PubMed] [Google Scholar]
- 31.Cummings BS, Peterson BL. A review of chromatographic methods for the assessment of phospholipids in biological samples. Biomed Chromatogr. 2006;20:227–43. doi: 10.1002/bmc.563. [DOI] [PubMed] [Google Scholar]
- 32.Minkler PE, Hoppel CL. Separation and characterization of cardiolipin molecular species by reverse-phase ion pair high-performance liquid chromatography-mass spectrometry. Journal of lipid research. 2010;51:856–865. doi: 10.1194/jlr.D002857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipids from animal tissues. The Journal of biological chemistry. 1957;226:497–509. [PubMed] [Google Scholar]
- 34.Tyurina YY, Kisin ER, Murray A, Tyurin VA, Kapralova VI, Sparvero LJ, Amoscato AA, Samhan-Arias AK, Swidin L, Lahesmaa R, Fadeel B, Shvedova AA, Kagan VE. Global phospholipidomics analysis reveals selective pulmonary peroxidation profiles upon inhalation of single-walled carbon nanotubes. ACS Nano. 2011;5:7342–53. doi: 10.1021/nn202201j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tyurina YY, Tyurin VA, kapralova VI, Wasserloos K, Mosher M, Epperly MW, Greenberger JS, Pitt BR, Kagan VE. Oxidative lipidomics of gamma radiation-induced lung injury: Mass sspectrometric characterization of cardiolipin and phosphatidylserine peroxidation. Radiation Res. 2011;175:610–621. doi: 10.1667/RR2297.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ji J, Kline AE, Amoscato A, Arias AS, Sparvero LJ, Tyurin VA, Tyurina YY, Fink B, Cheng JP, Alexander H, Clark RSB, Kochanke PM, Wipf P, Kagan VE, Bayir H. Global lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of acute brain injury (submitted) doi: 10.1038/nn.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schaich KM. Lipid Oxidation: Theoretical Aspects, Bailey's Industrial Oil and Fat Products. John Wiley & Sons, Inc.; 2005. [Google Scholar]
- 38.Niki E, Yoshida Y, Saito Y, Noguchi N. Lipid peroxidation: mechanisms, inhibition, and biological effects. Biochemical and biophysical research communications. 2005;338:668–676. doi: 10.1016/j.bbrc.2005.08.072. [DOI] [PubMed] [Google Scholar]
- 39.Fruhwirth GO, Hermetter A. Mediation of apoptosis by oxidized phospholipids. Sub-cellular biochemistry. 2008;49:351–367. doi: 10.1007/978-1-4020-8831-5_13. [DOI] [PubMed] [Google Scholar]