

Abstract
Primary microcephaly MCPH1 is an extremely rare autosomal recessive disorder associated with congenital microcephaly, mental retardation and a distinctive cellular phenotype of misregulated chromosome condensation. The MCPH1 gene encodes an 835-amino acid protein, microcephalin, which contains 1 N-terminal and 2 C-terminal BRCT (BRCA1 C-terminus) domains. BRCT domains are predominantly found in proteins involved in cell cycle control and DNA repair. Here we describe 1 novel and 1 previously reported MCPH1 missense mutation, p.Trp75Arg and p.Ser72Leu, respectively, in the N-terminal BRCT domain of microcephalin associated with severe congenital microcephaly. Both residues are entirely conserved in the MCPH1 orthologs of all vertebrate species and Drosophila. Proliferating lymphocytes of the patients with p.Trp75Arg and p.Ser72Leu show the unique cellular MCPH1 phenotype of misregulated chromosome condensation, indicating that these missense alterations disrupt the function of the N-terminal BRCT domain of the protein. Interestingly, both residues are strictly conserved in BRCT domains of BRCA1. ClustalW alignments show that the residue p.Ser72 of microcephalin corresponds to p.Ser1715 of the N-terminal BRCT domain of BRCA1, while the microcephalin residue p.Trp75 is analogous to p.Trp1718 in the N-terminal BRCT and to p.Trp1837 in C-terminal BRCT domains of BRCA1. Missense alterations for all 3 corresponding BRCA1 residues were described and are predicted to be deleterious resulting in the destabilization of the BRCA1 protein. Our data on the 2 MCPH1 missense alterations provide further evidence for the functional significance of these residues in BRCT domains.
Key Words: BRCA1, BRCT domains, MCPH1, Microcephaly, Premature chromosome condensation
Autosomal recessive primary microcephaly (MCPH) displays genetic heterogeneity (MCPH1–MCPH7) with 7 loci mapped to date [Jackson et al., 1998; Jamieson et al., 1999, 2000; Roberts et al., 1999; Moynihan et al., 2000; Pattison et al., 2000; Leal et al., 2003; Kumar et al., 2009]. Clinically, patients with mutations at any of the loci are indistinguishable, presenting with congenital microcephaly and mental retardation in absence of other severe congenital malformations or significant neurological deficits [Woods et al., 2005]. All underlying genes have been identified: MCPH1 encoding microcephalin [Jackson et al., 2002], MCPH2 encoding WDR62 [Nicholas et al., 2010], MCPH3 encoding CDK5RAP2 [Bond et al., 2005], MCPH4 encoding CEP152 [Guernsey et al., 2010], MCPH5 encoding ASPM [Bond et al., 2002], MCPH6 encoding CENPJ [Bond et al., 2005], and MCPH7 encoding STIL [Kumar et al., 2009]. It has been shown that all 7 proteins localize to the centrosome, suggesting that a centrosomal mechanism is responsible for controlling the neuron number in the developing human brain [Bond et al., 2005; Zhong et al., 2005, 2006; Kumar et al., 2009; Guernsey et al., 2010].
The MCPH1 gene encodes an 835 amino acid protein, microcephalin, which contains 1 N-terminal and 2 C-terminal BRCT domains (BRCA1 C-terminus). BRCT domains are predominantly found in many proteins which are involved in cell cycle regulation and DNA repair. The hallmark of mutations in the MCPH1 gene is the unique cellular phenotype with a high proportion of cells with prophase-like chromosomes due to premature chromosome condensation in the early G2 phase of the cell cycle (hence, ‘premature chromosome condensation’ syndrome) and delayed decondensation postmitosis [Neitzel et al., 2002; Trimborn et al., 2004]. Furthermore, patients with MCPH1 deficiency show an extremely poor metaphase resolution of approximately 250–300 bands per haploid genome in routine chromosome preparations even after synchronization techniques.
To date, 12 different homozygous mutations in 15 consanguineous families have been reported in MCPH1. Eight of them are predicted to result in protein truncation: 2 nonsense mutations c.74G>C, Ser25X, in exon 2 [Jackson et al., 2002] and c.302C>G, p.Ser101X, in exon 4 [Farooq et al., 2010]; 2 insertions of 1 base, c.427 428insA in exon 5 and c.566 567insA in exon 6 [Trimborn et al., 2004; Darvish et al., 2010]; 1 splice site mutation c.436 + 1G>T [Darvish et al., 2010]; and 3 gross deletions [Garshasbi et al., 2006; Darvish et al., 2010]. Only 4 missense mutations in MCPH1 have been reported: c.80C>G, p.Thr27Arg [Trimborn et al., 2005], c.147C>G, p.His49>Gln [Darvish et al., 2010], c.149T>G, p.Val50Gly [Leung et al., 2011], and c.215C>T, p.Ser72>Leu [Darvish et al., 2010; this report]. The characterization of missense mutations associated with loss of function is important in delineating protein regions with particular functional relevance.
We report here on 2 missense alterations in microcephalin: 1 alteration that was recently described but not characterized on the cellular level and 1 novel mutation. Both missense mutations affect residues in the N-terminal BRCT domain of microcephalin which are strictly conserved from Drosophila to human. Moreover, both residues are also strictly conserved in the BRCT domains of BRCA1 and other BRCT domain containing proteins. Clinically, both alterations were associated with severe microcephaly and at the cellular level with misregulated chromosome condensation.
Materials and Methods
Cytogenetics
Conventional cytogenetic analyses of cultured T-lymphocytes and lymphoblastoid cell lines (LCLs) were performed using standard techniques. LCLs were established as described elsewhere. At least 1,000 nuclei were counted and the percentage of prophase/prophase-like and metaphase nuclei was calculated among total nuclei.
Mutation Analysis and Bioinformatics
For mutation detection all 14 exons and exon-intron boundaries of the MCPH1 gene were PCR-amplified (primers and conditions are available upon request). The PCR products were cycle-sequenced using the Big Dye Terminator Ready Reaction Mix (Applied Biosystems) and analyzed on ABI 3730 DNA analyzer (Applied Biosystems). Sequences were compared with a reference cDNA sequence (GenBank accession: NM 024596). Primer sequences for the amplification of exon 3 were: MCPH1-Exon3F: TTCTGTCCTCTTTATCCTTGCAG and MCPH1-Exon3R: CCCCCTAGGCAGAGTTAGGTATT.
An alignment of MCPH1 proteins from multiple species (table 1) was prepared using ClustalX. The alignment of 18 human protein BCRT modules was prepared by extracting the corresponding protein sequences from the PFAM seed alignment for the BCRT family. The sequence log was generated using TEXshade [Beitz, 2000]. To predict the potential effect of the amino acid changes we used MutationTaster [Schwarz et al., 2010], SIFT [Ng and Henikoff, 2003] and PolyPhen [Ramensky et al., 2002]. The predictions are based on sequence annotations, alignments and structural information.
Table 1.
Cross species alignment of MCPH1
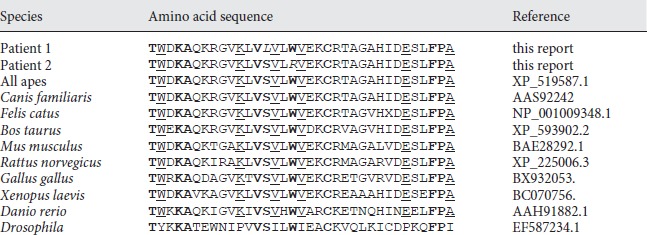
ClustalW alignment of amino acid residues 59-93 of the human microcephalin with the orthologs of different species. The substitutions of the patients at position Ser72 and Trp75 are given in italics. The amino acids of the partial N-terminal BRCT domain, which are conserved in all vertebrate model species listed, are underlined; those conserved in vertebrates and Drosophila are given in bold. The p. Trp75Arg alteration affects the Trp-X-X-X-Cys/Ser motif which is highly conserved in microcephalin and BRCT domains of various other BRCT containing proteins
Results
Clinical Description
Patient 1, a girl, was born after 42 weeks of an uneventful pregnancy to consanguineous Iraqi parents (first cousins). Her birth weight was 3,960 g (1 SD), length 50 cm (−1.5 SD) and head circumference 31 cm (−3.5 SD). The Apgar scores were 7/9/10. Early milestones were normal (sitting at 6 months, walking at 12 months, first words at 18 months). At the age of 5 years and 2 months she presented with a mild developmental delay, in particular affecting her fine motor skills. She has a mild muscular hypotonia. Her height was 98 cm (−2.9 SD), weight 14 kg (−2 SD) and head circumference 42.5 cm (−6.2 SD). MRI scan of the brain at the age of 6 months showed small frontal lobes and mild ventriculomegaly (fig. 1). EEG was normal.
Fig. 1.

Photograph of patient 1 at the age of 5 years and 2 months (A). She shows only mild facial dsymorphism with upslanting palpebral fissures and broad tip of the nose. MRI scan of the brain at the age of 6 months shows small frontal lobes and mild ventriculomegaly, while the sizes of brain stem and cerebellum were normal (B, C).
Patient 2, a boy, was referred for diagnostic evaluation at the age of 6 years and 9 months because of severe microcephaly and developmental delay. He is the second child of consanguineous Turkish parents (first cousins). He was born at 40 weeks of gestation with a birth weight of 2,990 g (−2.2 SD), length of 52 cm (−0.7 SD) and head circumference 30.5 cm (−5.7 SD). The Apgar scores were 6/9/9. Early milestones were in the normal range. At the age of 6 years and 9 months he presented with a height of 117 cm (10th percentile) and a head circumference of 46 cm (−4.8 SD). Both children are remarkably pleasant with good social skills.
Sequencing Data
Sequencing genomic DNA of patient 1 showed a homozygous missense mutation c.215C>T in exon 3 of the MCPH1 gene causing a serine-to-leucine substitution, p.Ser72Leu, which is a non-conservative change leading to an alteration in the polarity of the amino acid. The c.215C>T alteration was absent from 180 alleles of Turkish and 180 alleles of German control subjects.
Sequencing genomic DNA of patient 2 detected a homozygous missense mutation c.223T>C in exon 3 of the MCPH1 gene. The resulting amino acid substitution tryptophan to arginine (p.Trp75Arg) changes a highly lipophilic, non-polar residue to a positively charged, polar residue, located in a critical α3-helix of the N-terminal BRCT domain. The nucleotide alteration was heterozygous in both parents. The c.223T>C alteration was absent from 180 alleles of ethnically matched controls indicating that this is also not a common sequence polymorphism. Both the residues Ser72 and Trp75 are strictly conserved in microcephalin from Drosophila to human (table 1).
The program MutationTaster predicts both variants as disease causing (99.54% each) and SIFT classifies them as variants affecting the protein function. By PolyPhen the effect of p.Ser72Leu and p.Trp75Arg mutations was predicted as possibly (PSIC score 1.998) and probably damaging (PSIC score 3.757), respectively (score >1.7 is considered as damaging).
Cytogenetic Analysis
Chromosome preparations from patient 1 (p.Ser72Leu) showed an enhanced fraction of prophase-like cells (PLCs) in lymphocyte cultures. The percentage of 5.6% PLCs (fig. 2) was clearly less marked than that seen in patients with truncating mutations (8–18% PLCs) but significantly increased from that in normal controls (0.5–1% prophases). In chromosome preparations from lymphoblastoid cells of patient 2 (p.Trp75Arg), we observed an extremely elevated fraction of 8% PLCs comparable to that of patients with truncating mutations. As already demonstrated for patients with MCPH1 truncating mutations, chromosome preparations of both patients revealed the characteristic poor metaphase resolution of not more than 300 bands per haploid genome in routine chromosome preparations.
Fig. 2.

Percentage of PLCs and metaphases in LCLs of the patients with the missense alterations p.Ser72Leu and p.Trp75Arg compared to 1 patient with the truncating mutation Thr143AsnfsX5, 1 patient with the missense alteration p.Thr27Arg and 2 control subjects (A). Chromosome preparation from LCLs of the MCPH1 patient 2 displaying a high number of PLCs and highly condensed metaphase chromosomes (B).
Discussion
The majority of mutations described so far in the MCPH1 gene is predicted to result in protein truncation [Jackson et al., 2002; Trimborn et al., 2004; Garshasbi et al., 2006; Farooq et al., 2010]. The first reported MCPH1 missense mutation c.80C>G, p.Thr27Arg, has only a mild effect on chromosome condensation with 3–4% PLCs in lymphocytes. Clinically, the affected boy had a head circumference of −2.4 SD at birth and a very mild mental retardation with predominantly delayed motor skills but normal verbal IQ attainment [Trimborn et al., 2005]. Leung et al. [2011] reported aberrant chromosome condensation in a 6-month-old patient with primary microcephaly and antenatally detected cystic hygroma. The child of non-consanguineous parents carried the homozygous missense mutations c.149T>G and c.151A>G, resulting in amino acid substitutions p.Val50Gly and p.Ile51Val [Leung et al., 2011]. The 2 missense mutations c.147C>G, p.His49>Gln, and c.215C>T, p.Ser72 >Leu, described by Darvish et al. [2010] were not characterized at the cellular level.
Here, we describe 2 missense mutations in MCPH1 patients leading to a severe cellular phenotype of misregulated chromosome condensation. Chromosome preparations of both patients exhibit an elevated fraction of PLCs and an extremely poor metaphase resolution which is unique to MCPH1 microcephaly and microcephalin gene mutations. Both patients show a severe congenital microcephaly with head circumferences of −3.5 and −5.7 SD present at birth, respectively, in absence of other severe congenital malformations or significant neurological deficits. They present with developmental delay but with a pleasant personality and good social adaptation which is seemingly a characteristic of MCPH1 patients. Both amino acid substitutions, p.Ser72Leu and p.Trp75Arg, are localized in the N-terminal BRCT domain of the MCPH1 encoded protein microcephalin. Multiple sequence alignments of the microcephalin orthologs from diverse species indicate that both amino acids, Ser72 and Trp75, are entirely conserved in the orthologs of all vertebrate species listed and Drosophila (table 1). In addition, by the programs MutationTaster, PolyPhen and SIFT both amino acid substitutions p.Ser72Leu and p.Arg75Trp are predicted to be pathogenic.
BRCT domains, first described in the BRCA1 gene [Bork et al., 1997], are evolutionarily conserved modules of 85–95 amino acids which are found in a large number of cell cycle checkpoint proteins. BRCT repeats exist as either single modules or multiple tandem repeats separated by variable linker regions [Huyton et al., 2000]. The MCPH1 residue Trp75 corresponds to an almost invariable Trp residue of the Trp-X-X-X-Cys/Ser motif, which is localized within the helix-α3 near the C-terminus of various BRCT domains. The Trp-X-X-X-Cys/Ser motif, where the 2 variable residues after Trp are generally small hydrophobics, is highly conserved in the vast majority of BRCT domain containing proteins [Huyton et al., 2000] such as breast cancer 1 BRCA1, topoisomerase II binding protein TOPBP1, DNA ligase LIG4, X-ray repair complementing defective repair in Chinese hamster cells 1 XRCC1, and the human pescadillo homolog PESC (fig. 3). However, it is significantly divergent in PARP4 as well as in bacterial NAD+-dependent DNA ligases. We show here that the Trp-X-X-X-Cys/Ser motif is also strictly conserved in the N-terminal BRCT domain of all investigated microcephalin orthologs from Drosophila to human (table 1). In contrast, the residue Ser72, although strictly conserved in MCPH1/microcephalin, shows less conservation in BRCT domains of other proteins except for the C-terminal BRCT domain of BRCA1 and one of the BRCT domains of TOPBP1.
Fig. 3.

ClustalW alignments of BRCT domains for microcephalin, BRCA1 and various other BRCT domain containing proteins. The nomenclature of the proteins is according to the output file of PFAM (TOPB1 = TOPBP1; DNL4 = LIG4; PAXI1 = PAXIP1).
ClustalW alignments of BRCT domains for MCPH1 and BRCA1 show that the highly conserved MCPH1 Trp75 residue is analogous to Trp1718 in the N-terminal and Trp1837 in the C-terminal BRCT domain of BRCA1. Both Trp1718 and Trp1837 are residues in BRCA1 Trp-X-X-X-Cys/Ser motifs. The MCPH1 residue Ser72 corresponds to Ser1715 of the N-terminal BRCT domain of BRCA1 (fig. 3). For all 3 corresponding BRCA1 residues missense alterations have been described in the Breast Cancer Information Core Database (BIC) where several entries are listed for missense alterations of Ser1715 in BRCA1 (Ser1715Arg, Ser1715Cys, Ser1715Asn), 2 for Trp1718 (Trp1718Ser, Trp1718Cys), and 9 for Trp1837 (Trp1837Arg, Trp1837Gly, Trp1837Cys). BIC reports all these alterations as variants of unknown clinical significance. Contrarily, Abkevich et al. [2004] classified missense alterations of all 3 residues as deleterious on the basis of the chemical difference between the amino acids present at individual residues in combination with their evolutionary conservation in their vertebrate orthologs. Further evidence for their deleterious nature comes from segregation and functional studies as well as from computational methods applying specific algorithms. Furthermore, the Trp1837Arg mutation has been found in a woman with early onset breast cancer whose father also had breast cancer and the mutation was found to segregate with the disease [Montagna et al., 1996]. By using a functional transcription assay, Phelan et al. [2005] have shown that Trp1837Arg markedly interfered with the function of BRCA1 in transcriptional activation. Moreover, the substitution Trp1837Arg is known to be highly destabilizing resulting in aberrant folding of the BRCT repeat [Williams et al., 2003] and altered binding activity of p53 [Quaresima et al., 2006]. The pathogenic character of the BRCA1 Trp1837Arg mutation is further supported by the PolyPhen program, which predicted that the mutation is probably damaging with a PSIC score difference of 3.902. Applying computational prediction methods Karchin et al. [2007] have demonstrated that the residue Ser1715 forms hydrogen bonds in the normal BRCA1 structure with residue Trp1718, the invariable tryptophan in the Trp-X-X-X-Cys/Ser motif. Therefore, alterations of Ser1715 were judged to be destabilizing for the protein [Karchin et al., 2007].
In contrast to missense variants at the BRCA1 gene locus, which need to be biochemically assayed and classified by segregation analyses and sophisticated computational methods, clinically relevant alterations in MCPH1 can directly be assessed by the unique cellular phenotype that has not been described in any other human disorder. The severe cellular phenotype of both MCPH1 patients reported here demonstrates that alterations of the Trp-X-X-X-Cys/Ser (Trp75) motif and of the residue Ser72, which probably interacts with this motif, are deleterious mutations.
With respect to the aberrant chromosome condensation phenotype, it is of special interest that both missense mutations are not localized in the condensin II binding domain of microcephalin. In 2006, we reported that the misregulated chromosome condensation in MCPH1 is mediated by condensin II. In MCPH1 patient cells, siRNA-mediated depletions of condensin II subunits lead to a pronounced reduction of cells with the condensation defects in both G1 and G2 phases of the cell cycle [Trimborn et al., 2006]. Recently, Wood et al. [2008] have demonstrated that microcephalin and condensin II interact in vivo, mediated by the CAP-G2 subunit of condensin II binding to a region (residues 376–485) within the inter-BRCT-domain of microcephalin. By transfection experiments with various deletion mutants of MCPH1 into MCPH1−/– cells, Wood et al. [2008] showed that the region which is important for condensin II binding is not required for the rescue of the chromosome condensation defect. Considering that condensin II participates in an early stage of prophase chromosome condensation within the nucleus [Ono et al., 2004], this result is indeed surprising as especially the early chromosome condensation is severely disturbed in MCPH1 deficient cells. Further data of Wood et al. [2008] substantiate that deletion of the N-terminal BRCT domain alone failed to correct the chromosome condensation defect indicating that the N-terminal BRCT domain is required for rescue of the cellular PLC phenotype. This is in agreement with our results of the 2 missense mutations reported here. Both mutations affect highly conserved residues of the N-terminal BRCT domain of microcephalin leading to a severe chromosome condensation defect comparable to those previously described with premature truncating mutations of MCPH1, probably by aberrant folding of the BRCT domain. However, further studies will be necessary for the elucidation of the exact molecular interactions regulating chromosome condensation via the N-terminal BRCT domain of microcephalin.
Acknowledgements
We are indebted to the families for their participation in this study. We thank Mrs. Sylke Niehage for excellent technical assistance.
References
- 1.Abkevich V, Zharkikh A, Deffenbaugh AM, Frank D, Chen Y, et al. Analysis of missense variation in human BRCA1 in the context of interspecific sequence variation. J Med Genet. (2004);41:492–507. doi: 10.1136/jmg.2003.015867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beitz E. TEXshade: shading and labeling of multiple sequence alignments using LATEX2 epsilon. Bioinformatics. (2000);16:135–139. doi: 10.1093/bioinformatics/16.2.135. [DOI] [PubMed] [Google Scholar]
- 3.Bond J, Roberts E, Mochida GH, Hampshire DJ, Scott S, et al. ASPM is a major determinant of cerebral cortical size. Nat Genet. (2002);32:316–320. doi: 10.1038/ng995. [DOI] [PubMed] [Google Scholar]
- 4.Bond J, Roberts E, Springell K, Lizarraga SB, Scott S, et al. A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat Genet. (2005);37:353–355. doi: 10.1038/ng1539. [DOI] [PubMed] [Google Scholar]
- 5.Bork P, Hofmann K, Bucher P, Neuwald AF, Altschul SF, Koonin EV. A superfamily of conserved domains in DNA damage-responsive cell cycle checkpoint proteins. FASEB J. (1997);11:68–76. [PubMed] [Google Scholar]
- 6.Darvish H, Esmaeeli-Nieh S, Monajemi GB, Mohseni M, Ghasemi-Firouzabadi S, et al. A clinical and molecular genetic study of 112 Iranian families with primary microcephaly. J Med Genet. (2010);47:823–828. doi: 10.1136/jmg.2009.076398. [DOI] [PubMed] [Google Scholar]
- 7.Farooq M, Baig S, Tommerup N, Kjaer KW. Craniosynostosis-microcephaly with chromosomal breakage and other abnormalities is caused by a truncating MCPH1 mutation and is allelic to premature chromosomal condensation syndrome and primary autosomal recessive microcephaly type 1. Am J Med Genet A. (2010);152A:495–497. doi: 10.1002/ajmg.a.33234. [DOI] [PubMed] [Google Scholar]
- 8.Garshasbi M, Motazacker MM, Kahrizi K, Behjati F, Abedini SS, et al. SNP array-based homozygosity mapping reveals MCPH1 deletion in family with autosomal recessive mental retardation and mild microcephaly. Hum Genet. (2006);118:708–715. doi: 10.1007/s00439-005-0104-y. [DOI] [PubMed] [Google Scholar]
- 9.Guernsey DL, Jiang H, Hussin J, Arnold M, Bouyakdan K, et al. Mutations in centrosomal protein CEP152 in primary microcephaly families linked to MCPH4. Am J Hum Genet. (2010);87:40–51. doi: 10.1016/j.ajhg.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huyton T, Bates PA, Zhang X, Sternberg MJ, Freemont PS. The BRCA1 C-terminal domain: structure and function. Mutat Res. (2000);460:319–332. doi: 10.1016/s0921-8777(00)00034-3. [DOI] [PubMed] [Google Scholar]
- 11.Jackson AP, McHale DP, Campbell DA, Jafri H, Rashid Y, et al. Primary autosomal recessive microcephaly (MCPH1) maps to chromosome 8p22–pter. Am J Hum Genet. (1998);63:541–546. doi: 10.1086/301966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jackson AP, Eastwood H, Bell SM, Adu J, Toomes C, et al. Identification of microcephalin, a protein implicated in determining the size of the human brain. Am J Hum Genet. (2002);71:136–142. doi: 10.1086/341283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jamieson CR, Govaerts C, Abramowicz MJ. Primary autosomal recessive microcephaly: Homozygosity mapping of MCPH4 to chromosome 15. Am J Hum Genet. (1999);65:1465–1469. doi: 10.1086/302640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jamieson CR, Fryns JP, Jacobs J, Matthijs G, Abramowicz MJ. Primary autosomal recessive microcephaly: MCPH5 maps to 1q25–q32. Am J Hum Genet. (2000);67:1575–1577. doi: 10.1086/316909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karchin R, Monteiro AN, Tavtigian SV, Carvalho MA, Sali A. Functional impact of missense variants in BRCA1 predicted by supervised learning. PLoS Comput Biol. (2007);3:e26. doi: 10.1371/journal.pcbi.0030026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar A, Girimaji SC, Duvvari MR, Blanton SH. Mutations in STIL, encoding a pericentriolar and centrosomal protein, cause primary microcephaly. Am J Hum Genet. (2009);84:286–290. doi: 10.1016/j.ajhg.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leal GF, Roberts E, Silva EO, Costa SM, Hampshire DJ, Woods CG. A novel locus for autosomal recessive primary microcephaly (MCPH6) maps to 13q12.2. J Med Genet. (2003);40:540–542. doi: 10.1136/jmg.40.7.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leung JW, Leitch A, Wood JL, Shaw-Smith C, Metcalfe K, et al. SET nuclear oncogene associates with microcephalin/MCPH1 and regulates chromosome condensation. J Biol Chem. (2011);286:21393–21400. doi: 10.1074/jbc.M110.208793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Montagna M, Santacatterina M, Corneo B, Menin C, Serova O, et al. Identification of seven new BRCA1 germline mutations in Italian breast and breast/ovarian cancer families. Cancer Res. (1996);56:5466–5469. [PubMed] [Google Scholar]
- 20.Moynihan L, Jackson AP, Roberts E, Karbani G, Lewis I, et al. A third novel locus for primary autosomal recessive microcephaly maps to chromosome 9q34. Am J Hum Genet. (2000);66:724–727. doi: 10.1086/302777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neitzel H, Neumann LM, Schindler D, Wirges A, Tönnies H, et al. Premature chromosome condensation in humans associated with microcephaly and mental retardation: A novel autosomal recessive condition. Am J Hum Genet. (2002);70:1015–1022. doi: 10.1086/339518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. (2003);31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nicholas AK, Khurshid M, Desir J, Carvalho OP, Cox JJ, et al. WDR62 is associated with the spindle pole and is mutated in human microcephaly. Nat Genet. (2010);42:1010–1014. doi: 10.1038/ng.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ono T, Fang Y, Spector DL, Hirano T. Spatial and temporal regulation of condensins I and II in mitotic chromosome assembly in human cells. Mol Biol Cell. (2004);15:3296–3308. doi: 10.1091/mbc.E04-03-0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pattison L, Crow YJ, Deeble VJ, Jackson AP, Jafri H, et al. A fifth locus for primary autosomal recessive microcephaly maps to chromosome 1q31. Am J Hum Genet. (2000);67:1578–1580. doi: 10.1086/316910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Phelan CM, Dapic V, Tice B, Favis R, Kwan E, et al. Classification of BRCA1 missense variants of unknown clinical significance. J Med Genet. (2005);42:138–146. doi: 10.1136/jmg.2004.024711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quaresima B, Faniello MC, Baudi F, Crugliano T, Di Sanzo M, et al. Missense mutations of BRCA1 gene affect the binding with p53 both in vitro and in vivo. Oncol Rep. (2006);16:811–815. [PubMed] [Google Scholar]
- 28.Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. (2002);30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roberts E, Jackson AP, Carradice AC, Deeble VJ, Mannan J, et al. The second locus for autosomal recessive primary microcephaly (MCPH2) maps to chromosome 19q13.1–13.2. Eur J Hum Genet. (1999);7:815–820. doi: 10.1038/sj.ejhg.5200385. [DOI] [PubMed] [Google Scholar]
- 30.Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. (2010);7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 31.Trimborn M, Bell SM, Felix C, Rashid Y, Jafri H, et al. Mutations in microcephalin cause aberrant regulation of chromosome condensation. Am J Hum Genet. (2004);75:261–266. doi: 10.1086/422855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trimborn M, Richter R, Sternberg N, Gavvovidis I, Schindler D, et al. The first missense alteration in the MCPH1 gene causes autosomal recessive microcephaly with an extremely mild cellular and clinical phenotype. Hum Mutat. (2005);26:496. doi: 10.1002/humu.9382. [DOI] [PubMed] [Google Scholar]
- 33.Trimborn M, Schindler D, Neitzel H, Hirano T. Misregulated chromosome condensation in MCPH1 primary microcephaly is mediated by condensin II. Cell Cycle. (2006);5:322–326. doi: 10.4161/cc.5.3.2412. [DOI] [PubMed] [Google Scholar]
- 34.Williams RS, Chasman DI, Hau DD, Hui B, Lau AY, Glover JN. Detection of protein folding defects caused by BRCA1-BRCT truncation and missense mutations. J Biol Chem. (2003);278:53007–53016. doi: 10.1074/jbc.M310182200. [DOI] [PubMed] [Google Scholar]
- 35.Wood JL, Liang Y, Li K, Chen J. Microcephalin/MCPH1 associates with the condensin II complex to function in homologous recombination repair. J Biol Chem. (2008);283:29586–29592. doi: 10.1074/jbc.M804080200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Woods CG, Bond J, Enard W. Autosomal recessive primary microcephaly (MCPH): A review of clinical, molecular, and evolutionary findings. Am J Hum Genet. (2005);76:717–728. doi: 10.1086/429930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhong X, Liu L, Zhao A, Pfeifer GP, Xu X. The abnormal spindle-like, microcephaly-associated (ASPM) gene encodes a centrosomal protein. Cell Cycle. (2005);4:1227–1229. doi: 10.4161/cc.4.9.2029. [DOI] [PubMed] [Google Scholar]
- 38.Zhong X, Pfeifer GP, Xu X. Microcephalin encodes a centrosomal protein. Cell Cycle. (2006);5:457–458. doi: 10.4161/cc.5.4.2481. [DOI] [PubMed] [Google Scholar]