

Abstract
Introduction
The radiation doses used to treat unresectable lung cancer are often limited by the proximity of normal tissues. Overexpression of c-Met, a receptor tyrosine kinase occurs in about half of non-small cell lung cancers (NSCLCs) and has been associated with resistance to radiation therapy, and poor patient survival. We hypothesized that inhibiting c-Met would increase the sensitivity of NSCLC cells to radiation enhancing the therapeutic ratio, which may potentially translate into improved local control.
Methods
We tested the radiosensitivity of two high-c-Met-expressing NSCLC lines, EBC-1 and H1993, and two low-c-Met-expressing lines, A549 and H460, with and without the small-molecule c-Met inhibitor MK-8033. Proliferation and protein expression were measured with clonogenic survival assays and western blotting, respectively. γ-H2AX levels were evaluated by immunofluorescence staining.
Results
MK-8033 radiosensitized the high-c-Met–expressing EBC-1 and H1993 cells but not the low-c-Met–expressing cell lines A549 and H460. However, irradiation of A549 and H460 cells increased the expression of c-Met protein at 30 minutes after the irradiation. Subsequent targeting of this upregulated c-Met by using MK-8033 followed by a second radiation dose reduced the clonogenic survival of both A549 and H460 cells. MK-8033 reduced the levels of radiation-induced phosphorylated (activated) c-Met in A549 cells.
Conclusions
These results suggest that inhibition of c-Met could be an effective strategy to radiosensitize NSCLC tumors with high basal c-Met expression or tumors that acquired resistance to radiation due to up-regulation of c-Met.
Keywords: NSCLC, c-Met, radiosensitivity
Introduction
Despite advances in diagnosis and treatment over the past several years, unresectable lung cancer remains a highly lethal disease, with 5-year survival rate of only about 14-15% among patients selected for combined modality treatment in clinical trials.1 However, a larger percent of patients with unresectable disease are unable to tolerate combined-modality treatment, often receive either radiation or chemotherapy alone, and subsequently do even worse2, 3. While trails such as RTOG 9410 have demonstrated the benefits of concurrent external-beam radiation chemotherapy we are often limited by its toxicity to the surrounding normal structures, particularly normal lung and esophagus3, 4. Recently, receptor tyrosine kinases (RTKs) have been implicated in malignant progression and as such have become attractive targets for new therapeutics. Among them c-Met, a hepatocyte growth factor receptor (HGF) has been of great interest as many solid malignancies, including lung cancers, express high levels of c-Met that are associated with therapy resistance.
HGF, the only known ligand for c-Met, is a multifunctional heterodimeric protein typically produced by mesenchymal cells. Its pleiotropic activities are mediated through its cellular receptor c-Met, a transmembrane tyrosine kinase encoded by the proto-oncogene MET (mesenchymal-epithelial transition factor). HGF and c-Met are overexpressed in a broad spectrum of human solid tumors including lung, breast, and brain.5, 6 Activation of the HGF/c-Met pathway has been shown to confer protection against cell death induced by a variety of DNA-damaging agents, including radiation and topoisomerase inhibitors, both in vitro and in vivo.7, 8 HGF has also been shown to function as an autocrine or paracrine growth factor.9 The c-Met pathway activates a program of cell dissociation and motility coupled with increased protease production that has been shown to promote cellular invasion through extracellular matrices in a process that closely resembles tumor metastasis in vivo.6
In patients with non-small cell lung cancer (NSCLC), activation of the HGF/c-Met axis has been associated with poor survival.10 Amplification of c-Met has also been implicated in resistance to therapies targeting the epidermal growth factor receptor (EGFR).11 Combining the inhibition of both EGFR and c-Met is being explored to overcome this putative resistance feedback loop. This strategy was tested in a recent phase II study comparing the EGFR tyrosine kinase inhibitor erlotinib (Tarceva) plus placebo versus erlotinib plus a monoclonal antibody to c-Met for the treatment of NSCLC.12 The study found improvement in the progression-free survival among patients with high c-Met expression (6.4 weeks versus 12.4 weeks). The study also showed that 50% of NSCLC expressed elevated levels of c-Met, as measured by immunohistochemical staining and the data suggest an increase in overall survival (7.4 months versus 7.7 months) with the addition of Met antibody for patients with high c-Met expression tumors.12
The goal of the study reported here was to test whether inhibition of the HGF/c-Met axis with the small-molecule inhibitor MK-8033 is effective for sensitizing NSCLC cells to radiation. We further tested whether radiation influence c-Met expression as compensator stress response to cellular DNA damage.
Materials and Methods
Cell Cultures
The human NSCLC lines H1993, A549, and H460 were obtained from the American Type Culture Collection (Manassas, VA) and EBC-1 cells were purchased from Health Science Research Resources Bank (Japan Health Sciences Foundation, Tokyo, Japan). The four cell lines comprises of high c-MET expressing (H1993 and EBC-1) and low c-MET expressing (A549 and H460) cells. The cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin at 37°C in 5% carbon dioxide conditions. Cell lines were validated by MD Anderson's Characterized Cell Line Core facility by short tandem repeat (STR) DNA fingerprinting using the AmpF(x02113)STR Identifiler kit according to the manufacturer's recommendations (Applied Biosystems #4322288). The STR profiles were compared to known American Type Culture Collection fingerprints (ATCC), to the Cell Line Integrated Molecular Authentication database (CLIMA) version 0.1.200808 (http://bioinformatics.istge.it/clima/) (Nucleic Acids Research 37:D925-D932 PMCID: PMC2686526), and to MD Anderson's fingerprint database.
Treatment Conditions and Irradiation
The small-molecule c-Met inhibitor, MK-8033 (Merck Research Laboratory) was dissolved in dimethyl sulfoxide (DMSO) and used at final concentrations ranging from 0.1–10 μM. For clonogenic survival assay, cells were pre-treated with MK-8033 for 1 hour before irradiation. Control cells were treated with equal volumes of DMSO. A Mark I 137Cs irradiator (JL Shepherd and Associates, San Fernando, CA, USA) was used to irradiate the NSCLC cells at a dose rate of 3.5 Gy/min at room temperature.
Clonogenic Survival Assays
Clonogenic (colony-forming) survival assays were done as described previously.13 Briefly, the H460, A549, and H1993 NSCLC cells were seeded in 60-mm dishes and allowed to stabilize overnight. The next day, cells were treated with either vehicle control (DMSO) or 1 μM MK-8033 for 1 hour followed by irradiation to a dose of 0, 2 or 4 Gy. Cells were then counted, seeded in 60-mm dishes, and incubated for 12 days to allow macroscopic colony formation. Colonies were fixed and stained for 5 minutes with 0.5% crystal violet (Sigma, St. Louis, MO) in methanol. The number of colonies formed in each treatment group was counted, with a cutoff of 50 viable cells per colony. Survival were calculated relative to that of un-irradiated cells (Survival = (plating efficiency of treated cells)/(plating efficiency of control cells) where plating efficiency = (number of colonies formed by treated cells)/ (number of colonies formed by untreated cells).13 Because the EBC-1 cells did not form colonies on dishes, these cells were cultured in suspension by seeding them to ultralow attachment 6-well plates (Costar #3471) in Dulbecco's modified essential medium (Cell Gro #10-010-CV) with 1:50 B27 (Invitrogen #17504-0440), 20 ng/ ml beta fibroblast growth factor (Invitrogen #PHG0264), and 20 ng/ ml epidermal growth factor (Invitrogen #PHG0314).
To assess whether radiation could induce c-Met expression resulting in adaptive radioresistant and whether MK-8033 could revert the radiation sensitivity, we exposed low-c-MET expressing cells to 4 Gy and 2 h later treated them with MK-8033 (10 μM). One day after the first radiation dose, the cells were irradiated again with either 0, 2 or 4 Gy radiation and plated into 60-mm dishes and allowed to form colonies for 12 days as described above.
Protein Extraction and Immunoblotting
Cells were seeded on 10-cm petri dishes, allowed to stabilize for 24 hours, and then treated with MK-8033 (0.1-10 μM) for 2 hours, after which they were rinsed in ice-cold phosphate-buffered saline three times and lysed in lysis buffer containing 50 mmol/L HEPES (pH 7.9), 0.4 mol/L NaCl, 1 mmol/L ethylenediaminetetraacetic acid, 2 μg/mL leupeptin, 2 μg/mL aprotinin, 5 μg/mL benzamidine, 0.5 mmol/L phenylmethylsulfonyl fluoride, and 1% NP40. Lysates were subjected to electrophoresis on 4%-15% sodium dodecyl sulfate-polyacrylamide electrophoresis gels (Cat #161-1158, Bio-Rad, Hercules, CA), transferred to a polyvinylidene fluoride membrane, and blocked with 5% bovine serum albumin in Tris-buffered saline-Tween 20 (0.05%, vol/vol) for 1 hour at room temperature. The membrane was then incubated with primary antibody overnight at 4°C. Antibodies for c-Met (Cat #4560), p-c-Met (Cat #3121), pAKT (Cat4051), p-extracellular signal-regulated kinase 1/2 (pERK1/2) (Cat #2232), and β-actin (Cat #4967) were purchased from Cell Signaling. After the overnight incubation, the membrane was incubated with the appropriate horseradish peroxidase–conjugated secondary antibody (diluted 1:2,000; Amersham Biosciences) for 1 hour. The blots were developed by enhanced chemiluminescence (Amersham Biosciences).
To analyze the effect of MK-8033 on the radiation-induced phosphorylated c-Met, cells were irradiated with 4 Gy, incubated for 2 hours, and treated with MK-8033 (10 μM). One day after first irradiation, the cells were irradiated again with 4 Gy, incubated for 2 hours, and then harvested for protein extraction and immunoblotting.
Immunofluorescent staining for γ-H2AX
For this assay, 200,000 cells were plated on coverslip placed in 35-mm dish and allowed to attach overnight. Then cells were irradiated (4Gy) and 2 h later exposed to 10 μM MK-8033 for 24 h. After the incubation period, cells were again irradiated (4 Gy) and incubated for upto 6 hours. Then, the cells were fixed with 1% paraformaldehyde for 10 min followed by ethanol (70%) fixation for 10 minutes at room temperature. The cells were then treated with 0.1% NP40 in PBS for 20 min, washed in PBS four times and then blocked with 5% bovine serum albumin in PBS for 30 min. The cells were then incubated with anti–γ-H2AX (Millipore) antibody in 5% bovine serum albumin in PBS overnight. Next day, cells were incubated with FITC-labeled secondary antibody at a dilution of 1:300 (ྙ-H2AX) in 5% BSA in PBS for 30 min. Cells then were incubated in the dark with 4 4′,6-diamidino-2-phenylindole (DAPI, 1 mg/mL) in PBS for 5 min, and coverslips were mounted on a slide with an antifade solution (Molecular Probes). Slides were examined using a Leica fluorescence microscope, and images were captured by a CCD camera and imported into Advanced Spot Image analysis software package. For each treatment condition, the number of γ -H2AX foci were determined in at least 50 cells14.
Results
MK-8033 Affected c-Met–Induced Signaling in High-c-Met–Expressing EBC-1 and H1993 Cells
The activation of c-Met leads to cellular processes that are mediated in part by the ERK and Akt pathways. These pathways are known to be upregulated in various cancers and have been shown to promote cellular proliferation and anti-apoptosis. As shown in Figure 1, treating the high c-Met expressing EBC-1 and H1993 cells with 0.1-10 μM MK-8033 for 2 hours reduced the phosphorylation of c-Met, ERK, and Akt in a dose-dependent manner. However, this drug did not affect these signaling pathways in low c-Met expressing A549 or H460 cells (Fig. 1).
Figure 1.
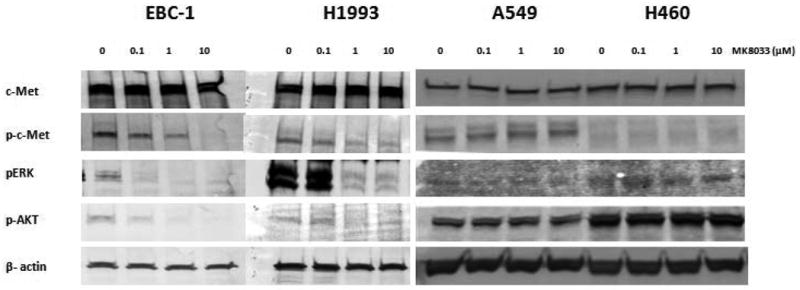
MK-8033 affects c-Met–induced signaling in EBC-1 and H1993 cells but not in A549 and H460 cells. MK-8033 downregulated the phosphorylation of c-Met, ERK, and Akt in EBC-1 and H1993 cells (cells with high basal c-Met levels) but not in A549 and H460 cells (cells with low-c-Met-expressing).
c-Met Levels Predicted Radiosensitizing Effect of MK-8033
Next, in analyzing the radiosensitizing effects of MK-8033, we found that treating the high-c-Met–expressing EBC-1 and H1993 cells with MK-8033 (1 μM) for 1 hour before irradiation sensitized these cells to radiation. However, this effect was not observed in the low-c-Met–expressing A549 and H460 cells (Fig. 2).
Figure 2.
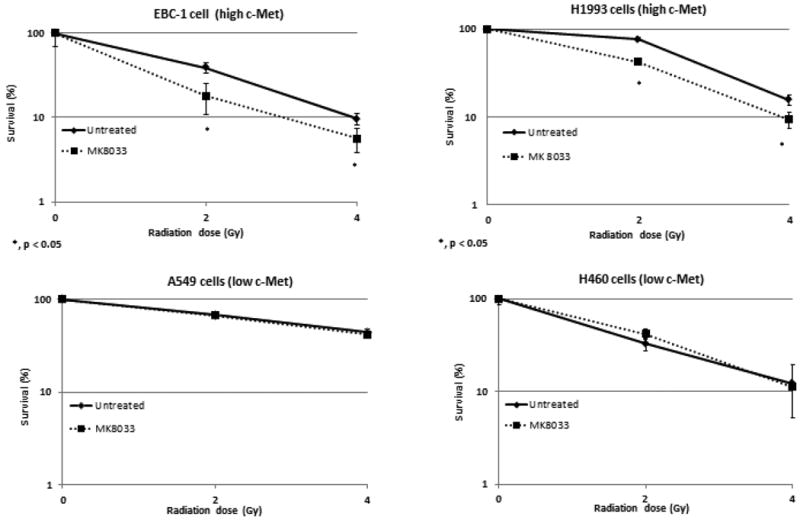
MK-8033 radiosensitizes EBC-1 and H1993 cells but not A549 and H460 NSCLC cells. A 1-hour pretreatment with MK-8033 (1 μM) radiosensitized NSCLC cells with high basal c-Met levels (EBC-1 and H1993) but not in low c-Met-expressing cells.
Radiation Induced c-Met Expression in Low-c-Met–Expressing A549 and H460 Cells and Sensitized them to MK-8033
Next we explored the effects of radiation on the induction or activation of c-Met in the low-c-Met–expressing NSCLC cells, A549 and H460. A one-time exposure to 4 Gy of radiation led to time-dependent increases in c-Met levels starting at 30 minutes and continuing for more than 24 hours in A549 cells but, not in H460 cells (Fig. 3A, B). However, we further found that adding MK-8033 (10 μM) at 2 hours after the first irradiation (4 Gy) and exposing the cells to a second dose of radiation reduced their colony-forming ability relative to two radiation exposures without the drug in both cell lines, more potently, in H460 cells (Fig. 3C, D). Hence, at least in A549 cells upregulation of c-Met made the cancer cells susceptible to MK-8033-induced radiosensitization. Finally, the combination of radiation followed by MK-8033 followed by a second irradiation reduced the levels of p-c-Met in A549 cells relative to two doses of radiation alone (Fig. 4A). Induction of c-Met appeared to be dose dependent and most prominent between 6-8Gy (data not shown).
Figure 3.
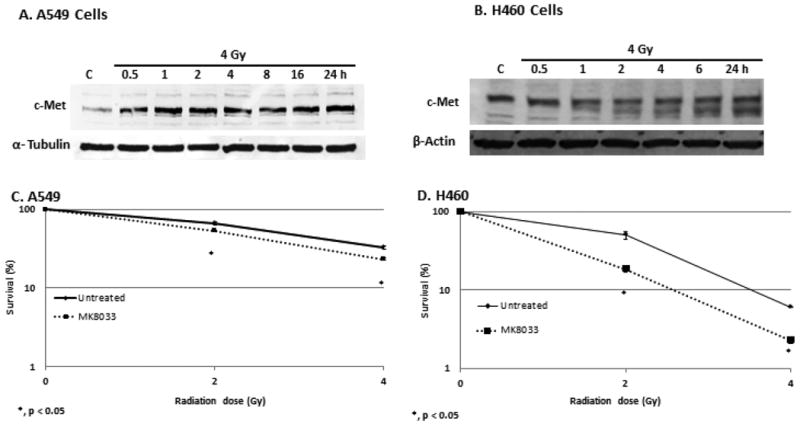
Radiation induces c-Met expression and radiosensitizes A549 and H460 cells. Treatment of A549 and H460 cells with a single 4-Gy dose of radiation induced c-Met expression starting 30 minutes after the irradiation and persisting for 24 hours (panels A and B). Further, adding MK-8033 to fractionated radiation (4Gy times two separated by 24hrs) reduced colony-forming ability, suggesting that upregulation of c-Met by the first irradiation further sensitized the cells to MK-8033.
Figure 4.
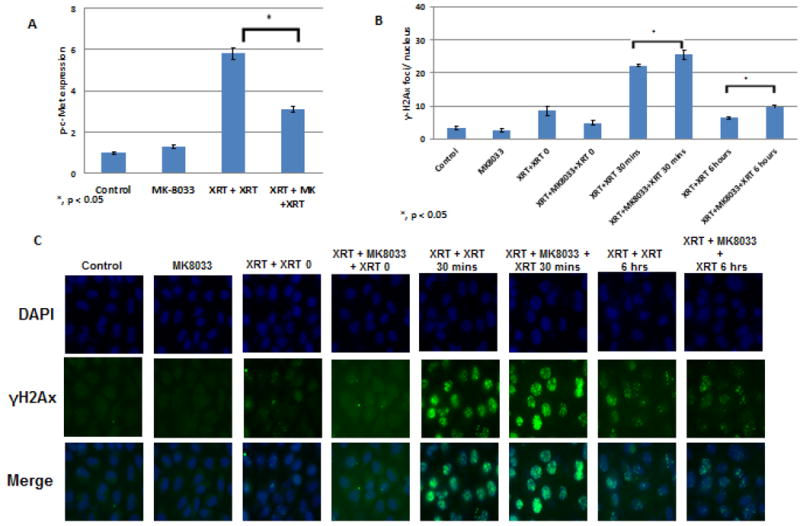
A, MK-8033 reduces p-c-Met levels in A549 cells after radiation. A single dose of radiation did not affect the expression of phosphorylated (activated) c-Met, but fractionated radiation (i.e., two doses) did. This increase was attenuated by the addition of MK-8033 (10μM, 2 hours after first radiation) between the two radiation doses. B and C, addition of MK-8033 enhances the levels of γ-H2Ax in A549 cells for upto 6 hours compared to double irradiation and MK-8033 treatment alone suggesting decrease in DNA repair.
c-Met inhibition prolonged the presence of nuclear γ-H2AX foci-induced by irradiation
To determine the effect of c-Met inhibition on the DNA damage/repair kinetics after irradiation, we analyzed the presence of nuclearγ-H2AX foci after two fractions of irradiation in the absence or presence of MK-8033, as mentioned above, in A549 cells. We found that treatment with MK-8033 between fractionated irradiation increases the number of γ-H2AX for upto 6 hours as compared to cells not treated with the drug (Fig. 4 B and C), suggesting the MK-8033 interfered with the DNA repair kinetics.
Discussion
Since there is a limitation on the dose of radiation which can be delivered to the malignant tissue/organ without affecting the surrounding normal tissues, efforts are made to increase the sensitivity of cancer cells to radiation. In this study, we analyzed if inhibition of c-Met activation would radiosensitize NSCLC cells. We found that inhibiting c-Met with MK-8033 can effectively sensitize cell lines that constitutively express high c-Met levels to radiation but not in cell lines with low constitutive c-Met expression. We also observed upregulation of c-Met protein after exposure to radiation, perhaps as a compensatory cellular stresses response. This radiation induced c-Met expression could be blocked using MK-8033 resulting in enhanced cell kill when combined with radiation. Our study suggests that combination of c-Met inhibitor and radiation should be further tested to confirm its clinical relevance.
The utilization of various kinase inhibitors in combination with radiation has also shown promise in the treatment of various forms of cancer, including lung cancer. Most such work has been done with EGFR inhibitors such as cetuximab and erlotinib. Erlotinib currently is approved by the US Food and Drug Administration for refractory NSCLC and may soon be approved as first-line therapy for NSCLC that has mutations in EGFR.15 EGFR has been shown to radiosensitize a variety of cancer cells in vitro and in animal models16 as well as in clinical settings in the treatment of head and neck cancers, where the addition of cetuximab to radiation increased the median survival time from 29 months with radiation alone to 49 months.17 This strategy has been evaluated for NSCLC in RTOG 0324, in which cetuximab was combined with radiation for unresectable disease; the encouraging median survival time of 22.7 months from this combination compares favorably to RTOG 9410, in which the median survival time was 17 months. However, in SWOG S0023, addition of EGFR TKI (gefitinib) post concurrent chemoradiation in unselected NSCLC patients negatively affected patient survival (35 months for placebo vs 23 months for genifitinib).18 Increase in toxicity due to gefitinib could be responsible for decreased survival, however, authors suggests 2% increase in toxicity should not explain the statistically significant difference in the survival. Another important factor could be patient's smoking, EGFR and K-ras status which may predict response to EGFR TKI therapy.18, 19
One of the mechanism underlying the resistance of cancer cells to EGFR inhibitors was recently shown to involve c-Met amplification.20 Thus c-Met inhibition may even have a greater role when used in combination with EGFR inhibition. The converse, that resistance to c-Met inhibition can be mediated through upregulation of EGFR, has also been demonstrated.21 Lastly, given the ability of c-Met kinase to drive the epithelial-to-mesenchymal transition, which is thought to encourage tumor cell invasion and metastatic spread, blocking c-Met may not only benefit local control through improved radiosensitization but could also may help in preventing systemic spread of the disease.22-24
Previously, De Bacco et al. observed that radiation induces c-Met levels in breast cancer cells.25 The increase in c-Met and its activation could be responsible for increased migratory and invasive potential of the cancer cells after irradiation.25 It would be interesting to see how irradiation affects c-Met levels in normal tissues and if blocking this might potentially reduce radiation sensitization in these tissues as well. Studies to further investigate this should be carried out before combining c-Met inhibitor and radiation in clinical settings. If the phenomenon is observed in all the tissues, c-Met inhibition may also sensitize normal tissues to radiation. It would be also interesting to see if induction of c-Met by radiation is also observed in c-Met mutated NSCLC which represent 2-4% of all cases.
In terms of the next steps to be taken, we are in the process of assessing the upregulation of c-Met in serum samples from patients being treated for unresectable NSCLC. If serum c-Met levels prove to be a useful biomarker of resistance, then, that would provide a relatively noninvasive way of identifying the patients who would most likely benefit from c-Met inhibition. Serum c-Met levels may also prove to be useful for evaluating both the response to c-Met-targeting agents and the induction of c-Met by radiation.
In conclusion, our findings suggest that c-Met inhibition may be an effective form of radiosensitization for the roughly 50% of NSCLC tumors with elevated basal level of c-Met protein expression. Other NSCLC tumors that express low levels of c-Met may show upregulation of c-Met after radiation therapy and perhaps respond to c-Met inhibition as well. Given the emerging role of EGFR inhibitors in combination with radiation therapy for the treatment of lung cancer and the implication of c-Met in resistance to EGFR inhibitors, c-Met inhibitors are effective agents for overcoming resistance to radiation therapy as well as resistance arising from other molecular feedback loops.
Acknowledgments
Supported by a grant from Merck, by the family of Mr. M. Adnan Hamed, National Institutes of Health through Cancer Center Support (Core) Grant CA016772 to MD Anderson Cancer Center, Lung SPORE (P50 CA070907) and R01 (1R01 CA168484-01).
References
- 1.Socinski MA, Blackstock AW, Bogart JA, et al. Randomized Phase II Trial of Induction Chemotherapy Followed by Concurrent Chemotherapy and Dose-Escalated Thoracic Conformal Radiotherapy (74 Gy) in Stage III Non-Small-Cell Lung Cancer: CALGB 30105. J Clin Oncol. 2008;26(15):2457–63. doi: 10.1200/JCO.2007.14.7371. [DOI] [PubMed] [Google Scholar]
- 2.Le Chevalier T, Arriagada R, Quoix E, et al. Radiotherapy alone versus combined chemotherapy and radiotherapy in nonresectable non-small-cell lung cancer: first analysis of a randomized trial in 353 patients. Journal of the National Cancer Institute. 1991;83(6):417–23. doi: 10.1093/jnci/83.6.417. [DOI] [PubMed] [Google Scholar]
- 3.Sause WT. The role of radiotherapy in non-small cell lung cancer. Chest. 1999;116(6 Suppl):504S–08S. doi: 10.1378/chest.116.suppl_3.504s. [DOI] [PubMed] [Google Scholar]
- 4.Curran WJ, Jr, Paulus R, Langer CJ, et al. Sequential vs. concurrent chemoradiation for stage III non-small cell lung cancer: randomized phase III trial RTOG 9410. Journal of the National Cancer Institute. 2011;103(19):1452–60. doi: 10.1093/jnci/djr325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maulik G, Shrikhande A, Kijima T, Ma PC, Morrison PT, S R. Role of the hepatocyte growth factor receptor, c-Met, in oncogenesis and potential for therapeutic inhibition. Cytokine Growth Factor Rev. 2002;13:41–59. doi: 10.1016/s1359-6101(01)00029-6. [DOI] [PubMed] [Google Scholar]
- 6.Birchmeier C, Birchmeier W, Gherardi E, Vande GFW. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–25. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 7.Fan S, Ma YX, Gao M, et al. The multisubstrate adapter gab1 regulates hepatocyte growth factor (scatter factor)-c-met signaling for cell survival and DNA repair. Mol Cell Biol. 2001;21(15):4968–84. doi: 10.1128/MCB.21.15.4968-4984.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qian LW, Mizumoto K, Inadome N, et al. Radiation stimulates HGF receptor/c-Met expression that leads to amplifying cellular response to HGF stimulation via upregulated receptor tyrosine phosphorylation and MAP kinase activity in pancreatic cancer cells. International journal of cancer Journal international du cancer. 2003;104(5):542–9. doi: 10.1002/ijc.10997. [DOI] [PubMed] [Google Scholar]
- 9.Danilkovitch-Miagkova A, Zbar B. Dysregulation of Met receptor tyrosine kinase activity in invasive tumors. J Clin Invest. 2002;109:863–67. doi: 10.1172/JCI15418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Siegfried JM, Weissfeld LA, Singh-Kaw P, Weyant RJ, Testa JR, Landreneau RJ. Association of Immunoreactive Hepatocyte Growth Factor with Poor Survival in Resectable Non-Small Cell Lung Cancer. Cancer Res. 1997;57(3):433–39. [PubMed] [Google Scholar]
- 11.Zhang YW, Staal B, Essenburg C, et al. MET kinase inhibitor SGX523 synergizes with epidermal growth factor receptor inhibitor erlotinib in a hepatocyte growth factor-dependent fashion to suppress carcinoma growth. Cancer Res. 2010;70(17):6880–90. doi: 10.1158/0008-5472.CAN-10-0898. [DOI] [PubMed] [Google Scholar]
- 12.Spigel D, Ervin T, Ramlau R, et al. Randomized multicenter double-blind placebo-controlled phase ii study evaluating metmab, an antibody to met receptor, in combination with erlotinib, in patients with advanced non-small-cell lung cancer. ESMO 2010. 2010 Abstract LBA15(Oct 8-12, 2010, Milan, Italy.) [Google Scholar]
- 13.Munshi A, Hobbs M, Meyn RE. Clonogenic cell survival assay. Methods in molecular medicine. 2005;110:21–8. doi: 10.1385/1-59259-869-2:021. [DOI] [PubMed] [Google Scholar]
- 14.Tanaka T, Munshi A, Brooks C, Liu J, Hobbs ML, Meyn RE. Gefitinib radiosensitizes non-small cell lung cancer cells by suppressing cellular DNA repair capacity. Clin Cancer Res. 2008;14(4):1266–73. doi: 10.1158/1078-0432.CCR-07-1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neal JW. The SATURN trial: the value of maintenance erlotinib in patients with non-small-cell lung cancer. Future Oncol. 2010;6(12):1827–32. doi: 10.2217/fon.10.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chinnaiyan P, Huang S, Vallabhaneni G, et al. Mechanisms of enhanced radiation response following epidermal growth factor receptor signaling inhibition by erlotinib (Tarceva) Cancer Research. 2005;65(8):3328–35. doi: 10.1158/0008-5472.CAN-04-3547. [DOI] [PubMed] [Google Scholar]
- 17.Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus Cetuximab for Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med. 2006;354(6):567–78. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 18.Kelly K, Chansky K, Gaspar LE, et al. Phase III trial of maintenance gefitinib or placebo after concurrent chemoradiotherapy and docetaxel consolidation in inoperable stage III non-small-cell lung cancer: SWOG S0023. J Clin Oncol. 2008;26(15):2450–6. doi: 10.1200/JCO.2007.14.4824. [DOI] [PubMed] [Google Scholar]
- 19.Keedy VL, Arteaga CL, Johnson DH. Does gefitinib shorten lung cancer survival? Chaos redux. J Clin Oncol. 2008;26(15):2428–30. doi: 10.1200/JCO.2008.16.0374. [DOI] [PubMed] [Google Scholar]
- 20.Engelman Jeffrey A, Janne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non small cell lung cancer. Clin Cancer Res. 2008;14(10):2895–99. doi: 10.1158/1078-0432.CCR-07-2248. [DOI] [PubMed] [Google Scholar]
- 21.McDermott U, Pusapati RV, Christensen JG, Gray NS, Settleman J. Acquired resistance of non-small cell lung cancer cells to MET kinase inhibition is mediated by a switch to epidermal growth factor receptor dependency. Cancer Res. 2010;70(4):1625–34. doi: 10.1158/0008-5472.CAN-09-3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mazzone M, Comoglio PM. The Met pathway: master switch and drug target in cancer progression. FASEB J. 2006;20(10):1611–21. doi: 10.1096/fj.06-5947rev. [DOI] [PubMed] [Google Scholar]
- 23.Bladt F, Riethmacher D, Isenmann S, Aguzzi A, Birchmeiert C. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature. 1995;376(6543):768–71. doi: 10.1038/376768a0. [DOI] [PubMed] [Google Scholar]
- 24.Zucali PA, Ruiz MG, Giovannetti E, et al. Role of cMET expression in non-small-cell lung cancer patients treated with EGFR tyrosine kinase inhibitors. Ann Oncol. 2008;19(9):1605–12. doi: 10.1093/annonc/mdn240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Bacco F, Luraghi P, Medico E, et al. Induction of MET by ionizing radiation and its role in radioresistance and invasive growth of cancer. J Natl Cancer Inst. 2011;103(8):645–61. doi: 10.1093/jnci/djr093. [DOI] [PubMed] [Google Scholar]