

Abstract
Memory and naive CD4 T cells have unique regulatory pathways for self/non-self discrimination. A memory cell specific regulatory pathway was revealed using superantigens to trigger the TCR. Upon stimulation by bacterial superantigens, like staphylococcal enterotoxin B (SEB), TCR proximal signaling is impaired leading to clonal tolerance (anergy). In the present report, we show that memory cell anergy results from the sequestration of the protein tyrosine kinase ZAP-70 away from the TCR/CD3ζ chain. During SEB-induced signaling, ZAP-70 is excluded from both detergent-resistant membrane microdomains and the immunological synapse, thus blocking downstream signaling. We also show that the mechanism underlying memory cell anergy must involve Fyn kinase, given that the suppression of Fyn activity restores the movement of ZAP-70 to the immunological synapse, TCR proximal signaling, and cell proliferation. Thus, toleragens, including microbial toxins, may modulate memory responses by targeting the organizational structure of memory cell signaling complexes.
Keywords: T lymphocytes, Immunological Memory, Superantigens, Anergy, Rodents, Signal Transduction, Immune Synapses, Lipid Rafts
INTRODUCTION
Upon exposure to foreign antigen, T lymphocytes are induced into clonal expansion and differentiation to become “antigen-experienced” cells (e.g.; effector and memory T cells) [1;2]. For both naive and memory CD4 T cells, stimulation through the TCR by peptide-MHC complexes involves multiple signal transduction pathways. Further, depending upon the nature of the initial signal (e.g.; foreign versus self-antigen, superantigen, anti-TCR Abs), different signaling pathways may be used and different functional outcomes (e.g.; cell activation, proliferation, or tolerance) may result [3–8]. Regardless of the stimulus, cell signaling is tightly regulated, in part through the defined organization of signaling molecules into complexes both on the T cell membrane (e.g.; lipid raft microdomains) [9–15], and, also, at the T cell-APC interface (e.g., immunological synapses) [16–20]. Studies using primary T cells (e.g. naive, memory) or cloned effector T cell lines (e.g.; Th1, Th2) showed that membrane organizational structures may be distinct, depending upon the specific T cell differentiation state [21–24]. However, the relationship of different membrane signaling complexes to specific cell function is unclear. Likewise, stimulatory or tolerogenic signals may lead to different signaling structures. For example, stimulation of resting T cells by agonist peptides promotes the formation of paradigm immune synapses, while tolerogenic peptides lead to incomplete synapses [18;25], where some critical signaling molecules move to the central supramolecular activation cluster (c-SMAC), whereas other critical molecules are excluded from the synapse.
We have previously examined responses of naive and memory CD4 T cells after stimulation by peptide antigens and microbial superantigens. While either stimulus elicited robust proliferation and cytokine secretion by naive cells, only peptide antigen promoted activation of resting memory CD4 cells [26]. In contrast, superantigens, such a Staphylococcal enterotoxin B (SEB) did not stimulate resting memory cells [26]. Indeed, SEB induced memory cells to become anergic, indicated by a failure of the cells to proliferate when subsequently exposed to an agonist peptide [8]. This observation supported the hypothesis that, after naive cells differentiate into memory cells, unique regulatory pathways are utilized. Specific memory cell regulation could facilitate both enhanced responses to recall antigens and, also, prevent untoward responses to self-antigens encountered by high avidity memory cells that traffic through tissues [2]. Exposure of memory cells to SEB revealed an anergy pathway that was characterized by impaired signaling through the TCR/CD3 complex [7]. Normally, the earliest signaling events during stimulation through the TCR include tyrosine phosphorylation of the TCR CD3ζ chain by the src kinase Lck, and, recruitment of the protein tyrosine kinase ZAP-70 to the plasma membrane to bind to pCD3ζ, so that ZAP-70 can then be phosphorylated and activated by Lck [27]. However, we found, during co-immunoprecipitation experiments, that when memory CD4 cells were exposed to SEB, there was a failure of association between the activated pCD3ζ molecule and ZAP-70 [7]. Hence, there was an absence of tyrosine phosphorylation of ZAP-70 and further downstream signaling was blocked. Additional studies showed that a specific hyperactivation of the src kinase Fyn was essential to SEB-induced memory cell anergy, as the inhibition or absence of Fyn restored both CD3ζ/ZAP-70 complex formation and cell activation [28].
Given that SEB promoted CD4 memory cell anergy with similar proximal signaling deficiencies as found in other tolerance models (e.g.; inactive ZAP-70 [29], Fyn kinase involvement [30]), and given that a failure of ZAP-70 recruitment was a controlling element in SEB-induced anergy, we hypothesized that productive signaling complexes were not formed when SEB was presented to memory T cells. In an earlier study we examined TCR signaling in immune synapses and lipid raft microdomains of memory cells presented with cognate peptide [22]. We identified several features that were distinct from the membrane signaling structures of naive cells responding to the same cognate peptide, suggesting structural bases for alternative activation pathways. In the present study, we investigated whether SEB could mediate a productive interaction between memory T cells and APCs and whether complete immunological synapses were formed. Since our earlier study showed that ZAP-70 did not bind to the TCR/CD3 complex [7], our current study specifically centered upon whether ZAP-70 localized to the same membrane regions as the TCR, when memory cells became exposed to SEB versus peptide antigen. We found that, in contrast to peptide stimulation, exposure of memory, but not naive, CD4 T cells to SEB resulted in the absence of ZAP-70 from both the immune synapse and, also, lipid rafts, suggesting that ZAP-70 and the TCR were physically separated. Given that the absence of Fyn did allow for ZAP-70 to migrate to the immune synapse, we conclude that SEB induces Fyn signaling which in turn leads to sequestration of ZAP-70 from the membrane compartments that contain the TCR, and thus prevents proximal signaling.
MATERIALS AND METHODS
Animals
The BALB/c ByJ, DO11.10 [31], and DO11.10 × Fyn−/− mice used in these experiments were bred and maintained at the Wadsworth Center Animal Core Facility under specific pathogen-free conditions. The majority of T cells in the DO11.10 and DO11.10 × Fyn−/− mice are CD4+ cells, which bear a TCR that recognizes a chicken ovalbumin-derived peptide, OVA323–339 (hereafter referred to as OVA), presented by I-Ad [31]. This TCR is encoded by transgenes encoding Vβ8.2/Vα13.1 chains and can be identified by the anti-clonotypic mAb, KJ1-26 [32]. We have previously characterized the DO11.10 memory cells and their responses to peptide antigen and superantigen [26;33]. Fyn−/− mice [34] were originally obtained from the Jackson Laboratory (Bar Harbor, ME) and backcrossed over 11 generations onto a BALB/c background, before they were bred to DO11.10 mice, to generate DO11.10 × Fyn−/− mice. For all experiments, cells were obtained from mice that were 10–12 weeks old. Cells from either male or female mice were used, in different experiments, with no discernible differences in the results. All mice used in these studies were bred and maintained in accordance with the guidelines of the Committee on Care and Use of Laboratory Animals of the Institute of Laboratory Resources, National Research Council (Washington, DC). All experiments were approved by the Wadsworth Center IACUC.
Reagents and antibodies
Mabs KJ1-26 (anti-DO11.10 clonotype) [32] and 23G2 (anti-CD45RB) [35] were prepared from the supernatants of hybridoma cell lines, as previously described. Additional antibodies and probes used for confocal microscopy were cholera toxin-B-rhodamine (CT-B-rhodamine) conjugate (List Biological Laboratories, Campbell, CA), anti-phospho-ZAP-70 (Tyr319) (Cell Signaling Technology, Beverly, MA), and anti-ZAP-70, goat anti-mouse IgG-HRPO, and goat anti-rabbit IgG-HRPO (Transduction Laboratories, San Diego, CA). The rabbit polyclonal Ab directed against phosphorylated Lck (pY394) [19] was obtained from A. Shaw (Washington University, St. Louis, MO). OVA peptide was synthesized and supplied by the Wadsworth Center Peptide Synthesis Core Facility. SEB (Toxin Technology, Sarasota, FL) was purchased.
Preparation of cells
In all experiments, enriched populations of CD4+ T cells were prepared by negative selection procedures as previously described [36], and were 90–95% CD4+ and <3% slg+, as determined by flow-cytometric analyses. Naive and memory cells were separated as previously described [8] based upon CD45RB expression using mAb 23G2 supernatant and MACS (Miltenyi Biotec, Auburn, CA). There were no discernible differences between DO11.10 and DO11.10 × Fyn−/− mice, in percentage of splenic cell populations, in number of total CD4+ cells, or in populations of naive and memory cells. OVA and SEB were presented to T cells using APCs prepared by T cell depletion of splenocytes using anti-Thy1-1.2 and complement, followed by anti-CD4 (mAb 2B6) and anti-CD8 plus complement [26].
Cell labeling and culture
For measurement of cell division using CFSE [37], DO11.10 and DO11.10 × Fyn−/− CD4 cells were labeled with CFSE (5 µM) prior to separation into naive and memory populations. DO11.10 naive and memory cells (1 × 105/well) were cultured in 96-well flat-bottom plates with APCs (2 × 105/well) in 0.2 ml RPMI-1640 medium supplemented with 10% FBS, 50 mM 2-mercaptoethanol, 100 U/ml penicillin, 100 mg/ml streptomycin, and 2 mM glutamine. Where indicated, SEB (20 µg/ml) or OVA323–339 (0.2 µg/ml) was added to the cultures. After 66 hours, the T cells were analyzed by flow cytometry, after staining with mAb KJ1-26, to identify the DO11.10 clonotype-bearing cells.
Confocal microscopy
DO11.10 and DO11.10 × Fyn−/− CD4 naive and memory cells were cultured in RPMI-1640 medium supplemented with 10% FBS, 50 µM 2-Me, 100 U/ml penicillin, 100 µg/ml streptomycin, and 2 mM glutamine. Where indicated, the TCR within conjugates was identified by staining with mAb KJ1-26, to identify the DO11.10 clonotype-bearing cells. Conjugates between T cells and APCs were formed by mixing of T cell and SEB-pulsed (20 µg/ml) or OVA-pulsed (1.0 µg/ml) APCs at a 1:2 ratio, with a brief centrifugation at 400 × g to initiate cell-cell contact [19]. Cells were incubated at 37°C, under 5% CO2, for various time intervals. The cells were fixed with freshly prepared 4% paraformaldehyde for 20 min at room temperature and allowed to adhere to poly-L-lysine coated slides at 4°C overnight or for 2 hr at 37°C [19]. Cells were permeabilized with 0.2% TX-100, blocked with 1% BSA/PBS, and stained with appropriate antibodies for 1 hr diluted in 1% BSA/PBS. Cells were washed 4 times with PBS, between the primary and secondary antibody incubations, and the before addition of mounting solution. Coverslips were mounted onto slides using the SlowFade Light Antifade Kit (Molecular Probes, Inc.) following the manufacturer’s specifications.
In experiments analyzing lipid rafts, CT-B-rhodamine was used to label the endogenous GM1 glycosphingolipids of the T cells prior to mixing with APCs [38]. Aggregation of lipid rafts, or patching, was induced in unstimulated cells by incubation of the cells with diluted anti-CT-B antibody (1/250 in PBS/0.1%BSA; Calbiochem-Novabiochem Corp.) for 30 min on ice, and then 20 min at 37°C [38]. For stimulated cells, CT-B rhodamine labeled T cells were mixed with OVA- or SEB-pulsed APCs at a 1:2 ratio and briefly centrifuged at 400 × g. Cells were incubated at 37°C, under 5% CO2, for 30 min on poly-L-lysine coated slides. Cells either were fixed with 4% paraformaldehyde for 20 min at room temperature and permeabilized with 0.2% TX-100, or else were permeabilized with 1% TX-100 followed by fixation with 4% paraformaldehyde. The latter protocol isolates lipid rafts based upon their detergent insolubility [38;39]. Slides were blocked with 1% BSA/PBS and were incubated with respective antibodies diluted in 1% BSA/PBS for 1hr at room temperature. Slides were washed 4 times with PBS, in between primary and secondary antibody incubations, and before the addition of mounting solution. Coverslips were mounted as above.
Confocal microscopy images were acquired with a Hamamatsu ORCA-ER digital CCD camera (Hamamatsu Photonics K.K., Hamamatsu City, Japan) attached to a Zeiss Axioskop 2 mot plus microscope (Carl Zeiss, Göttingen, Germany), using OpenLab software (Improvision Inc., Lexington, MA). Three-dimensional reconstruction of the T cell/APC interface was generated from 0.3 mM optical sections of x-y images along the z axis with subsequent processing with OpenLab and Velocity software (Improvision Inc.). For data representation, within an experiment a minimum of 50 conjugates per condition or time point were visualized; depicted data are representative of at least 67% of those conjugates. Unless otherwise indicated, each experiment was performed 3 times.
RESULTS
SEB-mediated conjugation of APCs with naive or memory CD4 T cells
Naive and memory CD4 T cells were obtained from DO11.10 mice. The expression of the clonotypic, KJ1-26, TCR allowed these cells to bind to either the OVA peptide or SEB. We have previously discussed the OVA-specific memory cells from these mice [26;33;28]. Of relevance to the current study, we extended our observations, originally made using non-transgenic memory cells [40;41], and showed that DO11.10 memory CD4 T cells were also hyporesponsive to SEB [26]. Further, not only did SEB fail to directly stimulate the DO11.10 memory cells to proliferate, but the cells that were exposed to SEB lost the ability to subsequently proliferate in response to OVA; i.e., they became anergic [8]. Both the initial failure to proliferate and subsequent anergy were due to impaired TCR proximal signaling, characterized by a failure of ZAP-70 to bind to the TCR/CD3 complex [7]. The lack of response to SEB was dependent upon Fyn activation as the absence of Fyn led to SEB-induced memory cell proliferation ([28] and Figure 1).
Figure 1. Fyn-deficient memory cells proliferate in response to SEB.

Memory CD4 T cells from DO11.10 and DO11.10 × Fyn−/− mice were labeled with CFSE and then stimulated in vitro with SEB, or OVA. After 3.5 days, the cells were stained for the DO11.10 clonotype (mAb KJ1-26) and proliferation was assessed by flow cytometry, gating upon the live lymphocyte population. Data are representative of five independent experiments.
In the present study, we determined if the different responses made by memory cells to peptide antigens vs superantigens, or the responses made by memory vs naive cells to superantigens, were due to the initial formation of membrane signaling complexes. Effective TCR-mediated signal transduction is regulated, in part, by the formation of a productive immunological synapse [18;20;42;43]. We previously showed that both naive and memory DO11.10 cells effectively form conjugates with APCs bearing OVA, and, in both cases, productive immunological synapses are formed [22]. Here we show that naive cells and memory cells are equally effective in forming SEB-mediated conjugates with APCs (Figure 2). Conjugate formation was determined using flow cytometry to identify fluorescently-labeled naive or memory T cells (CFSE) and APCs (DiD) [22]. Either T cell population was added into culture with SEB-pulsed APCs and cell conjugates were determined by couplets displaying both CFSE and DiD fluorescence. In the absence of SEB, no conjugates were observed. However, SEB-pulsed APCs effectively bound to either naive or memory cells with comparable frequencies. The conjugate frequency (~25% of total input T cells at most time points) was comparable to that previously observed when using OVA to mediate the interactions. Further, conjugate stability up throughout the 2-hr experiment was comparable for both cell types. Thus, the inability of memory T cells to proliferate in response to SEB appears not to be directly related to an inability to bind to the superantigen.
Figure 2. SEB promotes equivalent conjugate formation between APCs and naive and memory T cells.
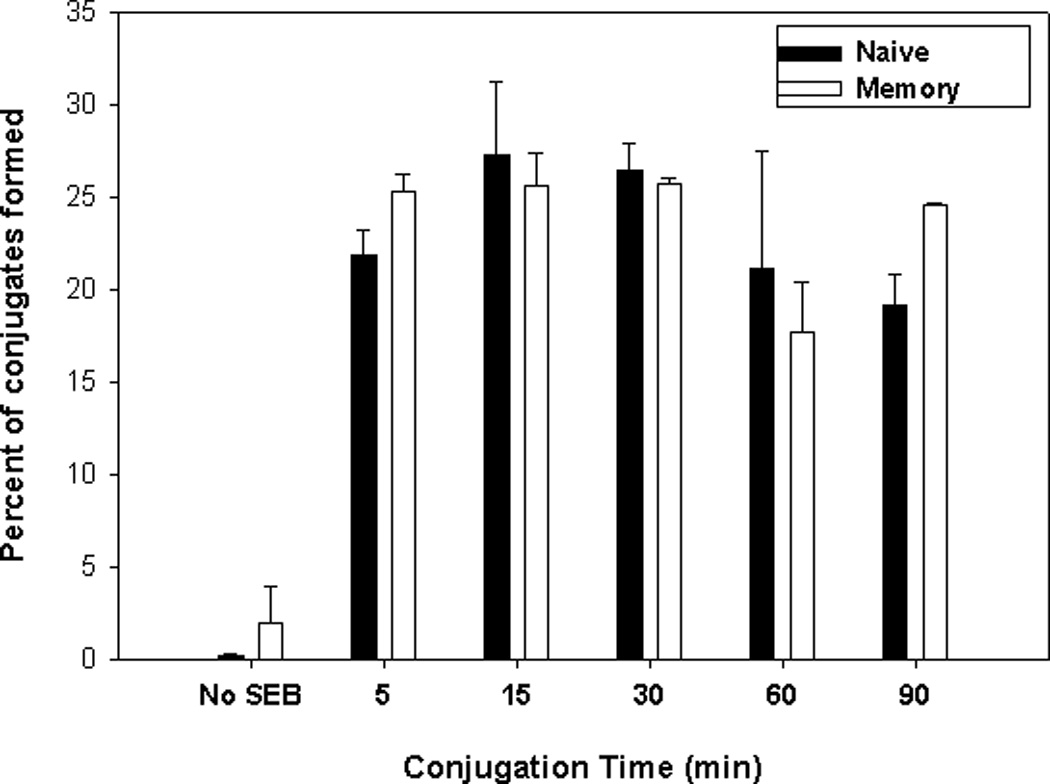
Naive (■) and memory (□) DO11.10 CD4+ T cells were labeled with CFSE. Splenic APCs were labeled with DiD and loaded with 20 µg/ml of SEB. T cells and APCs were quickly mixed and conjugated by a brief centrifugation step. They were then incubated at 37°C for the indicated lengths of time. The cultured cells were analyzed by flow cytometry and the percentage of T cells forming conjugates (both CFSE and DiD staining) was calculated.
Formation of immunological synapses by memory T cells and SEB-bearing APCs
We assessed SEB-mediated immunological synapse formation. In our previous study, which examined OVA-mediated conjugates, we showed that the mature synapse, defined here as the TCR molecules partitioned to the c-SMAC and LFA-1 molecules partitioned to the peripheral SMAC (p-SMAC), formed more quickly in memory cells than in naive cells [22]. In naive cells, the transition from the immature (LFA-1 in the c-SMAC, flanked by the TCR) to the mature immune synapse occurred over 30 min following initial cell:cell contact, whereas in memory cells mature immune synapses were formed within 5 min. Further, our previous study confirmed a report on Lck signaling [19] by demonstrating that, in naive cells, Lck was activated in the immature p-SMAC and its signaling was completed before the formation of the mature synapse [22]. This was shown by immuno-staining, using a site-specific Ab directed against a key phosphotyrosine residue (Y394) on Lck [19]. We also found that, in contrast, Lck kinase activity was retained in the mature synapse of memory cells. In the current study, we observed a similar pattern of protein segregation in cognate interactions mediated by SEB. Immuno-staining of TCR and LFA-1 showed that the TCR moved to the c-SMAC faster in SEB-mediated immune synapses of memory cells (5–10 min), as compared to naive cells (30 min) (data not shown). For both of these cell types, these kinetics were similar to that previously observed in OVA-mediated conjugates [22]. In subsequent experiments, the pattern of TCR localization was used to indicate maturation of the immune synapse.
SEB effectively elicits early TCR proximal signaling, including CD3ζ chain phosphorylation, and, Lck phosphorylation and activation [7]. When we examined conjugates formed between either naive or memory CD4 T cells and SEB-bearing APCS, we found that TCR and Lck both co-localized in the immune synapses (Figure 3). Further, in both T cell populations, immuno-staining with an anti-Y394 Ab indicated the presence of pLck, indicating the effective activation of Lck. Similar to the pattern that we had observed in OVA-mediated interactions, Lck signaling began before mature synapses had formed in naive cells, with co-localization of TCR, Lck, and pLck in the pSMAC. Lck signaling terminated prior to mature synapse formation (>15 min), so that only TCR and Lck were found in the c-SMAC. In contrast, the rapid formation of mature immune synapses in memory cells led to co-localization of TCR, Lck, and pLck in the SEB-induced mature c-SMAC.
Figure 3. SEB mediates early immune synapse formation in naive and memory CD4 T cells.

Conjugates were formed between DO11.10 naive and memory CD4 T cells and APCs pulsed with SEB. At the indicated times, the conjugates were assessed, to determine the presence of the TCR, Lck, and pLck. Shown are the fluorescence images along the x-y axis. The two far-right columns in both panels show fluorescence overlays and the DIC images of the conjugates, respectively. Data are representative of 3 separate experiments, examining a minimum of 50 conjugates per experiment.
Given that prior studies showed that SEB induced Lck activation but not ZAP-70 activation [7], we next examined whether ZAP-70 translocated with the TCR into the immune synapse. In immunological synapses formed by naive cells, SEB caused ZAP-70 to migrate to the immune synapse. Immuno-staining showed that both the TCR and ZAP-70 were initially found co-localized to the p-SMAC where tyrosine phosphorylation by Lck of CD3ζ and then of bound ZAP-70, presumably occurred (Figure 4). Additional immuno-staining with a site-specific mAb, specifically targeting phosphorylated ZAP-70 (Tyr319), showed that activation of ZAP-70 (pZAP-70) did begin in the immature p-SMAC, and that signaling continued through synapse maturation, resulting in TCR/pZAP-70 co-localization in the mature c-SMAC. The kinetics and immuno-staining patterns were again similar to those we had previously observed for naive cells responding to OVA [22]. In that study, we also found that TCR, ZAP-70, and pZAP-70 in the c-SMAC of OVA-stimulated memory T cells. In contrast, when we examined ZAP-70 in SEB-mediated memory cell conjugates, we found a different pattern. Although the TCR moved into the c-SMACs formed by memory cells, ZAP-70 did not appear to migrate to the synapse at all (Figure 4). Not surprisingly, given that ZAP-70 was not localized with TCR and Lck, activation was impaired, given that pZAP-70 was not detected in individual memory cells.
Figure 4. ZAP-70 is excluded from the immune synapses of SEB-mediated memory cell conjugates.

Conjugates were formed between DO11.10 naive and memory CD4 T cells, and APCs pulsed with SEB. At the indicated times, the conjugates were assessed for the presence of the TCR, ZAP-70 and pZAP-70. The two far-right columns in both panels show fluorescence overlays and the DIC images of the conjugates, respectively. Beneath the x-y images is a three-dimensional reconstruction along the x-z axis of a mature immune synapse. Data are representative of 3 separate experiments, examining a minimum of 50 conjugates per experiment.
A consequence of the failed ZAP-70 activation is the abrogation of downstream signaling. For example, we showed earlier that LAT and PLC-γwere not phosphorylated in memory cells responding to SEB, and consequently, PKCθ was not activated [7]. Likewise, in SEB-mediated memory cell conjugates, in contrast to OVA-mediated conjugates [22] or naive cell conjugates, PKC-θ was not recruited to the c-SMAC of the memory cell mature synapse (Figure 5). Note that in naive cells, where the immature synapse was seen at 5 minutes after stimulation, PKCθ had not migrated to the immune synapse. This was expected, given that ZAP-70 activation occurs in the mature synapse. As seen at 30 min following SEB stimulation, PKCθ did localize to mature immune synapse of naive cells. In contrast, PKCθ was not observed in the memory cell immune synapse at any time point. Hence, in SEB-mediated memory cell conjugates, immune synapse formation proceeds normally only until the point of ZAP-70 recruitment.
Figure 5. PKCθ translocation is impaired in memory CD4 cells exposed to SEB.

Conjugates were formed between DO11.10 naive and memory CD4 T cells, and APCs pulsed with SEB. At the indicated times, the conjugates were assessed for the presence of the TCR, LFA-1, and PKCθ. The two far-right columns in both panels show fluorescence overlays and the DIC images of the conjugates, respectively. Data are representative of 2 experiments, examining a minimum of 50 conjugates per experiment.
Immunological synapse formation by Fyn-deficient memory T cells and SEB-bearing APCs
SEB leads to a specific hyperactivation of Fyn kinase in mouse memory cells [7]. Further, the inhibition or absence of Fyn kinase is able to restore memory cell proliferation and prevent anergy [28]. Immunoblot studies showed that when Fyn was absent, complexes of pZAP-70 and CD3ζ could be precipitated from memory cells exposed to SEB [28]. We conclude from these studies that anergy induction is dependent upon Fyn signaling. Here, we determined whether the absence of Fyn also permitted ZAP-70 to migrate to the immunological synapse. Hence, we examined conjugates formed between SEB-bearing APCs and naive or memory T cells that we obtained from DO11.10 × Fyn−/− mice. The loss of Fyn did not interfere with normal immune synapse production, given that synapses in OVA or SEB-mediated conjugates, using wild-type or Fyn-deficient naive cells naive cells, were similar (data not shown). Similarly, we observed the same pattern and kinetics of immune synapse formation in memory cells from wild-type and Fyn-deficient DO11.10 mice in response to OVA (data not shown). However, in contrast to our observation with wild-type memory cells (Figure 4), we found that SEB could efficiently promote the recruitment of ZAP-70 to the immune synapse of Fyn-deficient memory cells (Figure 6). Further, ZAP-70 activation was restored by elimination of Fyn, given that we found both ZAP-70 and pZAP-70, along with the TCR, in the c-SMACs of the mature synapses of SEB-mediated conjugates (Figure 6). Hence, the increased activation of Fyn, observed during SEB stimulation of memory cells, prevents ZAP-70 from migrating to the immunological synapse, where it can bind to CD3ζ.
Figure 6. ZAP-70 migration to the immune synapse of SEB-mediated memory cell conjugates is regulated by fyn.

Conjugates were formed between DO11.10 × Fyn−/− memory CD4 T cells and APCs pulsed with SEB. At the indicated times, the conjugates were assessed for the presence of the TCR, ZAP-70 and pZAP-70. The two far-right columns show fluorescence overlays and the DIC images of the conjugates, respectively. Beneath the x-y images is a three-dimensional reconstruction along the x-z axis of a mature SEB-mediated immune synapse from naive or memory CD4 T cells. Data are representative of 3 separate experiments, examining a minimum of 50 conjugates per experiment.
Fyn itself was recruited to the c-SMAC and the TCR signaling complex during T cell-APC interactions, regardless of whether or not the T cell was naive or memory or if the stimulus was OVA or SEB (Figure 7). Further, the E3 ubiquitin ligase c-Cbl was also recruited to the synapse. This observation was of interest given that c-Cbl was strongly activated and associated with Fyn in anergic, but not stimulated memory cells [28]. Likewise, c-Cbl recruitment was more easily detected in the SEB-mediated memory cell synapses. For example, Figure 7 shows c-Cbl in the synapse of naive and memory cells using the same intensity and brightness settings. Finally, c-Cbl translocation to the c-SMAC was dependent upon Fyn, given that we were unable to detect c-Cbl in the synapses of DO11 × Fyn−/− memory cells (data not shown).
Figure 7. Fyn and c-Cbl migrate to the immune synapses of CD4 T cell conjugates.

Conjugates were formed between DO11.10 naive and memory CD4 T cells and APCs pulsed with SEB. After the 30 min (naive) or 5 min (memory), the mature synapses in the conjugates were assessed for the presence of the TCR, Fyn, and c-Cbl. The two far-right columns show fluorescence overlays and the DIC images of the conjugates, respectively. Data are representative of 2 separate experiments, examining a minimum of 50 conjugates per experiment.
Composition of lipid rafts of SEB-stimulated memory cells
TCR-mediated signal transduction is dependent, not only upon the specific molecular organization of the immunological synapses, but also upon the proper organization of lipid rafts. Several important molecules are either constitutively associated with, or else migrate to, lipid rafts, to create signaling platforms [9;11;38;44]. For example, the TCR on memory cells is constitutively associated with lipid rafts, whereas, the TCR on naive cells tightly associates with rafts only upon cell activation [22]. ZAP-70 is not associated with lipid rafts in membranes of resting naive cells or memory cells; however, if the cells are activated by anti-CD3 (naive cells) [11] or an agonist peptide, such as OVA (naive cells and memory cells), ZAP-70 migrates to lipid rafts (data not shown). Proteins associated with lipid rafts are typically identified by their resistance to extraction by nonionic detergents. We have previously used a microscopy technique [11] to identify lipid rafts and raft-associated proteins [22]. Single T cells or cognate T cell:APC conjugates were exposed to Triton X-100 (TX-100), and non-raft-associated proteins were then extracted from the cells. Lipid rafts, identified by rhodamine-cholera toxin B subunit (CTX-B), which binds to GM1 gangliosides [45;46], were visualized by fluorescence microscopy.
Using this approach in the current study, we examined the lipid rafts of naive and memory T cells that were exposed to SEB. By pre-labeling the cells with CTX-B, we observed that SEB induced the accumulation of lipid rafts in the immunological synapse of both T cell types. As previously noted, in resting memory CD4 cells, the TCR is a constitutive raft-associated protein ([22], and Figure 8). Hence, without stimulation the TCR was resistant to detergent extraction and could be immuno-stained after membrane solubilization with TX-100. In contrast, we did not observe TCR staining on unstimulated naive cells after detergent extraction. For both cell types, stimulation by OVA [22] or SEB (Figure 8), resulted in tight association of the TCR with lipid rafts in the immune synapses. We next examined the relationship between ZAP-70 and lipid rafts in response to stimulation with superantigen. In SEB-mediated naive cell conjugates, ZAP-70 partitioned to the lipid rafts and became resistant to detergent extraction (Figure 8). In contrast, while ZAP-70 translocated to lipid rafts, when memory cells were stimulated by OVA (data not shown), it remained excluded from lipid rafts during stimulation by SEB (Figure 8). Hence, exposure of memory CD4 cells to SEB prevented the movement of ZAP-70 to both the lipid raft platform and the synapse microdomain that contained the TCR/CD3 complex and Lck.
Figure 8. ZAP-70 is excluded from membrane lipid rafts of SEB-treated memory CD4 T cells.

DO11.10 naive and memory CD4 T cells were labeled with rhodamine-CTX-B (GM1) and were analyzed before or after conjugation with SEB-pulsed APCs. The cells either were fixed and then permeabilized for staining (control) or permeabilized with 1% Triton X-100, to solubilize and remove non-raft proteins. The conjugates were assessed to determine the presence of the TCR and ZAP-70. The two far-right columns in both panels show fluorescence overlays and the DIC images of the conjugates, respectively. Data are representative of 3 separate experiments, examining a minimum of 50 cells or conjugates per experiment.
Finally, we determined if Fyn regulated the association between ZAP-70 and lipid rafts, when memory cells were stimulated by SEB. Naive and memory cells were obtained from DO11.10 × Fyn−/− mice and conjugates were formed with APCs presenting SEB. As shown above (Figure 6), the absence of Fyn restored that ability of ZAP-70 to co-localize with the TCR in the c-SMAC of the immune synapse. Further, in both naive and memory cells ZAP-70 was recruited to the lipid rafts, given that it was resistant to detergent extraction (Figure 9). Hence, heightened stimulation of Fyn by SEB appears to prevent ZAP-70 recruitment to both lipid rafts and the immune synapse. Given that the absence of Fyn leads to a functional response to SEB by memory cells [28], we conclude that regulation of ZAP-70 recruitment is the primary mechanism underlying anergy induction.
Figure 9. Fyn controls ZAP-70 recruitment to membrane lipid rafts of SEB-treated memory CD4 T cells.

DO11.10 × Fyn−/− naive and memory CD4 T cells were labeled with rhodamine-CTX-B (GM1) and were analyzed before or after conjugation with SEB-pulsed APCs. The cells either were fixed and then permeabilized for staining (control) or permeabilized with 1% Triton X-100, to solubilize and remove non-raft proteins. The conjugates were assessed to determine the presence of the TCR and ZAP-70. The two far-right columns in both panels show fluorescence overlays and the DIC images of the conjugates, respectively. Data are representative of 2 experiments, examining a minimum of 50 cells or conjugates per experiment.
DISCUSSION
The current study extends our previous demonstrations of novel regulatory processes in CD4 memory T cells. Using both normal and TCR transgenic mice, we earlier identified a mechanism contributing to SEB-induced anergy [7]. TCR proximal signaling was impaired because ZAP-70 did not bind to the activated TCR/CD3 complex (pCD3ζ) and, therefore, was not phosphorylated/activated by Lck [7]. This, in turn, led to an abrogation of downstream signaling. We now show that membrane TCR signaling complexes are likewise unique on SEB-treated memory cells and that the major defect also lies in the ability of ZAP-70 to migrate to the same membrane compartments as the TCR/CD3 complex. We conclude that the sequestration of ZAP-70 is a central control point for superantigen-induced anergy.
We note that in our examinations of naive DO11.10 cells we find that peptide (OVA) and superantigen elicit similar rates of immune synapse formation and a similar synapse structure. Further, other than a more rapid synapse formation, peptide-elicited memory cell synapses are also similar. We observed aberrant synapse formation only when memory cells were exposed to superantigen, consistent with consistent with the hypothesis that formation of signaling clusters regulates activation versus anergy. A main structural difference between naive and memory cell synapses was that, for either peptide [22] or SEB (unpublished observations), the CD45 molecule co-localized with the TCR in the c-SMAC of memory cell synapses while remaining in the p-SMAC of naive cell synapses. Further, CD45 was constitutively associated with lipid rafts in memory cells but it was excluded from lipid rafts in naive cells. We previously speculated that co-localization of the TCR and CD45 (and CD4) might differentially regulate memory cell activation to peptide [22]. The constitutive clustering of the three signaling molecules is a main factor in failure of memory cells to respond to stimulation with soluble anti-CD3 [24;47–49]. Considering that Fyn is also a constitutive raft protein, it is tempting to speculate that the closer proximity to raft-associated CD45 in memory cells facilitates increased Fyn activation in response to SEB. How the superantigen, as opposed to peptide antigen, might favor Fyn activation also remains unclear.
Productive immune synapses are clearly necessary for cell activation [50] and altered synapses have been described in a variety of cell inactivation, ignorance, and anergy models. While our observations on synapses of anergic memory CD4 cells are largely consistent with earlier studies, we do note the some differences, suggesting that synapse formation can be impaired at a number of levels. Hence, in contrast to our study, others have shown that anergy can be characterized by a reduction in conjugate formation (binding), failed recruitment of the TCR or lck, or even lipid rafts to the synapse [18;25;51] [52]. Such differences may be attributed to different points in blocking TCR proximal signaling by the different stimuli used to induce anergy in each study. Or, different blocks in synapse formation may be a function of the different cell type (CD8 versus CD4) or differentiation state (naive versus memory). Our model may be unique in that the anergic signal is applied directly to resting memory cells, whereas in most other studies the tolerized cells were naive cells at the time of anergy induction. The distinct lipid raft and normal c-SMAC structures formed by memory cells might facilitate novel signaling molecule interactions leading to hyperactivation of Fyn and anergy.
Our observation that ZAP-70 is excluded from both of the c-SMAC and lipid rafts is consistent with our previous finding that CD3ζand ZAP-70 did not co-immunoprecipitate upon stimulation by SEB [7]. We suggest that anergy is induced and maintained by this spatial separation. At this time, we have not determined if ZAP-70 first associates with the lipid rafts and then migrates to the immune synapse or whether it binds first to CD3ζand thus becomes raft associated. At least one study has shown that anergy is initiated by modulating lipid raft structure. Hundt et observed segregation of the adapter protein LAT from lipid rafts, via depalmitoylation, in CD4 cells induced to become anergic by ionomycin [53]. It is tempting to speculate that excessive Fyn signaling, prompted by SEB, causes exclusion of ZAP-70 from lipid rafts, preventing movement of ZAP-70 into the immune synapse where it can bind to CD3ζ.
Memory cell hyporesponsiveness to SEB, and the causative signaling defects are reversed by inhibition or elimination of the src kinase Fyn [28]. Here we provide data to suggest that Fyn signaling regulates compartmentalization of ZAP-70, since Fyn−/− memory cells form productive immune synapses in SEB-mediated cell conjugates. Further, stimulation of Fyn−/− memory cells with SEB led to the translocation of ZAP-70 to lipid rafts. We conclude that anergy is imposed through segregation of this important TCR proximal signaling protein. To our knowledge, this is the first report the Fyn activation can lead to alterations in immune synapse structure or determine the lipid raft attachment of a major signaling protein. How SEB specifically promotes Fyn signaling and compartmentalization of ZAP-70 is unclear at present. It may be that a Fyn-mediated regulatory pathway alters the attachment of ZAP-70 to membrane cytoskeletal components or directly to lipid rafts. A candidate for a regulator of ZAP-70 dynamics is c-Cbl [28]. C-Cbl is inducibly phosphorylated by Fyn [54–56] and can, in turn, modulate ZAP-70 through a number of mechanisms, including preventing its binding to pCD3ζ [57]. Further, there is a strong interaction between Fyn and c-Cbl specifically in memory cells that have been exposed to SEB [28]. This interaction is also observed at the membrane level as there is a strong, Fyn-dependent translocation of c-Cbl to the cSMAC in SEB-mediated memory cell conjugates. A recent study also observed an accumulation of c-Cbl (and also Cbl-b) at the anergic synapse [58]. We have not examined Cbl-b, which appeared to play a more prominent role in the study by Doherty et al, but we do note that memory cells from Cbl-b-deficient mice, like their wild type counterpart, become anergic in response to SEB (unpublished observations). Hence, c-Cbl likely plays the important role in our model. We speculate that c-Cbl prevents the recruitment of ZAP-70 to the TCR through ubiquitinylation and by interfering with its access to the synapse.
In summary, based upon our current data, we propose that triggering of the memory CD4 cell TCR leads to heightened Fyn activation. The excessive Fyn signaling changes the accessibility of ZAP-70 to the same membrane compartments containing the TCR/CD3 complex and thus prevents the formation of productive proximal signaling complexes. Consequently, neither SEB nor, subsequently, OVA can complete the signaling paths essential for cell proliferation and the cells are thus anergic. It is tempting to speculate that Fyn activates c-Cbl and that ubiquitinylation of ZAP-70 causes its exclusion from lipid rafts, preventing movement into the immune synapse where ZAP-70 can bind to CD3ζ.
HIGHLIGHTS.
A bacterial toxin (SEB) causes Fyn kinase-dependent memory CD4 T cell inactivation (anergy)
We show that a memory cell anergy is associated with a failure to build productive immune synapses
SEB-mediated TCR signaling promotes TCR and Lck kinase recruitment to the mature immune synapse
We show a defect in the recruitment of ZAP-70 to the c-SMAC and a failure of ZAP-70 to migrate to lipid rafts
Fyn-deficient memory cells are able to recruit ZAP-70 to lipid rafts and to the c-SMAC
ACKNOWLEDGMENTS
The authors would like to thank Mr. K. Moynehan, Mr. G. Pasos, and Ms. E. Filippelli for their expert technical assistance. We also acknowledge the Wadsworth Center Immunology Core, the Wadsworth Center Peptide Synthesis Core, and the David Axelrod Institute Microscopy Core. We especially thank Dr. A. Ramsingh for her critical reading and Ms. A. Verschoor for her help in the preparation of this manuscript. This work was supported by National Institutes Health grant # AI35583.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Abbreviations used: SEB, staphylococcal enterotoxin B; SMAC, supramolecular, activation cluster; TX-100, Triton X-100; CT-B, cholera toxin-B subunit
DISCLOSURES
The authors declare no conflict of interest or financial interests.
REFERENCES
- 1.Sprent J, Surh CD. T cell memory. Annu.Rev.Immunol. 2002;20:551–579. doi: 10.1146/annurev.immunol.20.100101.151926. [DOI] [PubMed] [Google Scholar]
- 2.Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu.Rev.Immunol. 2004;22:745–763. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz RH. T cell anergy. Annu.Rev.Immunol. 2003;21:305–334. doi: 10.1146/annurev.immunol.21.120601.141110. [DOI] [PubMed] [Google Scholar]
- 4.Farber DL. Biochemical signaling pathways for memory T cell recall. Semin.Immunol. 2009;21:84–91. doi: 10.1016/j.smim.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farber DL. T cell memory: heterogeneity and mechanisms. Clin.Immunol. 2000;95:173–181. doi: 10.1006/clim.2000.4858. [DOI] [PubMed] [Google Scholar]
- 6.Zamoyska R. Superantigens: supersignalers? Sci.STKE. 2006;2006:e45. doi: 10.1126/stke.3582006pe45. [DOI] [PubMed] [Google Scholar]
- 7.Watson AR, Lee WT. Defective T cell receptor-mediated signal transduction in memory CD4 T lymphocytes exposed to superantigen or anti-T cell receptor antibodies. Cell Immunol. 2006;242:80–90. doi: 10.1016/j.cellimm.2006.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watson AR, Mittler JN, Lee WT. Staphylococcal enterotoxin B induces anergy to conventional peptide in memory T cells. Cell Immunol. 2003;222:144–155. doi: 10.1016/s0008-8749(03)00117-5. [DOI] [PubMed] [Google Scholar]
- 9.Xavier R, Seed B. Membrane compartmentation and the response to antigen. Curr.Opin.Immunol. 1999;11:265–269. doi: 10.1016/s0952-7915(99)80043-0. [DOI] [PubMed] [Google Scholar]
- 10.Brown DA, London E. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J.Biol.Chem. 2000;275:17221–17224. doi: 10.1074/jbc.R000005200. [DOI] [PubMed] [Google Scholar]
- 11.Janes PW, Ley SC, Magee AI, Kabouridis PS. The role of lipid rafts in T cell antigen receptor (TCR) signalling. Semin.Immunol. 2000;12:23–34. doi: 10.1006/smim.2000.0204. [DOI] [PubMed] [Google Scholar]
- 12.Montixi C, Langlet C, Bernard AM, Thimonier J, Dubois C, Wurbel MA, Chauvin JP, Pierres M, He HT. Engagement of T cell receptor triggers its recruitment to low-density detergent-insoluble membrane domains. EMBO J. 1998;17:5334–5348. doi: 10.1093/emboj/17.18.5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bi K, Altman A. Membrane lipid microdomains and the role of PKCtheta in T cell activation. Semin.Immunol. 2001;13:139–146. doi: 10.1006/smim.2000.0305. [DOI] [PubMed] [Google Scholar]
- 14.Langlet C, Bernard AM, Drevot P, He HT. Membrane rafts and signaling by the multichain immune recognition receptors. Curr.Opin.Immunol. 2000;12:250–255. doi: 10.1016/s0952-7915(00)00084-4. [DOI] [PubMed] [Google Scholar]
- 15.Dykstra M, Cherukuri A, Sohn HW, Tzeng SJ, Pierce SK. Location is everything: lipid rafts and immune cell signaling. Annu.Rev.Immunol. 2003;21:457–481. doi: 10.1146/annurev.immunol.21.120601.141021. [DOI] [PubMed] [Google Scholar]
- 16.Bromley SK, Burack WR, Johnson KG, Somersalo K, Sims TN, Sumen C, Davis MM, Shaw AS, Allen PM, Dustin ML. The immunological synapse. Annu.Rev.Immunol. 2001;19:375–396. doi: 10.1146/annurev.immunol.19.1.375. [DOI] [PubMed] [Google Scholar]
- 17.Delon J, Germain RN. Information transfer at the immunological synapse. Curr.Biol. 2000;10:R923–R933. doi: 10.1016/s0960-9822(00)00870-8. [DOI] [PubMed] [Google Scholar]
- 18.Monks CR, Freiberg BA, Kupfer H, Sciaky N, Kupfer A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature. 1998;395:82–86. doi: 10.1038/25764. [DOI] [PubMed] [Google Scholar]
- 19.Lee KH, Holdorf AD, Dustin ML, Chan AC, Allen PM, Shaw AS. T cell receptor signaling precedes immunological synapse formation. Science. 2002;295:1539–1542. doi: 10.1126/science.1067710. [DOI] [PubMed] [Google Scholar]
- 20.Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, Dustin ML. The immunological synapse: a molecular machine controlling T cell activation. Science. 1999;285:221–227. doi: 10.1126/science.285.5425.221. [DOI] [PubMed] [Google Scholar]
- 21.Dianzani U, Luqman M, Rojo J, Yagi J, Baron JL, Woods A, Janeway CA, Jr, Bottomly K. Molecular associations on the T cell surface correlate with immunological memory. Eur.J.Immunol. 1990;20:2249. doi: 10.1002/eji.1830201014. [DOI] [PubMed] [Google Scholar]
- 22.Watson AR, Lee WT. Differences in signaling molecule organization between naive and memory CD4+ T lymphocytes. J.Immunol. 2004;173:33–41. doi: 10.4049/jimmunol.173.1.33. [DOI] [PubMed] [Google Scholar]
- 23.Balamuth F, Leitenberg D, Unternaehrer J, Mellman I, Bottomly K. Distinct patterns of membrane microdomain partitioning in Th1 and th2 cells. Immunity. 2001;15:729–738. doi: 10.1016/s1074-7613(01)00223-0. [DOI] [PubMed] [Google Scholar]
- 24.Leitenberg D, Balamuth F, Bottomly K. Changes in the T cell receptor macromolecular signaling complex and membrane microdomains during T cell development and activation. Semin.Immunol. 2001;13:129–138. doi: 10.1006/smim.2000.0304. [DOI] [PubMed] [Google Scholar]
- 25.Ehrlich LI, Ebert PJ, Krummel MF, Weiss A, Davis MM. Dynamics of p56lck translocation to the T cell immunological synapse following agonist and antagonist stimulation. Immunity. 2002;17:809–822. doi: 10.1016/s1074-7613(02)00481-8. [DOI] [PubMed] [Google Scholar]
- 26.Lee WT, Cole-Calkins J, Street NE. Memory T cell development in the absence of specific antigen priming. J.Immunol. 1996;157:5300–5307. [PubMed] [Google Scholar]
- 27.Chan AC, Desai DM, Weiss A. The role of protein tyrosine kinases and protein tyrosine phosphatases in T cell antigen receptor signal transduction. Annu.Rev.Immunol. 1994;12:555–592. doi: 10.1146/annurev.iy.12.040194.003011. [DOI] [PubMed] [Google Scholar]
- 28.Watson AR, Janik DK, Lee WT. Superantigen-induced CD4 memory T cell anergy. I. Staphylococcal enterotoxin B induces Fyn-mediated negative signaling. Cell Immunol. 2012 doi: 10.1016/j.cellimm.2012.02.003. (In press), (PM:22386537). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sloan-Lancaster J, Shaw AS, Rothbard JB, Allen PM. Partial T cell signaling: Altered phospho-zeta and lack of zap70 recruitment in APL-induced T cell anergy. Cell. 1994;79:913–922. doi: 10.1016/0092-8674(94)90080-9. [DOI] [PubMed] [Google Scholar]
- 30.Salmond RJ, Filby A, Qureshi I, Caserta S, Zamoyska R. T-cell receptor proximal signaling via the Src-family kinases, Lck and Fyn influences T-cell activation, differentiation, and tolerance. Immunol.Rev. 2009;228:9–22. doi: 10.1111/j.1600-065X.2008.00745.x. [DOI] [PubMed] [Google Scholar]
- 31.Murphy KM, Heimberger AB, Loh DY. Induction by antigen of intrathymic apoptosis of CD4+CD8+TCRlo thymocytes in vivo. Science. 1990;250:1720–1722. doi: 10.1126/science.2125367. [DOI] [PubMed] [Google Scholar]
- 32.Haskins K, Kubo R, White J, Pigeon M, Kappler J, Marrack P. The major histocompatibility complex-restricted antigen receptor on T cells. I. Isolation with a monoclonal antibody. J.Exp.Med. 1983;157:1149–1169. doi: 10.1084/jem.157.4.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee WT, Shiledar-Baxi V, Winslow GM, Mix D, Murphy DB. Self-restricted dual receptor memory T cells. J.Immunol. 1998;161:4513–4519. [PubMed] [Google Scholar]
- 34.Stein PL, Lee HM, Rich S, Soriano P. pp59fyn mutant mice display differential signaling in thymocytes and peripheral T cells. Cell. 1992;70:741–750. doi: 10.1016/0092-8674(92)90308-y. Ref Type: Generic. [DOI] [PubMed] [Google Scholar]
- 35.Birkeland ML, Metlay J, Sanders VM, Fernandez-Botran R, Vitetta ES, Steinman RM, Pure E. Epitopes on CD45R (T200) molecules define differentiation antigens on murine B and T lymphocytes. J.Mol.Cell.Immunol. 1988;4:71–85. [PubMed] [Google Scholar]
- 36.Lee WT, Yin X-M, Vitetta ES. Functional and ontogenetic analysis of murine CD45Rhi and CD45Rlo CD4+ T cells. J.Immunol. 1990;144:3288–3295. [PubMed] [Google Scholar]
- 37.Lyons AB, Parish CR. Determination of lymphocyte division by flow cytometry. J.Immunol.Methods. 1994;171:131–137. doi: 10.1016/0022-1759(94)90236-4. [DOI] [PubMed] [Google Scholar]
- 38.Janes PW, Ley SC, Magee AI. Aggregation of lipid rafts accompanies signaling via the T cell antigen receptor. J.Cell Biol. 1999;147:447–461. doi: 10.1083/jcb.147.2.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kozak SL, Heard JM, Kabat D. Segregation of CD4 and CXCR4 into distinct lipid microdomains in T lymphocytes suggests a mechanism for membrane destabilization by human immunodeficiency virus. J.Virol. 2002;76:1802–1815. doi: 10.1128/JVI.76.4.1802-1815.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee WT, Vitetta ES. Memory T cells are anergic to the superantigen, staphylocoocal enterotoxin B. J.Exp.Med. 1992;176:575–580. doi: 10.1084/jem.176.2.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee WT, Thrush GR, Vitetta ES. Staphylococcal enterotoxin B induces the expression of activation markers on murine memory T cells in the absence of proliferation or lymphokine secretion. Cell.Immunol. 1995;162:26–32. doi: 10.1006/cimm.1995.1047. [DOI] [PubMed] [Google Scholar]
- 42.Delon J, Bercovici N, Liblau R, Trautmann A. Imaging antigen recognition by naive CD4+ T cells: compulsory cytoskeletal alterations for the triggering of an intracellular calcium response. Eur J Immunol. 1998;28:716–729. doi: 10.1002/(SICI)1521-4141(199802)28:02<716::AID-IMMU716>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 43.Van der Merwe PA. Formation and function of the immunological synapse. Curr.Opin.Immunol. 2002;14:293–298. doi: 10.1016/s0952-7915(02)00350-3. [DOI] [PubMed] [Google Scholar]
- 44.Viola A, Schroeder S, Sakakibara Y, Lanzavecchia A. T lymphocyte costimulation mediated by reorganization of membrane microdomains. Science. 1999;283:680–682. doi: 10.1126/science.283.5402.680. [DOI] [PubMed] [Google Scholar]
- 45.Fra AM, Williamson E, Simons K, Parton RG. Detergent-insoluble glycolipid microdomains in lymphocytes in the absence of caveolae. J.Biol.Chem. 1994;269:30745–30748. [PubMed] [Google Scholar]
- 46.Harder T, Scheiffele P, Verkade P, Simons K. Lipid domain structure of the plasma membrane revealed by patching of membrane components. J.Cell Biol. 1998;141:929–942. doi: 10.1083/jcb.141.4.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leitenberg D, Boutin Y, Lu DD, Bottomly K. Biochemical association of CD45 with the T cell receptor complex: regulation by CD45 isoform and during T cell activation. Immunity. 1999;10:701–711. doi: 10.1016/s1074-7613(00)80069-2. [DOI] [PubMed] [Google Scholar]
- 48.Farber DL. Cutting edge commentary: Differential TCR signaling and the generation of memory T cells. J.Immunol. 1998;160:535–539. [PubMed] [Google Scholar]
- 49.Farber DL, Acuto O, Bottomly K. Differential T cell receptor-mediated signaling in naive and memory CD4 T cells. Eur J Immunol. 1997;27:2094–2101. doi: 10.1002/eji.1830270838. [DOI] [PubMed] [Google Scholar]
- 50.Huppa JB, Gleimer M, Sumen C, Davis MM. Continuous T cell receptor signaling required for synapse maintenance and full effector potential. Nat.Immunol. 2003;4:749–755. doi: 10.1038/ni951. [DOI] [PubMed] [Google Scholar]
- 51.Zambricki E, Zal T, Yachi P, Shigeoka A, Sprent J, Gascoigne N, McKay D. In vivo anergized T cells form altered immunological synapses in vitro. Am.J.Transplant. 2006;6:2572–2579. doi: 10.1111/j.1600-6143.2006.01517.x. [DOI] [PubMed] [Google Scholar]
- 52.Ise W, Nakamura K, Shimizu N, Goto H, Fujimoto K, Kaminogawa S, Hachimura S. Orally tolerized T cells can form conjugates with APCs but are defective in immunological synapse formation. J.Immunol. 2005;175:829–838. doi: 10.4049/jimmunol.175.2.829. [DOI] [PubMed] [Google Scholar]
- 53.Hundt M, Tabata H, Jeon MS, Hayashi K, Tanaka Y, Krishna R, De GL, Liu YC, Fukata M, Altman A. Impaired activation and localization of LAT in anergic T cells as a consequence of a selective palmitoylation defect. Immunity. 2006;24:513–522. doi: 10.1016/j.immuni.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 54.Deckert M, Elly C, Altman A, Liu YC. Coordinated regulation of the tyrosine phosphorylation of Cbl by Fyn and Syk tyrosine kinases. J.Biol.Chem. 1998;273:8867–8874. doi: 10.1074/jbc.273.15.8867. [DOI] [PubMed] [Google Scholar]
- 55.Hunter S, Burton EA, Wu SC, Anderson SM. Fyn associates with Cbl and phosphorylates tyrosine 731 in Cbl a binding site for phosphatidylinositol 3-kinase. J.Biol.Chem. 1999;274:2097–2106. doi: 10.1074/jbc.274.4.2097. [DOI] [PubMed] [Google Scholar]
- 56.Tezuka T, Umemori H, Fusaki N, Yagi T, Takata M, Kurosaki T, Yamamoto T. Physical and functional association of the cbl protooncogen product with an src-family protein tyrosine kinase, p53/56lyn, in the B cell antigen receptor-mediated signaling. J.Exp.Med. 1996;183:675–680. doi: 10.1084/jem.183.2.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thien CB, Bowtell DD, Langdon WY. Perturbed regulation of ZAP-70 and sustained tyrosine phosphorylation of LAT and SLP-76 in c-Cbl-deficient thymocytes. J.Immunol. 1999;162:7133–7139. [PubMed] [Google Scholar]
- 58.Doherty M, Osborne DG, Browning DL, Parker DC, Wetzel SA. Anergic CD4+ T cells form mature immunological synapses with enhanced accumulation of c-Cbl and Cbl-b. J.Immunol. 2010;184:3598–3608. doi: 10.4049/jimmunol.0902285. [DOI] [PMC free article] [PubMed] [Google Scholar]