

‘Dual antiplatelet therapy’, comprising aspirin and a P2Y12 receptor inhibitor, is firmly established for the secondary prevention of thrombotic events with the rationale that they inhibit thromboxane A2- (TxA2) and ADP-P2Y12-dependent pathways of platelet activation, respectively. We have recently reported that strong P2Y12 receptor blockade alone, however, can provide inhibition of platelet aggregation to a broad range of agonists that is not further enhanced by aspirin [1]. While the clinical relevance of these observations is unclear, we have speculated that administration of aspirin to individuals achieving sufficiently strong P2Y12 receptor blockade, has the potential to produce effects secondary to inhibition of cyclo-oxygenase at non-platelet sites, without providing additional antithrombotic activity [2]. The degree of P2Y12 pathway blockade that is achieved in clinical practice, however, is quite variable [3], reflecting both the choice of drug and large inter-individual differences in drug metabolism [4]. Here, we have extended our previous observations of the interactions between aspirin and strong P2Y12 blockade [1] by considering what additional anti-aggregatory effects aspirin provides when only partial P2Y12 blockade is achieved, which may better reflect the clinical reality of these drugs.
We measured aggregation responses of platelet-rich plasma (PRP), using 96-well plate light transmission aggregometry, as previously described [1]. Blood was collected by venepuncture into tri-sodium citrate (0.32% final) from healthy volunteers who had abstained from non-steroid anti-inflammatory drug consumption for 14 days. To model the effects of P2Y12 blockade and cyclo-oxygenase inhibition in vitro, PRP was incubated with the irreversible thienopyridine P2Y12 inhibitor, prasgurel-active metabolite (PAM; 0.1–10 μmol L−1), the reversible, cyclo-pentyl-triazolo-pyrimidine P2Y12 antagonist, ticagrelor (0.1–10 μmol L−1) and/or aspirin (1–100 μmol L−1) for 30 min at 37 °C before addition of the agonist. Additional methodological details are provided as online supplementary information.
Using this approach we determined the inhibitory potencies of ticagrelor, PAM and aspirin against aggregations induced by ADP (0.625–20 μmol L−1), the thromboxane-mimetic U46619 (0.1–30 μmol L−1) and arachidonic acid (0.1–1 mmol L−1). Both ticagrelor and PAM caused concentration-dependent inhibition of aggregations induced by ADP (Fig. S1), with ticagrelor displaying greater potency than PAM (log IC50 values for inhibition of aggregation to 20 μmol L−1 ADP: ticagrelor, −6.46; PAM, −5.64). Notably, the potency of ticagrelor, but not PAM, varied with the concentration of ADP (e.g. log IC50 values for inhibition of aggregation to 2.5 μmol L−1 ADP: ticagrelor, −7.05; PAM, −5.63). Aspirin, at concentrations up to 100 μmol L−1, was without significant effect upon ADP-induced aggregations. Ticagrelor and PAM, but not aspirin, produced complete, concentration-dependent inhibition of platelet aggregations induced by U46619 (Fig. S2) with similar potency as for inhibition of ADP-induced aggregations (log IC50 values for inhibition of aggregation to 30 μmol L−1 U46619; ticagrelor, −6.24; PAM, −5.25). This is consistent with earlier reports that the second, irreversible wave of platelet aggregation that follows TP receptor activation is dependent upon platelet-derived ADP acting upon platelet P2Y12 receptors [5]. Ticagrelor and PAM, as well as aspirin, also produced complete, concentration-dependent inhibitions of platelet aggregations induced by AA (Fig. S3; log IC50 values for inhibition of aggregation to 1 mmol L−1 AA: ticagrelor, −6.88; PAM, −6.00; aspirin, −5.20). When the production of TxA2 accompanying platelet aggregation induced by AA was measured (by immunoassay for the levels of TxB2), ticagrelor, PAM and aspirin were all found to cause concentration-dependent reductions in TxA2 formation (Fig. S3; log IC50 values for inhibition of aggregation to 1 mmol L−1 AA: ticagrelor, −6.88; PAM, −5.985; aspirin, −5.51). This is in agreement with our early findings [1, 6], and indicates that P2Y12 receptors are important in supporting both the activation mechanisms of platelets that drive TxA2 formation and pathways downstream of the TP receptor.
To explore further the interactions between P2Y12 receptors and the TxA2 system in platelets, we examined the effect of aspirin on aggregation induced by a range of agonists in the presence of concentrations of ticagrelor or PAM producing different degrees of partial P2Y12 blockade. From the inhibitor curves to ADP described above, concentrations of ticagrelor showing approximate IC5 (0.03 μmol L−1), IC10 (0.1 μmol L−1), IC50 (0.3 μmol L−1) and IC90 effects (3 μmol L−1) were combined with 30 μmol L−1 aspirin, a concentration approximately equivalent to the peak plasma levels following ingestion of a 75–100 mg dose of aspirin. In these experiments, responses to AA (Fig. 1A) were found to be completely inhibited by ticagrelor at the higher two concentrations (representing ∼IC50 and IC90 for ADP-induced aggregation) without the need for aspirin. The lower two concentrations of ticagrelor (representing ∼IC5 and IC10 for ADP-induced aggregation) also produced substantial, but incomplete, inhibitions (Table S1). Ticagrelor also inhibited aggregations induced by ADP, collagen, epinephrine, the PAR-1 activating peptide, TRAP-6 (SFLLRN-amide) and U46619, in a concentration-dependent manner (Fig. 1).
Figure 1.
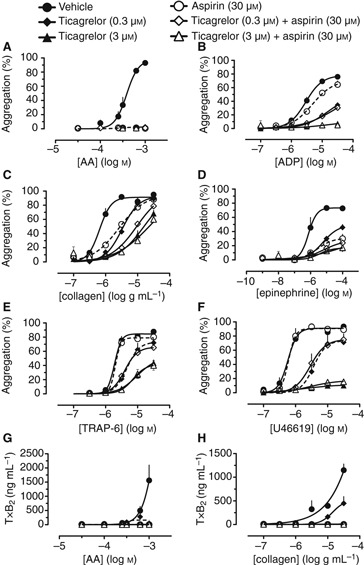
Concentration-response curves for the inhibition by combinations of ticagrelor (0.3 or 3 μmol L−1) and aspirin (30 μmol L−1) of platelet aggregations induced by (A) arachidonic acid (AA), (B) ADP, (C) collagen, (D) epinephrine, (E) TRAP-6 or (F) U46619, and of platelet TxB2 formation induced by (G) AA and (H) collagen. n = 4.
When applied alone, aspirin inhibited aggregations induced by AA (Fig. 1A), collagen (Fig. 1C) and epinephrine (Fig. 1D), and showed a weak effect against ADP (Fig. 1B) but did not alter aggregations induced by TRAP-6 (Fig. 1E) or U46619 (Fig. 1F). In contrast, aspirin did augment the anti-aggregatory effects of the lower three concentrations of ticagrelor (achieving incomplete P2Y12 inhibition) against both collagen (Fig. 1C) and epinephrine (Fig. 1D). In the presence of the highest tested concentration of ticagrelor (3 μmol L−1; ∼IC90 for ADP-induced aggregation), aspirin provided no additional anti-aggregatory effects to those of ticagrelor against aggregations to any agonist (Fig. 1). In agreement with earlier experiments, production of TxA2 induced by either AA (Fig. 1G) or collagen (Fig. 1H) was partially inhibited by 0.3 μmol L−1 ticagrelor (∼IC50 for ADP-induced aggregation) and abolished by 3 μmol L−1 ticagrelor (∼IC90 for ADP-induced aggregation). We have previously reported that TxA2 production in response to epinephrine is inhibited by P2Y12 blockade in the same manner for AA and collagen [1]. Aspirin (30 μmol L−1) either alone or in combination with ticagrelor also completely inhibited TxA2 production to either agonist (Fig. 1G,H). A similar pattern of results was obtained using equivalent inhibitory concentrations of PAM in place of ticagrelor (Table S2), and when the concentration of aspirin was increased to 120 μmol L−1 (Tables S1 and S2).
These studies show that ticagrelor and PAM inhibit platelet aggregation induced by a range of platelet agonists through a mechanism consistent with blockade of platelet P2Y12 receptors and that ticagrelor is more potent than PAM in this regard. As well as inhibiting aggregation following from direct activation of P2Y12 receptors by the addition of exogenous ADP, ticagrelor and PAM inhibited aggregations resulting from stimulation of platelets with AA, a response which is well characterized as being TxA2 dependent. In addition to inhibiting platelet responses to endogenously produced TxA2, ticagrelor and PAM also inhibited the production of TxA2 by platelets [6]. Interestingly, ADP itself is a poor stimulus for TxA2 production [1], suggesting that released ADP, acting on the P2Y12 receptor, acts to potentiate the stimulation of TxA2 synthesis by other signaling pathways activated in parallel. These results are consistent with the idea that whereas aspirin may inhibit just the TxA2-dependent pathway of platelet activation, ticagrelor and PAM can inhibit both the ADP-P2Y12-dependent and the TxA2-dependent pathways of platelet aggregation. The observation that aspirin adds anti-aggregatory effects to partial, but not complete, P2Y12 receptor blockade, further supports this idea.
Taken together these results demonstrate that rather than ADP-P2Y12 and TxA2 pathways acting independently, the TxA2-dependent pathway is dependent upon the ADP-P2Y12 pathway both for the production of TxA2 and fundamentally for the irreversible aggregation that follows activation of TP receptors. If these data accurately model the situation in vivo, this may have important implications for the use of dual antiplatelet therapy using potent P2Y12 antagonists in clinical practice [2]. For example, one could postulate that addition of aspirin could produce side-effects secondary to inhibition of cyclo-oxygenase at non-platelet sites, as has recently become apparent for non-steroid anti-inflammatory drugs, while providing little additional anti-aggregatory effect [7, 8]. Clearly, the validity of this hypothesis remains to be determined by clinical studies.
Disclosure of Conflict of Interests
T.D. Warner has received honoraria and research grants from AstraZeneca; S. Nylander is an employee of AstraZeneca. The other authors state that they have no conflict of interest.
Supporting Information
Figure S1. Concentration-response curves forthe inhibition by (A) ticagrelor(0.1–10 μmol L−1), (B)prasugrel active metabolite (PAM;0.1–10 μmol L−1) and (C)aspirin (1–100 μmol L−1) ofplatelet aggregation induced by ADP(0.625–20 μmol L−1).n = 4.
Figure S2. Concentration-response curves forthe inhibition by (A) ticagrelor(0.1–10 μmol L−1), (B)prasugrel active metabolite (PAM;0.1–10 μmol L−1) and (C)aspirin (1–100 μmol L−1) ofplatelet aggregation induced by U46619(0.1–30 μmol L−1).n = 4.
Figure S3. Concentration-response curves forthe inhibition by (A, B) ticagrelor(0.1–10 μmol L−1), (C, D)prasugrel active metabolite (PAM;0.1–10 μmol L−1) and (E, F)aspirin (1–100 μmol L−1) ofplatelet aggregation (A, C, E) and TxA2 release (B, D,F; measured as TxB2) induced by arachidonic acid (AA;0.03–1 mmol L−1).n = 4.
Table S1. Area under the concentration-responsecurve summary data for the effect of combinations of ticagrelor(0.03, 0.1, 0.3 and 3 μmol L−1) andaspirin (30 and 120 μmol L−1) onplatelet aggregation (A–C) and platelet TxA2release (D; measured as TxB2).
Table S2. Area under the concentration-responsecurve summary data for the effect of combinations ofprasugrel-active metabolite (PAM; 0.5, 1, 2 and10 μmol L−1) and aspirin (30 and120 μmol L−1) on platelet aggregation(A–C) and platelet TxA2 release (D; measured asTxB2).
References
- 1.Armstrong PC, Leadbeater PD, Chan MV, Kirkby NS, Jakubowski JA, Mitchell JA, Warner TD. In the presence of strong P2Y12 receptor blockade, aspirin provides little additional inhibition of platelet aggregation. J Thromb Haemost. 2011;9:552–61. doi: 10.1111/j.1538-7836.2010.04160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Warner TD, Armstrong PC, Curzen NP, Mitchell JA. Dual antiplatelet therapy in cardiovascular disease: does aspirin increase clinical risk in the presence of potent P2Y12 receptor antagonists? Heart. 2010;96:1693–4. doi: 10.1136/hrt.2010.205724. [DOI] [PubMed] [Google Scholar]
- 3.Angiolillo DJ. Variability in responsiveness to oral antiplatelet therapy. Am J Cardiol. 2009;103:27A–34A. doi: 10.1016/j.amjcard.2008.11.020. [DOI] [PubMed] [Google Scholar]
- 4.Simon T, Verstuyft C, Mary-Krause M, Quteineh L, Drouet E, Meneveau N, Steg PG, Ferrieres J, Danchin N, Becquemont L the French Registry of Acute STEaN-STEMII. Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med. 2009;360:363–75. doi: 10.1056/NEJMoa0808227. [DOI] [PubMed] [Google Scholar]
- 5.Paul BZS, Jin J, Kunapuli SP. molecular mechanism of thromboxane A2-induced platelet aggregation. Essential role for P2Tac and alpha 2a receptors. J Biol Chem. 1999;274:29108–14. doi: 10.1074/jbc.274.41.29108. [DOI] [PubMed] [Google Scholar]
- 6.Armstrong PC, Dhanji AR, Tucker AT, Mitchell JA, Warner TD. Reduction of platelet thromboxane A2 production ex vivo and in vivo by clopidogrel therapy. J Thromb Haemost. 2010;8:613–5. doi: 10.1111/j.1538-7836.2009.03714.x. [DOI] [PubMed] [Google Scholar]
- 7.Warner TD, Mitchell JA. COX-2 selectivity alone does not define the cardiovascular risks associated with non-steroidal anti-inflammatory drugs. Lancet. 2008;371:270–3. doi: 10.1016/S0140-6736(08)60137-3. [DOI] [PubMed] [Google Scholar]
- 8.White WB. Cardiovascular effects of the cyclooxygenase inhibitors. Hypertension. 2007;49:408–18. doi: 10.1161/01.HYP.0000258106.74139.25. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.