

Abstract
Chemical purity of RNA samples is important for high-precision studies of RNA folding and catalytic behavior, but photodamage accrued during ultraviolet (UV) shadowing steps of sample preparation can reduce this purity. Here, we report the quantitation of UV-induced damage by using reverse transcription and single-nucleotide-resolution capillary electrophoresis. We found photolesions in a dozen natural and artificial RNAs; across multiple sequence contexts, dominantly at but not limited to pyrimidine doublets; and from multiple lamps recommended for UV shadowing. Irradiation time-courses revealed detectable damage within a few seconds of exposure for 254 nm lamps held at a distance of 5 to 10 cm from 0.5-mm thickness gels. Under these conditions, 200-nucleotide RNAs subjected to 20 seconds of UV shadowing incurred damage to 16-27% of molecules; and, due to a ‘skin effect’, the molecule-by-molecule distribution of lesions gave 4-fold higher variance than a Poisson distribution. Thicker gels, longer wavelength lamps, and shorter exposure times reduced but did not eliminate damage. These results suggest that RNA biophysical studies should report precautions taken to avoid artifactual heterogeneity from UV shadowing.
As studies of RNA behavior seek greater quantitative precision and explanatory power, experimenters have uncovered numerous unexpected phenomena. Multiple folding pathways, molecular individuality, highly heterogeneous kinetics, and long-lived metastable states have been detected for numerous RNA and RNA/protein systems – especially through single-molecule approaches – and have important implications for fully understanding the biological and evolutionary behavior of RNA.1,2,3,4,5,6,7,8,9 Nevertheless, few mechanisms for these molecule-to-molecule variations have been established, and there remain concerns, in some cases confirmed10, that some of the observed variations stem from impurities or misfolding arising during synthesis or subsequent handling.11,12
One point of entry for chemical damage might be polyacrylamide-gel-based purification of RNA preparations, which is routine in biochemistry labs. In particular, purification protocols typically involve localizing samples in gels by illumination with hand-held ultraviolet (UV) lamps, which results in shadows on fluorescent screens. UV-induced lesions in nucleic acids, particularly photo-dimerization of proximal pyrimidines to produce cyclobutane-like or other linkages (Figure 1A), have been studied for decades due to their adverse biological and medical effects on genomic DNA and non-coding RNA (refs13,14,15 and refs. therein). To reduce such damage during routine nucleic acid purification, researchers can use long-wavelength UV lamps, minimize exposure times, and use higher RNA concentrations and thicker gels (see below). Perhaps the safest precaution involves loading side-by-side replicate samples onto gels, some of which are ‘sacrificed’ to UV shadowing so that the other un-shadowed samples can be approximately located and then excised without risk of UV exposure. Nevertheless, several protocols and handbooks for UV shadowing do not prescribe such precautions.16,17,18,19,20
Figure 1. Characterizing RNA damage during UV shadowing by reverse transcription read out by capillary electrophoresis.

(A) Cyclobutane dimerization product of two uracil bases initiated by UV absorption. (B) Capillary electropherograms of reverse-transcription products for seven RNAs that were or were not UV-shadowed during polyacrylamide gel purification. Sequence is as given except for reverse transcription binding site at 3′ end (see Methods). Boldface letters and asterisks mark second nucleotides of pyrimidine-pyrimidine doublets. Exposure time was 100 seconds by a hand-held UVG-54 lamp (Ultraviolet Products) at 10 cm distance to samples. (C) Design of sequences UV1 to UV5 to confirm damage predominantly at pyrimidine-pyrimidine (UU, UC, CU, and CC) sites. Gray circles mark second nucleotides of pyrimidine-pyrimidine doublets. (D) Capillary electropherograms of gel-purified sequences UV1 to UV5. (E) Histograms of UV reactivities at AA, AU, and UU sites from RNAs in (B); reactivities are relative to mean UV reactivity seen for each construct. (F) Box plot of reactivities across all dinucleotide types, shown as medians (white symbols), 25th–75th percentile (interquartile) ranges (black boxes), most extreme data points that are outside the interquartile range by no more than 1.5 times this range (whiskers), and values beyond the whisker range (small black symbols). (G) Effects of different UV handheld lamps, gel thickness, and loaded RNA amounts on the P4–P6 RNA (P4–P6 domain of the Tetrahymena group I ribozyme).
The effects of UV shadowing have not been quantitatively characterized, partially due to the difficulty of lesion detection. Photodimers connecting sequence-adjacent residues do not change an RNA's mass and negligibly change its gel mobility. Thus, UV damage would not be readily observed in experiments involving mass spectroscopy or denaturing gel electrophoresis (see, e.g., refs5,10,21). Quantitative RT-PCR22 and nuclease-based tests10,14 have not given the sequence dependence or time-course of the damage in RNAs of interest.
Here, we report single-nucleotide-resolution experiments that quantify RNA damage incurred during UV shadowing for numerous RNAs, several commercially available UV lamps, a series of exposure times, varying gel thicknesses, and different RNA concentrations. Our data reveal conditions in which significant covalent damage occurs, affecting 20% or greater of purified molecules, and the factors that can diminish or increase the damage rate. The results also indicate unanticipated molecule-by-molecule lesion distributions that may complicate quantitative RNA biophysical analyses.
Results
Survey of several RNAs confirms damage
In initial studies by our laboratory investigating chemical modification of RNAs23,24, we found that lesions from exposure to ultraviolet lamps were readily detected by reverse transcription with a fluorescent primer, a rapid readout for chemical structure mapping or footprinting experiments. Figure 1B presents the results of this procedure on seven RNA molecules visualized by UV shadowing after denaturing polyacrylamide gel electrophoresis (PAGE; 8% polyacrylamide with 7 M urea and 1x TBE electrophoresis buffer, 89 mM Tris-borate, pH 8.3; 1 mM EDTA). The seven sequences included riboswitch ligand-binding domains (adenine riboswitch from Vibrio vulnificus25; a cyclic diGMP riboswitch from Vibrio cholerae26; and a glycine-binding domain from Fusobacterium nucleatum27); other natural RNAs (the P4-P6 domain of the Tetrahymena ribozyme28, unmodified tRNAphe29 and 5S rRNA30 from Escherichia coli); and an artificial hairpin construct, the Medloop RNA23,24. The samples were exposed to UV light with a hand-held lamp (Ultraviolet Products UVG-54, 254 nm, 6 W) at a distance of 10 to 20 cm from the sample, resulting in an exposure rate of approximately 0.2–0.8 mJ cm–2 s–1. In all seven cases, samples exposed to the lamp for at least one second showed striking patterns of reverse transcriptase stops compared to samples that were excised from the same gels without UV exposure. For illustration, the data in Figure 1B were taken on samples of 50–100 μg exposed for 100 seconds within gels of thickness 0.5 mm. Damage was also detected with smaller exposure times, other loaded RNA amounts, and increased gel thicknesses; quantitation of the total lesion rate required fits to time-courses and are discussed below.
To investigate whether our PAGE protocol produced other covalent effects aside from UV-induced lesions, we also carried out control measurements with RNAs purified by non-gel means (phenol/chloroform extraction and gel filtration; MagMax RNA-binding beads; and hybridization of 3′ ends to complementary DNA oligonucleotides bound to magnetic beads). These experiments revealed no reproducible effects from UV-free PAGE purification aside from modest damage traced to oxidation from ammonium persulfate used to polymerize acrylamide; see SI Figure S1. This damage occurred primarily at guanosines, consistent with known oxidative lesions31, and affected 0.02–0.04% of RNA molecules. Nevertheless, this measurement and the measurements below should be considered lower bounds, since the reverse transcriptase used herein (SuperScript III) may bypass some types of lesions at a non-negligible rate.
Sequence Dependence of UV lesions
UV damage from photodimerization is expected to occur predominantly at sequence segments with neighboring pyrimidines (YY = UU, CU, UC, and UU) through cyclobutane-type linkages (Figure 1A)13,14. To test this expectation, we assigned the sequences of the capillary electrophoretic traces by repeating the reverse transcription with 2′-3′-dideoxyguanosine triphosphate (ddGTP) to obtain reference peaks whose positions corresponded to C positions (see also below). Sequence annotation of the electropherograms verified that UV-induced damage occurred predominantly at YY positions (boldface in Figure 1B).
To further confirm the general sensitivity of RNA sequences to UV damage, we designed five artificial RNA sequences UV1 to UV5 containing pyrimidine-pyrimidine sites arrayed in a pattern spelling out a warning message, “UV BAD” (Figure 1C). After these RNAs were purified with UV shadowing and subjected to reverse transcription, the resulting capillary electropherograms displayed the expected message (Figure 1D).
Given the visually striking prevalence of pyrimidine-pyrimidine sequences at UV-induced stops, we investigated whether the extent of damage could be quantitatively correlated with sequence. We assessed the degree of uniformity of the reactivity across pyrimidine-pyrimidine (YY) positions and whether damage occurred at other sites by quantifying capillary electrophoretic traces at single-nucleotide resolution using the HiTRACE software32.
As shown in Figures 1E & 1F, UV reactivities across all studied RNAs were not strongly stereotyped. For example, the distribution at UU positions was not strongly peaked; it spanned a ten-fold range of reactivities and was approximately flat within this range. One explanation for this heterogeneity is residual structure in the RNA within the polyacrylamide gel despite the presence of 7 M urea; UV experiments in non-denaturing conditions have shown modulation or enhancement of photodimerization due to secondary or tertiary structure formation (data not shown; see also, e.g., ref.33). One example supporting the role of residual structure is apparent in the UV1-UV5 RNAs (Fig. 1D). The first 18 nucleotides of UV1 through UV4 are the same, and the first pyrimidine doublet (U11-C12; left-hand edge of the letter ‘U’ in “UV BAD”) occurs within this constant region. However, this doublet only gives a strong damage signal in UV4 and not UV1-UV3. The cause of this differential effect is unclear, but must involve sequences at least 6 nucleotides downstream of the site. Regardless of its origin, the spread in reactivities even for specific sequences currently precludes a strongly predictive quantitative model for UV damage from sequence.
The quantification of reactivities also showed that many segments of the RNA sequences that contained a pyrimidine after a purine, such as GU or AU, were covalently modified by UV exposure. Less reactivity was observed at other positions. See Figure 1E for sample histograms of AU vs. UU and AA; and Figure 1F for summary of median and ranges over all sequence doublets. Again, a possible explanation for effects at non-YY sequences is residual structure, e.g., base pairs or transient contacts that enable photodimerization of pyrimidines distant in sequence from one another. Alternatively or in addition, these positions may correspond to UV-induced lesions that are not the common cyclobutane-type photodimers (Figure 1A)14,34. The isolation and characterization of these lesions through nuclease digestion and mass spectrometry may reveal further information on the damage but is currently challenging; in the following instead we continue to focus on practical considerations of how photodamage might be avoided.
UV wavelength dependence
Photodimerization of pyrimidines is promoted by UV radiation at shorter wavelengths35 and so might be avoided by using UV lamps mainly emitting radiation at longer wavelengths and/or filtering out shorter wavelengths. A variety of available lamps with different emission wavelengths were tested for their effects on the P4-P6 RNA during gel purification, including 254 nm emitters recommended by several published protocols16,17,18. A lamp emitting higher wavelength radiation (UVP 3UV-34 three-setting lamp at 302 nm) gave detectable but substantially less damage (12±6%), compared to the 254-nm irradiated samples (Figure 1G). However, with 0.5-mm-thick gels, UV shadow bands were not clearly visible with the amount of RNA used (50–100 μg), and were not visible at all with less RNA (20 μg; see also below for variation of gel thickness). Lamps emitting longer wavelength radiation (UVP 3UV-34 three-setting lamp with 365 nm setting; and UVP UVL-56, 366 nm) gave no detectable damage on the RNA (Supporting Information Figure S2) after 100 seconds of exposure, but they also did not give detectable UV shadows during gel purification. Another two lamps (UVP UVG-54 lamps, 254 nm) all gave detectable shadows but also UV-induced damage bands in the reverse transcription assay (Supporting Information Figure S2).
Time-course of UV damage
One route to reducing covalent RNA damage during UV shadowing would be to lessen the UV dose illuminating the sample. This reduction might be accomplished in two ways. Moving the source further from the sample might diminish UV exposure, but risks reducing the clarity of the RNA band's UV shadow and precluding visual detection. Another strategy to diminish UV exposure would be to reduce the exposure time to the minimum required for finding and marking the desired band. We tested the latter strategy by measuring the rate of UV damage by carrying out a time-course of UV exposure. Some protocols suggest exposures up to 30 seconds.17 We therefore exposed gel samples of the P4-P6 RNA to UV lamp radiation (254 nm, 6 W) from one second to several minutes (to obtain an endpoint) and quantified UV-induced damage as above.
The results of this timecourse are shown in Figure 2A. For the earliest time points, the UV damage pattern was detectable above background within 3 seconds. Such early damage was seen for experiments with the lamp placed 5 cm, 10 cm, and 20 cm from the sample. For time points between 20 seconds up to 600 seconds, the data approached an apparent ‘steady-state’ pattern. At the longest time points, 40 and 60 minutes, the reverse transcription signal was sharply attenuated for extension lengths beyond 34 nucleotides, corresponding to the end of an 8-nucleotide segment containing six double-pyrimidine sites.
Figure 2. Time-course of UV damage and a ‘skin effect model’.

(A) Ultraviolet damage for the P4-P6 RNA shadowed during gel purification by a UVG-54 lamp at a distance of 5 cm. Arrows mark damage appearance at 3 second time point. (B) Quantitation of reverse transcribed products. (C) Fit of data in (B) to a skin effect model in which the UV lesion rate in the 0.5 mm gel slice is 0.032 s–1 but attenuated exponentially by absorption with skin depth λ = 0.1 mm. (D), (E), (F), and (G): Predictions of the skin effect model for the distribution of damage (here, the fraction of RNAs with at least one lesion) at different depths of the gel slice (vertical axis) at different points in the observed time-course. (H) Molecule-by-molecule distribution of lesions in the Poisson and skin effect models when the average number of lesions per molecule is 1 [corresponding to the 60 second timepoint (E)].
Accounting for these data required taking into account the heterogeneity in radiation dose due to the absorbance of UV radiation as it passes through the RNAs (Figures 2A–H). RNAs at the top of the gel slice (here, closest to the UV lamp) receive higher exposure than those at the bottom, where the radiation is attenuated by the RNA molecules' absorption. Because different populations of RNAs receive different exposures, a Poisson process with a single rate of modification does not hold. Instead, the UV damage rate is attenuated exponentially with parameter λ as a function of the depth through the gel slice. Mathematical modeling of the resulting ‘skin effect’ model led to excellent fits of the experimental measurements [Figures 2B and 2C; see equations (1)-(4)], with λ = 0.12 ± 0.05 mm and with the damage rate at the top of the gel as k = 0.03 ± 0.01 s–1 (summed over all nucleotides of the RNA). Further descriptions of the skin effect model compared to a simpler Poisson model, independent verification with ddGTP-doped measurements, and estimates of RNA concentration are given in the Supporting Information.
One unanticipated implication of the skin effect model is that a population of UV-shadowed RNAs gives, on a molecule-by-molecule basis, a more heterogeneous distribution of damage than would be predicted from the Poisson model. Figure 2H illustrates the fraction of molecules containing 0, 1, 2, ... lesions for RNA populations with average lesion number of 1.0 (seen at 60 seconds of UV exposure under the tested conditions in Figure 2). The variance in the lesion rate in the skin effect model is 3.8, substantially greater than the variance in the Poisson model (1.0).
Damage attenuation at higher RNA concentration
In a final experiment, we tested qualitative predictions of the skin effect model with practical implications for reducing UV-induced damage. In this model, higher concentrations of RNA should protect the overall population from UV-induced damage, since radiation should penetrate less deeply into the population. We varied the gel RNA concentration in two ways. First, we varied the amount of RNA loaded onto an 8% acrylamide gel by five-fold, from 20 μg to 100 μg (lower concentrations did not give clear bands). After irradiation by a 254 nm lamp held 10 cm from the gel for 100 seconds, the sample with more RNA gave somewhat less damage (reduction by 16±9%; Figure 2A). However, we noticed that the larger amount of RNA was spread into a wider band and therefore the concentration in the gel did not actually increase five-fold. We also loaded these RNA amounts onto a 6-fold thicker gel, using 3 mm vs. 0.5 mm separators, hypothesizing that more of the RNA molecules would be protected by the ‘skin’ layer. A reduction of the damage by 41±6% was observed, supporting the skin effect model. Similar reductions in damage with thicker gels were observed with 302 nm irradiation (Figure 1G), although this protocol did not permit visualization of the smaller RNA amount in the 0.5-mm-thick gel or either RNA amount in the 3-mm-thick gel. (In our hands, the thicker gels appeared to absorb some UV radiation and thus reduce contrast of the UV shadow.) We also tested RNA samples irradiated in solution, as they offered better control of RNA concentration than gel-based measurements. After 60 seconds of UV treatment, samples with low RNA concentrations (0.05 mg/mL) gave damage products (SI Figure S4), while samples with 10-fold higher RNA concentrations gave reverse transcription patterns with no detectable damage, again supporting the skin effect model.
Discussion
UV damage of nucleic acids is a well-appreciated biological phenomenon and a known risk during gel purification of RNA, but the magnitude of this damage during routine applications has not been characterized. We have now quantified the effects of UV shadowing using reverse transcription read out by capillary electrophoresis. Across several natural and artificial RNAs, across multiple sequence contexts (dominantly at but not limited to pyrimidine doublets), and with multiple lamps that are recommended for UV shadowing, we have detected substantial RNA damage occurring in routine UV shadowing protocols.
To more precisely assess the UV-induced damage, we measured and modeled the lesions occuring within polyacrylamide gels versus time. Unexpectedly, the time course of UV damage presented a long phase (10 seconds to 600 seconds) in which the reverse transcription pattern reached an apparent steady state. A skin effect model provided an explanation for this behavior (Figures 2D–G). At early timepoints, the only damaged RNA molecules were those closest to the lamp, at the top of the gel slice (Figure 2D). At later timepoints, this very top layer became so damaged that its RNAs did not contribute to the observed UV photodimer pattern read out by reverse transcription (Figure 2E). RNA molecules at the very bottom of the gel also did not contribute to the damage pattern, but for a different reason: they were unmodified due to protection from the top layer. Instead the observed pattern of reverse transcription products was due to an intermediate ‘active’ layer with skin depth ~λ (Figure 2E). This active layer traveled down the gel slice slowly; its starting depth increased logarithmically with time (Figure 2F) while its thickness stayed nearly constant. The RNAs in this active layer contributed a constant UV damage pattern [Methods eq. (5)]. Once the active layer reached the bottom of the gel slice, all the RNAs in the gel succumbed to numerous damage events, and their reverse transcription was strongly attenuated (Figure 2G).
Our data gave a lesion rate for the P4–P6 RNA (202 nucleotides) of 0.03–0.04 s–1 at the top of the gel, decreasing exponentially with a skin depth of λ = 0.10−0.12 mm under the tested conditions, a 254 nm lamp held 5 cm from the band. This lesion rate was derived from fitting the full time-course data to the skin effect model and is thus corrected for background reverse transcription stops and heterogeneity in damage for RNAs at different gel depths. If locating and marking an RNA gel band by UV shadowing requires 10 to 20 seconds, the mean number of lesions per RNA under these conditions is calculated to be 0.16 to 0.27. (For comparison, numerous single-molecule folding and catalysis studies have uncovered sub-populations of kinetically distinct molecules at frequencies of 10–30%.)
Five factors besides exposure time can change the fraction of damaged molecules. First, higher concentrations of RNA decrease the overall lesion rate since the skin depth becomes shorter. However, the practical use of this effect may be modest; in our hands, higher loaded amounts spread into wider bands with similar RNA concentrations. Second, setting gels with thicker spacers permit the skin layer to protect more RNA. Our experiments with 3 mm thick gels gave reduced damage by 40%. Third, using longer wavelength lamps (e.g, 302 nm) gave weaker damage, by nearly 10-fold, but permitted visualization only in the thinner gels (0.5 mm) with high RNA amounts (100 μg). Fourth, the distance of the lamp to the sample can be increased, from 5 to 10 cm, resulting in 2 to 4-fold less radiation dose and proportionally less damage. Finally, the lesion rate per RNA varies with the length of nucleotides of the molecule. RNAs under 20 nucleotides as are used for, e.g., duplex thermodynamic studies36 should give low mean numbers of lesions (less than 0.02) per RNA in our tested conditions. Conversely, preparations of long RNAs, e.g., the Tetrahymena group I ribozyme (388 nts), under our tested conditions would have a mean fraction of greater than 0.5 lesions after 20 seconds of UV shadowing.
None of the variations described above eliminate UV damage or heterogeneities due to the skin effect (Figure 2G), which may cause counterintuitive behavior in single molecule experiments. UV damage can be completely avoided if a small amount (20 μg) of a replicate sample or an edge of the gel lane of interest is sacrificed to 254 nm UV irradiation to enable localization of nearby RNA that remains undamaged. Alternatively, several groups have proposed non-gel-based purification methods for crystallography, NMR, and high-throughput biophysics applications [see, e.g., refs.11,37,38,39,40,41], and these methods either give sub-second UV doses during chromatographic detection or bypass UV exposure altogether. By avoiding gel electrophoresis, these strategies may also reduce oxidative damage (ref.42 and Supplemental Figure S1), heat treatment10, and potential contamination from acrylamide or polyacrylamide.
Our results suggest that UV shadowing may be causing underappreciated covalent damage in high-precision and single-molecule-resolution RNA biophysical measurements. Future studies may thus wish to report precautions taken against UV-induced damage or set experimental bounds on the damage through primer extension.
Methods
Polyacrylamide gel electrophoresis and UV shadowing
All RNAs were prepared as in prior work24,41 by in vitro transcription with T7 RNA polymerase from PCR products (with the 20 bp T7 promoter sequence TTCTAATACGACTCACTATA included at the 5′ end), with transcription volumes up to 1.5 mL. Transcriptions were precipitated by adding 1/10 volume of sodium acetate (pH 5.2) and 3 volumes of cold ethanol (taken out of −20°C storage), cooling on dry ice for at least 15 minutes, and centrifuging at 14,000 g for 1.5 hours. After removal of supernatant, pellets were rinsed with 1 mL 70% cold ethanol twice, dried in air for at least 30 minutes, and resolubilized in deionized water at volumes equal to 1/10 of the original transcription. A half volume of denaturing loading buffer (90% formamide, 0.1% xylene cyanol, 0.1% bromophenol blue) was added, and the samples were loaded onto polyacrylamide gels. The gels were 0.5 mm in thickness, 20 cm in height (direction of electrophoresis), and 27 cm in width. The gel mix contained 1x TBE (89 mM Tris-Borate, 1 mM EDTA), 8% polyacrylamide (29:1 acrylamide:bis, Sigma), and 7 M urea, and were polymerized by the addition of 1/100 volume of 10% ammonium persulfate and 1/1000 volume of TEMED (N,N,N′,N′-Tetramethylethylenediamine); after pouring between glass plates, the gels were given at least 1.5 hours to polymerize. Variations with longer polymerization times and use of flavin mononucleotide as the polymerization activating reagent are discussed in SI Figure S1. Gels were run at 25 W or less for 1 to 3 hours (temperatures remained less than 40°C under electrophoresis conditions).
Gels were transferred from gel plates onto UV-transparent plastic wrap (Saran), covered with wrap on both sides, and placed on a fluorescent TLC plate (Life Technologics). Samples were exposed to UV hand-held lamps (Ultraviolet Products UVG-54, 254 nm, 6 W; unless specified otherwise) and boxes were marked on plastic wrap around band locations with Sharpie markers. In most cases, half of the lanes were exposed, with the other half being covered with aluminum foil; the halves were excised separately, with the covered portions serving as UV-untreated controls. The radiation exposure was estimated assuming that the radiation was reflected into one hemisphere underneath the lamp, decreasing as distance squared; this is an underestimate since the radiating tubes are not point sources but extend over approximately 10 cm. For time course measurements (Fig. 2), early timepoints were acquired by turning on the lamp for a few seconds (for warm-up) and transiently removing the foil for the presented times. Gel slices were excised with sterile, disposable scalpels (BD) after peeling back plastic wrap and placed in 1.5 mL Eppendorf tubes with 200 μL deionized water. RNAs passively eluted into the water during incubation overnight at 4°C, and concentrations were estimated by absorption measurements at 260 nm on a Nanodrop spectrophotometer.
Reverse transcription
RNA sequences are presented in Fig. 1B (for non-coding RNAs) and Fig. 1C (for UV1-UV5); all sequences included an additional 20 nt sequence AAAGAAACAACAACAACAAC at the 3′ end as a common reverse transcription binding site. Reverse transcription with Superscript III reverse transcriptase (Invitrogen) and capillary electrophoresis on ABI 3100 and 3730 machines were carried out as previously described, using poly(A) purist magnetic beads (Life Technologies) to accelerate purification steps23,24,41. For reverse transcriptions with ‘doping’ from 2′-3′-dideoxyguanosine triphosphate (ddGTP), concentrations of nucleotides were 0.05 mM ddGTP and 1 mM dATP, dCTP, dTTP, and dITP (2′-deoxyinosine triphosphate). Data were aligned and quantitated with the HITRACE software32. Data have been deposited as entries UVBAD5 UVP 0001 and TRP4P6 UVP 0001 in the RNA Mapping Database (http://rmdb.stanford.edu).
Model for UV damage timecourses
Data for ultraviolet damage timecourses were fit to an extension of the basic Poisson model, in which modifications at different sites occur independently and stochastically [see, e.g., references23,43 for prior applications to chemical mapping data]. Assume that the total rate of modification at all sites is k, and the fractional modification rate each site i is given as ki = αi k. The sum over the fractional modification rates αi is unity. The skin effect model is different from the simple Poisson model in that the damage rates are not constant for all RNAs but are attenuated at depth z with characteristic length λ:
The lesion probability at site i at time t is then:
where bi is the (UV-independent) background rate of stopping reverse transcription. The fraction of molecules Fn with n lesions is given by the Poisson formula, but averaged through different depths:
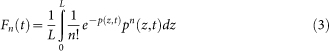 |
The fraction of reverse transcription product fi at site i after traversal through sites 1, 2, … i–1 is:
Note that i indexes the position in the reverse transcribed cDNA (5′ to 3′), with i = 1, 2, … corresponding to primers extended by one, two, … nucleotides. The equations (1)–(3) were computed in MATLAB (Mathworks), with the integrals numerically approximated by subdividing the gel slice into subslices of length 0.001 L. The site-dependent background stopping probability bi was measured by the reverse transcription experiment without UV exposure. The site-specific UV-damage rate αi was estimated based on measurements with 600 seconds of UV exposure, subtracting bi and normalizing to unity. Similar parameter fits were obtained for αi estimated from later timepoints and also by iterating the fits so that new αi were obtained based on the ratio of data at late timepoints with predictions based on the original estimates of αi. Data at the very 5′ and 3′ ends gave strong signals that saturated the capillary electrophoresis fluorescence detector and were not used in the fits (see Fig. 2); so the total modification rates given in the text are slight underestimates. Note that a standard Poisson model can be obtained from equations (1)–(4) above by setting 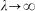 (no attenuation of radiation by absorption).
(no attenuation of radiation by absorption).
To model measurements that included ddGTP for internal controls, the ‘background’ stopping probabilities bi were adjusted based on reverse transcription measurements without UV but including ddGTP. Suppose that there is a ddGTP signal nucleotide at h, and the next UV-induced signal is at i>h, such that there is no cross contamination (kh = 0,
bi = 0, and no bands in between h and i); examples of these pairs are shown in SI Figures 2D and 2E. For the standard Poisson model, the ratio of the two signals is given by 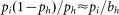 with the complex attenuation factor
with the complex attenuation factor 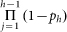 canceling out. For times up to saturation, this ratio increases linearly with time since bh is constant and pi increases as in eq. (2), which is linear for pi < 1. This relation was used in the text to make comparisons between different conditions (UV wavelength, loaded RNA amount, thickness of gel). Specifically, the ratio of the UV-damage signal at nucleotides 167, 181, 205, and 224 in the P4-P6 RNA was computed, after normalization to ddGTP reference peaks; the mean and standard deviation of these numbers is reported.
canceling out. For times up to saturation, this ratio increases linearly with time since bh is constant and pi increases as in eq. (2), which is linear for pi < 1. This relation was used in the text to make comparisons between different conditions (UV wavelength, loaded RNA amount, thickness of gel). Specifically, the ratio of the UV-damage signal at nucleotides 167, 181, 205, and 224 in the P4-P6 RNA was computed, after normalization to ddGTP reference peaks; the mean and standard deviation of these numbers is reported.
For determining absolute rates of damage, the general skin effect model was numerically calculated as above and fit to the data. (In principle, the overall damage rate could be estimated based on measuring the amount of fully reverse-transciption extended products compared to partially extended products, and subtracting values from UV-untreated samples as controls. However, attempts to measure these values through dilution series gave large absolute errors in damage rates, around 30%.)
For a large range of times and negligible background bi, the reverse transcription pattern [equation (4)] reaches an approximate ‘steady state’ in which the top of the gel slice is saturated with lesions, the bottom of the gel slice to remain undamaged, and an intermediate subslice of RNAs (‘active’ layer) dominates the contribution to the reverse transcription signal. The steady state is given by the simple formula:
 |
The equation is derived in the Supporting Text.
Author Contributions
RD designed research; WK, JH, and RD collected experimental results; and RD wrote the manuscript. All authors reviewed the manuscript.
Supplementary Material
Supporting Information for
Acknowledgments
We thank M. Greenfeld & D. Herschlag for useful discussions and sharing of their manuscript before publication; the Stanford Biochemistry Department for sharing ultraviolet lamps; and M. Zhang and H. Mabuchi for use of a hand-held UV detector. This work was supported by the Burroughs-Wellcome Foundation (CASI to RD).
References
- Zhuang X. et al. Correlating structural dynamics and function in single ribozyme molecules. Science 296, 1473–1476 (2002). [DOI] [PubMed] [Google Scholar]
- Tan E. et al. A four-way junction accelerates hairpin ribozyme folding via a discrete intermediate. Proc Natl Acad Sci U S A 100, 9308–9313 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z., Srividya N., Sosnick T. R., Pan T. & Scherer N. F. Single-molecule studies highlight conformational heterogeneity in the early folding steps of a large ribozyme. Proc Natl Acad Sci U S A 101, 534–539 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemay J. F., Penedo J. C., Tremblay R., Lilley D. M. & Lafontaine D. A. Folding of the adenine riboswitch. Chemistry & biology 13, 857–868 (2006). [DOI] [PubMed] [Google Scholar]
- Korennykh A. V., Plantinga M. J., Correll C. C. & Piccirilli J. A. Linkage between substrate recognition and catalysis during cleavage of sarcin/ricin loop RNA by restrictocin. Biochemistry 46, 12744–12756 (2007). [DOI] [PubMed] [Google Scholar]
- Karunatilaka K. S., Solem A., Pyle A. M. & Rueda D. Single-molecule analysis of Mss116-mediated group II intron folding. Nature 467, 935–939 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomatin S. V., Greenfeld M., Chu S. & Herschlag D. Multiple native states reveal persistent ruggedness of an RNA folding landscape. Nature 463, 681–684 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z. et al. One RNA aptamer sequence, two structures: a collaborating pair that inhibits AMPA receptors. Nucleic acids research 37, 4022–4032 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neupane K., Yu H., Foster D. A., Wang F. & Woodside M. T. Single-molecule force spectroscopy of the add adenine riboswitch relates folding to regulatory mechanism. Nucleic acids research 39, 7677–7687 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfeld M., Solomatin S. V. & Herschlag D. Removal of covalent heterogeneity reveals simple folding behavior for P4-P6 RNA. J Biol Chem 286, 19872–19879 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira M. J., Behera V. & Walter N. G. Nondenaturing purification of co-transcriptionally folded RNA avoids common folding heterogeneity. PLoS One 5, e12953 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomatin S. V., Greenfeld M. & Herschlag D. Implications of molecular heterogeneity for the cooperativity of biological macromolecules. Nat struct mol biol 18, 732–734 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenstock C. L., Brown I. H., Hunt J. W. & Johns H. E. Photodimerization of pyrimidine nucleic acid derivatives in aqueous solution and the effect of oxygen. Biochem Biophys Res Commun 27, 431–436 (1967). [DOI] [PubMed] [Google Scholar]
- Sinha R. P. & Hader D. P. UV-induced DNA damage and repair: a review. Photochem Photobiol Sci 1, 225–236 (2002). [DOI] [PubMed] [Google Scholar]
- Wurtmann E. J. & Wolin S. L. RNA under attack: cellular handling of RNA damage. Crit Rev Biochem Mol Biol 44, 34–49 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann R. K., Bindereif A., Schön A. & Westhof E. (eds.) Handbook of RNA Biochemistry. (Wiley-VCH, Morlenbach, Germany; 2005).
- Hendry P. & Hannan G. Detection and quantitation of unlabeled nucleic acids in polyacrylamide gels. Biotechniques 20, 258–264 (1996). [DOI] [PubMed] [Google Scholar]
- Andrus A. & Kuimelis R. G. Polyacrylamide gel electrophoresis (PAGE) of synthetic nucleic acids. Current protocols in nucleic acid chemistry/edited by Serge, L. Beaucage, ... [et al.] Chapter 10, Unit 10 14 (2001). [DOI] [PubMed] [Google Scholar]
- Golden B. L. in Methods in Molecular Biology, Vol. 363: Macromolecular Crystallography Protocols. (ed. S.R. Doublié) 247 (Humana Press, TotowaNJ; 2007).
- Clarke P. A. in Methods in Molecular Biology: RNA-Protein Interaction Protocols, Vol. 118. (ed. S.R. Haynes) 9 (Humana Press, TotowaNew Jersey; 1999).
- Ditzler M. A., Rueda D., Mo J., Hakansson K. & Walter N. G. A rugged free energy landscape separates multiple functional RNA folds throughout denaturation. Nucleic acids research 36, 7088–7099 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong X., Tao R. & Li Z. Quantification of RNA damage by reverse transcription polymerase chain reactions. Anal Biochem 357, 58–67 (2006). [DOI] [PubMed] [Google Scholar]
- Kladwang W., Vanlang C. C., Cordero P. & Das R. Understanding the errors of SHAPE-directed RNA structure modeling. Biochemistry 50, 8049–8056 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kladwang W., Cordero P. & Das R. A mutate-and-map strategy accurately infers the base pairs of a 35-nucleotide model RNA. RNA 17, 522–534 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal M. & Breaker R. R. Adenine riboswitches and gene activation by disruption of a transcription terminator. Nat struct mol biol 11, 29–35 (2004). [DOI] [PubMed] [Google Scholar]
- Sudarsan N. et al. Riboswitches in eubacteria sense the second messenger cyclic di-GMP. Science 321, 411–413 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal M. et al. A glycine-dependent riboswitch that uses cooperative binding to control gene expression. Science 306, 275–279 (2004). [DOI] [PubMed] [Google Scholar]
- Murphy F. L. & Cech T. R. An independently folding domain of RNA tertiary structure within the Tetrahymena ribozyme. Biochemistry 32, 5291–5300 (1993). [DOI] [PubMed] [Google Scholar]
- Byrne R. T., Konevega A. L., Rodnina M. V. & Antson A. A. The crystal structure of unmodified tRNAPhe from Escherichia coli. Nucleic acids research 38, 4154–4162 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correll C. C., Freeborn B., Moore P. B. & Steitz T. A. Metals, motifs, and recognition in the crystal structure of a 5S rRNA domain. Cell 91, 705–712 (1997). [DOI] [PubMed] [Google Scholar]
- Chiari M., Michelettie C., Righetti P. G. & Poli G. Polyacrylamide gel polymerization under non-oxidizing conditions, as monitored by capillary zone electrophoresis. J Chromatography 598, 287–297 (1992). [Google Scholar]
- Yoon S. et al. HiTRACE: high-throughput robust analysis for capillary electrophoresis. Bioinformatics 27, 1798–1805 (2011). [DOI] [PubMed] [Google Scholar]
- Sawa H. & Abelson J. Evidence for a base-pairing interaction between U6 small nuclear RNA and 5′ splice site during the splicing reaction in yeast. Proc Natl Acad Sci U S A 89, 11269–11273 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravanat J. L., Douki T. & Cadet J. Direct and indirect effects of UV radiation on DNA and its components. J Photochem Photobiol B 63, 88–102 (2001). [DOI] [PubMed] [Google Scholar]
- Saitou M. & Hieda K. Dithymine photodimers and photodecomposition products of thymidylyl-thymidine induced by ultraviolet radiation from 150 to 300 nm. Radiat Res 140, 215–220 (1994). [PubMed] [Google Scholar]
- Xia T. et al. Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson-Crick base pairs. Biochemistry 37, 14719–14735 (1998). [DOI] [PubMed] [Google Scholar]
- Lukavsky P. J. & Puglisi J. D. Large-scale preparation and purification of polyacrylamide-free RNA oligonucleotides. Rna 10, 889–893 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieft J. S. & Batey R. T. A general method for rapid and nondenaturing purification of RNAs. RNA 10, 988–995 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batey R. T. & Kieft J. S. Improved native affinity purification of RNA. RNA 13, 1384–1389 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kladwang W. & Das R. A mutate-and-map strategy for inferring base pairs in structured nucleic acids: proof of concept on a DNA/RNA helix. Biochemistry 49, 7414–7416 (2010). [DOI] [PubMed] [Google Scholar]
- Kladwang W., VanLang C. C., Cordero P. & Das R. A two-dimensional mutate-and-map strategy for non-coding RNA structure. Nat Chem 3, 954–962 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W., Muller J. G., Rachlin E. M. & Burrows C. J. Characterization of spiroiminodihydantoin as a product of one-electron oxidation of 8-Oxo-7,8-dihydroguanosine. Org Lett 2, 613–616 (2000). [DOI] [PubMed] [Google Scholar]
- Aviran S. et al. Modeling and automation of sequencing-based characterization of RNA structure. Proc Natl Acad Sci U S A 108, 11069–11074 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information for