

Abstract
The two most common genetic developmental disorders that cause intellectual disability are Down syndrome (DS) and Fragile X syndrome (FXS). Although the genetics and behavioral hallmarks of these two disorders are distinct, common underlying defects in neural development may lead to the cognitive impairment characteristic of both. Human neural progenitor cells (hNPCs) enable the study of prenatal human brain development in these developmental disorders. We therefore tested whether there are common affected molecular pathways in FXS and DS hNPCs that may be indicators of the fundamental developmental causes of intellectual disability. Comparison of gene expression data from FXS and DS (disorder group) hNPCs to unaffected hNPCs indicated genes in specific signal transduction cascades are dysregulated. Importantly, altered expression of genes in these signaling pathways did not emerge when the two disorder hNPCs were analyzed separately. Specifically, genes in the mitogen-activated protein kinases (MAPK/ERK) and calcium signaling pathways are mis-expressed in disorder hNPCs. These results suggest that DS and FXS hNPCs do not communicate or respond appropriately to extracellular cues during neural development. These results validate the use of hNPCs as a tool to assess complex cell functions during neural development and suggest that defects in the pathways identified could have profound effects on how neural progenitor cells survive, proliferate and differentiate, thereby leading to intellectual disability.
Keywords: Down syndrome, Fragile X syndrome, developmental disorders, intellectual disability, mental impairment, neurogenesis, neural progenitor cells, human, transcriptional analysis, MAPK
Introduction
Intellectual disability (ID) is defined as limited mental capacity and difficulty with adaptive behaviors; the prevalence in the U.S. is approximately 3% [1]. ID characteristic of many neurodevelopmental disorders is caused by mistakes made during the formation of the brain that lead to malfunctions in crucial neurological pathways, at the molecular, cellular and connectivity levels. The two most common genetic developmental disorders characterized by ID are Down syndrome (DS or trisomy 21) and Fragile X syndrome (FXS), which are caused by very different genetic mutations. Down syndrome is caused by triplication of chromosome 21 (HSA21) while Fragile X syndrome is a single gene disorder.
HSA21 is the smallest human chromosome comprised of 225 to 400 genes [2,3]. It is not clear how trisomy 21 causes the mental impairment associated with DS. Most lines of research have focused on the premise that specific genes located on chromosome 21 are responsible for DS symptoms and that these genes may exert direct effects that lead to manifestations of DS, or have secondary effects on other genes. Known HSA21 genes include those that encode cell adhesion molecules (e.g. NCAM2, DSCAM), signaling molecules/kinases (e.g. DYRK1a), and transcription factors (e.g. ETS2). Several genes on HSA21 have been implicated in DS and other disorders: familial Alzheimer’s disease (APP), familial amyotrophic lateral sclerosis (SOD1), and a predisposition for leukemia (AML1, GATA1). Based on the phenotypic variability of DS individuals, genetic, environmental, and stochastic influences are likely to play a role in DS [4-6].
In contrast, FXS is caused by mutational expansion of a CGG repeat in a single gene, FMR1, which leads to hypermethylation, and transcriptional silencing [7,8]. FMR1 encodes the Fragile X Mental Retardation Protein (FMRP) which is an mRNA binding protein involved in control of the timing and location of translation of specific mRNAs [9-11]. FXS-associated translational dysregulation causes wide-ranging neurological deficits including severe impairments of biological rhythms, learning processes, and memory consolidation [12].
Because the bulk of brain formation in humans occurs prenatally, it is challenging to define the neurodevelopmental mistakes that occur in these developmental disorders. Human stem and progenitor cells enable the study of specific aspects of brain formation in developmental disorders caused by known mutations and have been reported for both FXS and DS [13-21]. Previous studies using human neural progenitor cells (hNPCs) have revealed that these cells have attributes that corroborate in vivo aspects of both of these disorders [13,14,19-23] Yet, it is still not well understood how the DS or FXS mutations cause neurodevelopmental defects, nor how defects in neural development in these disorders lead to ID.
It is possible that common cellular and molecular pathways are affected during neural development in both DS and FXS that lead to aspects of cognitive impairment. We tested this hypothesis by comparing gene expression in FXS and DS hNPCs (disorder group) to unaffected control hNPCs and found that genes in specific signal transduction cascades are misexpressed in the disorder group. Specifically, genes in the MAPK/ERK and calcium signaling pathways are misex-pressed in disorder hNPCs. Our results suggest that DS and FXS hNPCs do not communicate or respond appropriately to extracellular cues during neural development. As both of these pathways are crucial in the regulation of proliferation and differentiation, changes in these pathways may alter neural progenitor cells’ ability to function properly.
Materials and methods
Cells
Isolation and characterization of DS and FXS human neural progenitor cells (hNPCs) have been previously described [19,14,24]. Briefly, two DS hNPC lines (from 13 weeks gestation cerebral cortical tissue), one FXS hNPC line (from 14 weeks gestation cerebral cortical tissue) and three gestationally age matched unaffected controls were used to generate the data used in this study.
Affymetrix GeneChip data analysis
Analysis of gene expression differences in DS hNPC and FXS hNPC compared separately to unaffected control hNPCs has been previously reported [19,14]. For this study, transcriptional expression data from these previous studies were combined to form a disorder group, which was compared to control. Log transformed GeneChip data was analyzed and statistical analysis conducted using the GeneSifter software (http://www.geospiza.com/Products/AnalysisEdition.shtml). Student t-tests were conducted for each data set with only genes with a p value < 0.05 being considered in the statistical analysis.
Stimulation of MAPK pathway
Control, FXS, and DS hNPCs were starved overnight and then stimulated with Epidermal Growth Factor (EGF, 100ng/ml, Peprotech, Rocky Hill NJ) for 30 minutes. Quantitative reverse transcriptase-polymerase chain reaction was used to quantitate c-FOS expression after stimulation.
Real-time quantitative reverse transcriptase-polymerase chain reaction
Real-time quantitative reverse transcriptase-polymerase chain reaction (RT-qPCR) was performed using SYBR Green for several genes that were found to have biological significance. In brief, RNA was extracted from two DS hNPC lines, one FXS hNPC line and three euploid control hNPC lines using RNeasy Mini Kit (Qiagen, Valencia, CA). Total RNA was additionally cleaned up and concentrated using the RNeasy MinElute Cleanup Kit (Qiagen, Valencia, CA). RNA from two DS lines was pooled prior to reverse transcription. RNA from three age-matched control cell lines was pooled prior to reverse transcription. Equal amounts of RNA from each sample were pooled. Reverse transcription was performed for control, FXS and DS samples using the Invitrogen SuperScript® III First-Strand Synthesis System (Invitrogen, Carlsbad, CA). Quantitative PCR was performed using SYBR® Green PCR Master Mix (Applied Biosystems, Foster City, CA). Immediate detection of the PCR product is measured by the increase in fluorescence caused by the binding of SYBR® Green dye to the control, FXS or DS double-stranded DNA. Beta-actin was used as internal control in PCR amplification, and the comparative Ct method was used for relative quantification of expression. For each gene, at least two qPCR runs were completed using separate cDNAs from separate RNA pools.
Results
Previous studies have shown that human neural progenitor cells (hNPCs) are a valuable tool to study the influence of genetic factors on early neural development in Down syndrome (DS) and Fragile X syndrome (FXS) [13,14,19-23]. In this study, we compared gene expression changes in DS and FXS hNPCs (Disorder) to unaffected control hNPCs. The results implicate aberrant MAPK and calcium signaling in both FXS and DS during human neural development.
Gene expression profiling was previously performed using Affymetrix U133 Plus 2.0 arrays and reported comparing one FXS hNPC line or a pool of two DS hNPC lines, respectively, to a pool of three control hNPC lines [19,14]. Transcriptional expression data from these previous studies were combined and analyzed to determine whether there are common gene expression differences in these two disorders when compared to controls. In order to obtain information about the relationship of the three groups (FXS, DS and control), the groups were analyzed separately and then the FXS and DS data was combined to create a disorder group that was used for subsequent analysis. Pairwise comparisons of the overall gene expression levels in each group were compared to each other (Figure 1A). This analysis yielded Pearson correlation coefficients (r) close to 1 for all comparisons and is highest (0.99) when comparing the control to disorder (FXS+DS) data. These results indicate that the datasets are nearly identical and suggest that gene expression differences that we identify are significant and not random. Hierarchical clustering shows that the control and FX data are more similar to each other than to the DS group (Figure 1B), which is not unexpected as FXS hNPCs have fewer gene expression changes when compared to controls than DS hNPCs [19,14].
Figure 1.
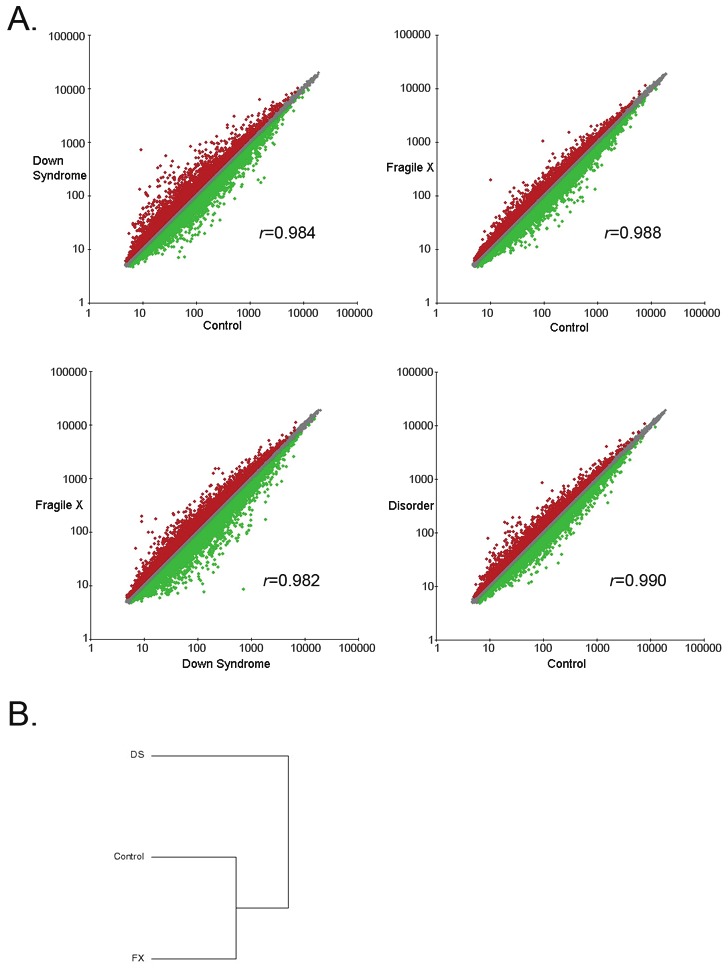
Pairwise comparisons of overall gene expression levels in FXS and DS hNPCs. Microarray expression data of the three groups (FXS, DS and control) were analyzed using Genesifter expression analysis software. A) Pairwise comparisons of the overall gene expression levels in each group were compared to each other and the Pearson correlation coefficients (r) were calculated. B) Hierarchical clustering shows that the control and FX data are more similar to each other than to the DS.
Gene expression changes greater than 1.2 fold with a p value of ≤ 0.05 in the disorder group compared to controls were considered. Transcriptional regulation was affected throughout the genome, with nearly equal transcript expression being upregulated as downregulated in the disorder hNPCs. Gene ontology analysis of genes whose expression changed in the disorder group revealed that many of the genes are involved in cell growth and proliferation and brain development which are expected in an ID model (Table 1). KEGG analysis revealed that particular signaling cascades showed transcriptional dysregulation in the disorder group hNPCs including expression of genes involved in focal adhesion, many components of the mitogen-activated protein kinase, extracellular signal regulated kinase (MAPK/ERK), and calcium signaling genes (Table 2).
Table 1.
Gene ontology analysis of genes whose expression is changed in disorder hNPCs.
| Gene Identifier | Number of genes | Biological process |
|---|---|---|
| ADRA1A, API5, ATP7A, BAG5, BCL10, BCL6, BRAF, BTG1, CARD8, CASP6, CLN8, CYCS, DLC1, DOCK1, EDAR, EGLN3, ELMO2, ERBB3, ERC1, EYA1, FAIM3, FGF2, GADD45A, HIP1, HIPK3, IFIH1, IL4, ISG20L1, KCNIP3, MAPK8IP2, MICA, MMP9, MNT, NF1, NME3, NOTCH2, NRAS, P2RX4, PHLDA1, PHLDA2, PIK3R2, PPP3R1, PRKAA1, PSEN1, PTGER3, RABEP1, RFFL, RUNX3, SCG2, SELS, SFRP1, SON, SST, TBX3, TNFAIP3, TNFRSF1B, TP63, TRIM69, TTBK2, UACA | 60 | Apoptosis, Cell death, Death, Programmed cell death |
| ACHE, ADRA1A, APBB2, APC, ATP7A, ATP8A2, AXIN2, BCAT1, BCL10, BCL6, BLM, BMPR1B, BTG1, BTG3, CABLES1, CACNB2, CALM3, CAPN3, CCNB2, CCND1, CDK6, CDK9, CHRNA7, CLIP1, CLN8, COL4A4, CROCC, DAB2, DAZL, DIRAS3, DLC1, DOCK2, DST, EGF, EID2, EMP1, EPHB3, ERBB2, ERBB3, ESR2, EYA1, FES, FGF2, FGFR2, FHL1, FLT4, FNTB, FTH1, GADD45A, GAP43, GAS2L1, GHR, GHRHR, GJC1, GLI1, GRIN2A, GSTM3, H1FNT, H1FOO, HHEX, HOXC8, HUS1, ICOSLG, IFITM1, IGFBP5, IGSF11, IL4, INSR, IRS2, KANK1, KITLG, KLF4, KRT7, LEF1, LEPRE1, LGR4, LZTS1, MAP9, MDM2, MNT, MT3, MYCBP2, MYH11, NBN, NEDD9, NF1, NOTCH2, NRAS, NUMBL, OSGIN2, PARD6G, PGF, PMS1, PPARG, PPP3CA, PRKG1, PSEN1, RBL1, RIF1, ROBO1, RORB, RUNX2, RUNX3, SCG2, SEMA5A, SENP5, SEPT11, SERPINF1, SESN3, SLC29A2, SLIT1, SMAD1, SPAG6, SST, SYCP1, TBX3, TIMP2, TMOD1, TNN, TNS3, TOB1, TP63, TYR, WISP1, WNT7A, ZEB2 | 126 | Cell cycle, Cell division, Cell growth, Cell proliferation |
| ACHE, ADAM22, AFF2, AHNAK, APBA2, ARHGAP2, ATP7A, AVIL, AXIN2, BCL10, BCL6, BLM, BMPR1B, BTG1, CABLES1, CACNB2, CAND1, CAPN3, CCND1, CDK6, CLN8, COL4A4, DAZ2, DAZL, DLC1, DLX2, DOCK2, DOK5, DSCAM, EDAR, EGF, EGR1, EGR2, EID2, EPHB3, ERBB2, ERBB3, EYA1, FGF2, FHL1, FOS, GAL, GAP43, GGNBP2, GHRHR, GJC1, GLI1, GRIN2A, GSTM3, H1FNT, HECTD1, HEY2, HHEX, HOXC8, HRB, IGSF10, IL4, IRS2, KALRN, KLF4, LAMA3, LECT1, LEF1, LEP, MMP9, MPPED2, MT3, MYH11, NEUROD6, NF1, NLGN2, NOTCH2, NR2C2, NRAS, NUMBL, ODF3, OLFM1, OSGIN2, PBX1, PCDHA6, PCDHB11, PCDHB12, PCDHB13, PCDHB3, PCDHB5, PCDHB6, PGF, PPARG, PPP3CA, PRKG1, PSEN1, PURB, ROBO1, RORB, RUNX2, S100A13, SEMA4A, SEMA5A, SERPINF1, SFRP1, SLFN5, SLIT1, SMA4, SMAD1, SMAD2, SPAG6, SPATA19, SPATA5, SRY, ST8S1A4, TBCB, TBX3, TIMP2, TMOD1, TMOD2, TNN, TOB1, UPK1B, WNT7A, WWTR1, ZEB2 | 121 | Brain development, Cell differentiation, cell fate, Neurogenesis, Glial cell differentiation |
| AFF2, CHRNA7, CLN8, GRIA1, GRIN2A, ITGA5, MAN2B1, NF1, NRAS, PSEN1, TMOD2 | 11 | Learning and Memory |
Table 2.
KEGG pathway analysis of gene expression changes in disorder hNPCs.
| Gene Identifier | KEGG pathway |
|---|---|
| ADCY2, ADRA1A, CACNA1B, CACNA1G, CALM3, CHP, CHRNA7, ERBB2, ERBB3, GNA14, GRIN2A, GRM5, P2RX2, P2RX4, P2RX6, PDE1A, PLCB1, PLCD1, PLCG2, PPP3CA, PPP3R1, PRKCB1, PTGER3, SLC8A1 | Calcium Signaling Pathway |
| ARHGAP5, BRAF, CCND1, COL11A1, COL4A4, DOCK1, EGF, ERBB2, IBSP, ITGA5, ITGA6, LAMA3, PARVA, PGF, PIK3R2, PRKCB1, PXN, RAPGEF1, SOS1, TNN, VAV3 | Focal Adhesion Pathway |
| BRAF, CACNA1B, CACNA1G, CACNA2D2, CACNB2, CACNG3, CACNG8, CHP, EGF, GADD45A, MAP3K71P1, MAPK81P2, NRAS, PLA2G4A, PLA2G5, PPP3CA, PPP3R1, PRKCB1, RPS6KA5, SOS1 | MAPK Signaling Pathway |
We focused our attention on the expression changes in the disorder group that involve genes in the MAPK/ERK pathway because this pathway is particularly crucial during neural development where it controls many signaling events. KEGG analysis of the microarray data revealed that gene expression changes in disorder hNPCs occur at all levels in the MAP/ERK pathway: ligand (EGF, FGF), receptor (FGFR), cytoplasmic effector (SOS, Ras, NF1, RafB) and target gene (c-FOS) (Figure 2). To validate that genes in the MAP/ERK pathway are misexpressed in disorder hNPCs, quantitative PCR (qPCR) of selected genes was carried out on DS and FXS hNPCs and confirmed that most of the MAP/ERK pathway genes are misexpressed in the FXS hNPCs and in DS hNPCs (Figure 3). The FXS and DS hNPCs behave differently in terms of the directionality (upregulated vs. downregulated) of gene expression changes, but the entire pathway is affected in each disorder cell type.
Figure 2.
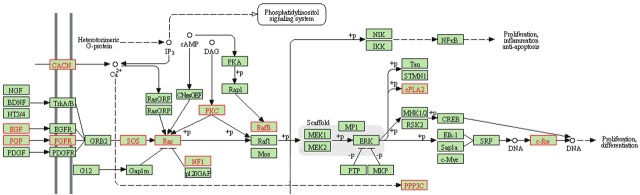
Gene expression changes in disorder hNPCs that are in the MAPK/ERK signal transduction pathway. Microarray expression data of disorder hNPCs was compared to control and analyzed using Genesifter expression analysis software. KEGG analysis revealed that many components of the mitogen-activated protein kinase, extracellular signal regulated kinase (MAPK/ERK) pathway are misexpressed. Misexpressed genes are highlighted in red text.
Figure 3.
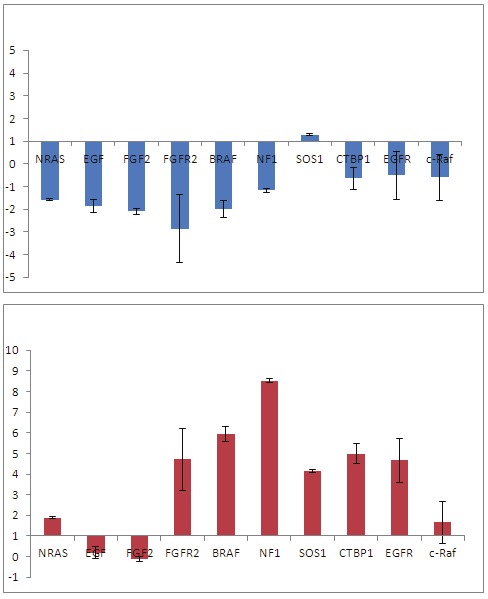
Quantitative PCR (qPCR) validation of gene expression changes in MAPK/ERK pathway in disorder hNPCs. qPCR of selected genes in the MAPK/ERK pathway was carried out on DS and FXS hNPCs. Most of the MAP/ERK pathway genes are misexpressed in FXS hNPCs and in DS hNPCs.
To determine whether the gene expression differences observed in the MAP/ERK pathway result in cellular changes in disorder hNPCs, we stimulated the pathway in cells and assayed downstream gene expression. Epidermal Growth Factor (EGF) stimulates the MAP/ERK pathway and hNPCs are responsive to EGF [25-27]. EGF binds epidermal growth factor receptor (EGFR) on the cell surface, initiating a signal transduction cascade of phosphorylation events of cytoplasmic proteins that ultimately results in transcription of c-fos, an immediate early gene in the nucleus of the cell. The expression of the MAP/ERK pathway target gene, c-FOS, was assayed. Figure 4 shows that both control and disorder (FXS and DS) hNPCs respond to EGF stimulation by expression of c-FOS. However, the disorder hNPCs have a slower response. Together with the gene expression data, these results indicate that disorder hNPCs respond aberrantly to stimulation of the MAPK/ERK pathway.
Figure 4.
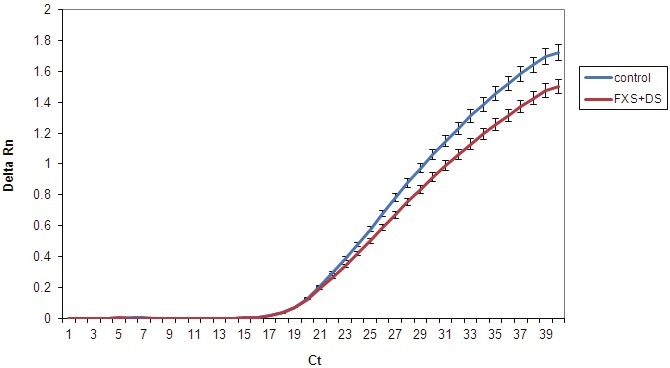
Differential response of Disorder hNPCs to MAPK/ERK pathway stimulation. hNPCs were stimulated with Epidermal Growth Factor (EGF) and assayed for downstream gene expression of c-FOS by qPCR. Graph shows the detection of c-FOS (Delta Rn) at each cycle of PCR (Ct). All hNPCs responded to EGF with expression of c-FOS but disorder (FXS+DS) hNPCs showed a trend that was slower and with reduced expression of c-FOS.
Discussion
The two most common genetic developmental disorders characterized by ID are DS and FXS, which are initiated by very different genetic mutations. DS is caused by trisomy 21 while FXS is a single gene disorder. The many genes on Hsa21 are varied in their function and the biological pathways that they are involved in are diverse [2,28,29,4]. In contrast, the single gene affected in FXS, FMR1, encodes for an RNA binding protein whose function is to regulate the translation of specific mRNAs throughout development of the nervous system [9-11].
Cross-disorder comparisons that focus on molecular and cellular components of neural development are rare in the study of ID [30,31]. Consequently, there has been little evaluation of the aberrant cellular and molecular events that occur early in neural development that may be common to both FXS and DS. We took advantage of human neural progenitor cells (hNPCs) to study the influence of genetic factors on early neural development and asked whether there are common gene expression differences in these two disorders that may be indicators of the underlying causes of mental impairment and deficiencies in learning and memory. Our results indicate that genes in specific signal transduction cascades are misexpressed in the disorder group. Our results suggest that DS and FXS cells do not communicate or respond appropriately to extracellular cues during neural development. The gene expression changes occur primarily in signaling pathways that are important for growth, proliferation, and differentiation of hNPCs and may affect the function of these cells. Furthermore, continued misregulation of these genes as the progenitor cells differentiate and mature could have an impact on the formation and functioning of the brain. Miscommunication can be particularly destructive in the brain where it can lead to mental impairment.
The MAPK/ERK pathway is a signal transduction pathway that couples intracellular responses to the binding of growth factors to cell surface receptors [32]. This pathway transduces a large variety of external signals, leading to a wide range of cellular responses, including growth, differentiation, inflammation and apoptosis and so is particularly crucial during neural development. Disruption in MAPK/ERK signaling has been linked to many neurodevelopmental disorders and MAPK/ERK signaling is crucial during neuronal maturation and plasticity and, ultimately, to learning and memory [33-36]. In DS in particular, the overexpression of HSA21 genes does in fact predict perturbation of MAPK/ERK signaling [37,38]. Therefore, our results fit well with the idea that MAPK/ERK signaling is crucial for neural progenitor development and that mis-expression of genes in this pathway is a characteristic of neurodevelopmental disorders.
It is critical to note that dysregulation of the identified signaling pathways did not emerge when the two disorder hNPCs were analyzed separately. Furthermore, the direction of expression changes in individual genes in each disorder was not consistent. If common pathways are being sought, it may be necessary to pool data from different disorders rather than comparing the data separately and to focus on pathway analysis rather than specific genes. For FXS and DS, comparisons of these two disorders has been carried out in terms of their behavioral phenotypes and it has been recently appreciated that the two syndromes share common mechanisms of dysfunction that may be therapeutic targets [39-41]. Our results taken in this context highlight the importance of cross-disorder comparisons to identify affected pathways for both basic study and for potential therapeutic strategies.
Our results also validate the use of hNPCs to identify complex cell functions that go awry during neural development. However, the advent of induced pluripotent stem cell (iPSC) technology permits the establishment of disorder-specific pluripotent cells that can be used to study more aspects of brain formation in developmental disorders caused by known mutations. It will be crucial to test whether neural progenitor cells differentiated from DS and FXS iPSCs have similar dysregulation in these pathways and, more importantly, whether other aspects of neural development are affected.
Acknowledgements
We thank Elizabeth Capowski for technical advice and critical reading of the manuscript. This work was supported by a Charles Epstein Research Award from the National Down Syndrome Society (A.B.), a FRAXA Foundation Research Award (A.B. and C.N.S.), the Charles and MaryClaire Phipps Foundation and in part by a core grant from the National Institute of Child Health and Human Development (P30 HD03352). The authors declare that there are no conflicts of interest.
References
- 1.Larson SA, Lakin KC, Anderson L, Kwak N, Lee JH, Anderson D. Prevalence of mental retardation and developmental disabilities: estimates from the 1994/1995 National Health Interview Survey Disability Supplements. Am J Ment Retard. 2001;106:231–252. [PubMed] [Google Scholar]
- 2.Gardiner K, Davisson M. The sequence of human chromosome 21 and implications for research into Down syndrome. Genome Biol. 2000;1:REVIEWS0002. doi: 10.1186/gb-2000-1-2-reviews0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gardiner K, Slavov D, Bechtel L, Davisson M. Annotation of human chromosome 21 for relevance to Down syndrome: gene structure and expression analysis. Genomics. 2002;79:833–843. doi: 10.1006/geno.2002.6782. [DOI] [PubMed] [Google Scholar]
- 4.Antonarakis SE. Chromosome 21: from sequence to applications. Curr Opin Genet Dev. 2001;11:241–246. doi: 10.1016/s0959-437x(00)00185-4. [DOI] [PubMed] [Google Scholar]
- 5.Reeves RH, Baxter LL, Richtsmeier JT. Too much of a good thing: mechanisms of gene action in Down syndrome. Trends Genet. 2001;17:83–88. doi: 10.1016/s0168-9525(00)02172-7. [DOI] [PubMed] [Google Scholar]
- 6.Roper RJ, Reeves RH. Understanding the basis for Down syndrome phenotypes. PLoS Genet. 2006;2:e50. doi: 10.1371/journal.pgen.0020050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra BA, Caskey CT, Nelson DL. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. 1991;66:817–822. doi: 10.1016/0092-8674(91)90125-i. [DOI] [PubMed] [Google Scholar]
- 8.Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, Eussen BE, van Ommen GJB, Blonden LAJ, Riggins GJ, Chastain JL, Kunst CB, Galjaard H, Caskey CT, Nelson DL, Oostra BA, Warren ST. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 9.Darnell J. Defects in translational regulation contributing to human cognitive and behavioral disease. Curr Opin Genet Dev. 2011;21:465–473. doi: 10.1016/j.gde.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bassell GJ, Warren ST. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008;60:201–214. doi: 10.1016/j.neuron.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bear MF, Dolen G, Osterweil E, Nagarajan N. Fragile X: translation in action. Neuropsychopharmacology. 2008;33:84–87. doi: 10.1038/sj.npp.1301610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hagerman RJ, Hagerman PJ. Fragile X syndrome. 2nd 2002. [Google Scholar]
- 13.Bhattacharyya A, Svendsen CN. Human neural stem cells: a new tool for studying cortical development in Down's syndrome. Genes Brain Behav. 2003;2:179–186. doi: 10.1034/j.1601-183x.2003.00025.x. [DOI] [PubMed] [Google Scholar]
- 14.Bhattacharyya A, McMillan E, Chen SI, Wallace K, Svendsen CN. A critical period in cortical interneuron neurogenesis in Down syndrome revealed by human neural progenitor cells. Developmental Neuroscience. 2009;31:497–510. doi: 10.1159/000236899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Biancotti JC, Narwani K, Buehler N, Mandefro B, Golan-Lev T, Yanuka O, Clark A, Hill D, Benvenisty N, Lavon N. Human embryonic stem cells as models for aneuploid chromosomal syndromes. Stem Cells. 2010;28:1530–1540. doi: 10.1002/stem.483. [DOI] [PubMed] [Google Scholar]
- 16.Eiges R, Urbach A, Malcov M, Frumkin T, Schwartz T, Amit A, Yaron Y, Eden A, Yanuka O, Benvenisty N, Ben Yosef D. Developmental study of fragile X syndrome using human embryonic stem cells derived from preimplantation genetically diagnosed embryos. Cell Stem Cell. 2007;1:568–577. doi: 10.1016/j.stem.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 17.Lu J, Delli-Bovi LC, Hecht J, Folkerth R, Sheen VL. Generation of neural stem cells from discarded human fetal cortical tissue. J Vis Exp. 2011;(51) doi: 10.3791/2681. pii: 2681. doi: 10.3791/2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, Lensch MW, Cowan C, Hochedlinger K, Daley GQ. Disease-specific induced pluripotent stem cells. Cell. 2008;134:877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bhattacharyya A, McMillan E, Tubon T, Wallace K, Capowski EE, Svendsen CN. Normal neurogenesis but abnormal gene expression in human Fragile X cortical progenitor cells. Stem Cells and Development. 2008;17:107–117. doi: 10.1089/scd.2007.0073. [DOI] [PubMed] [Google Scholar]
- 20.Esposito G, Imitola J, Lu J, De Filippis D, Scuderi C, Ganesh VS, Folkerth R, Hecht J, Shin S, Iuvone T, Chesnut J, Steardo L, Sheen V. Genomic and functional profiling of human Down syndrome neural progenitors implicates S100B and Aquaporin 4 in cell injury. Hum Mol Genet. 2008;17:440–457. doi: 10.1093/hmg/ddm322. [DOI] [PubMed] [Google Scholar]
- 21.Bahn S, Mimmack M, Ryan M, Caldwell MA, Jauniaux E, Starkey M, Svendsen CN, Emson P. Neuronal target genes of the neuronrestrictive silencer factor in neurospheres derived from fetuses with Down's syndrome: a gene expression study. Lancet. 2002;359:310–315. doi: 10.1016/S0140-6736(02)07497-4. [DOI] [PubMed] [Google Scholar]
- 22.Cairney CJ, Sanguinetti G, Ranghini E, Chantry AD, Nostro MC, Bhattacharyya A, Svendsen CN, Keith WN, Bellantuono I. A systems biology approach to Down syndrome: identification of Notch/Wnt dysregulation in a model of stem cells aging. Biochim Biophys Acta. 2009;1792:353–363. doi: 10.1016/j.bbadis.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 23.Lu J, Esposito G, Scuderi C, Steardo L, Delli-Bovi LC, Hecht JL, Dickinson BC, Chang CJ, Mori T, Sheen V. S100B and APP Promote a Gliocentric Shift and Impaired Neurogenesis in Down Syndrome Neural Progenitors. PLoS One. 2011;6:e22126. doi: 10.1371/journal.pone.0022126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Svendsen CN, ter Borg MG, Armstrong RJ, Rosser AE, Chandran S, Ostenfeld T, Caldwell MA. A new method for the rapid and long term growth of human neural precursor cells. J Neurosci Methods. 1998;85:141–152. doi: 10.1016/s0165-0270(98)00126-5. [DOI] [PubMed] [Google Scholar]
- 25.Caldwell MA, He X, Wilkie N, Pollack S, Marshall G, Wafford KA, Svendsen CN. Growth factors regulate the survival and fate of cells derived from human neurospheres. Nat Biotechnol. 2001;19:475–479. doi: 10.1038/88158. [DOI] [PubMed] [Google Scholar]
- 26.Nelson AD, Suzuki M, Svendsen CN. A high concentration of epidermal growth factor increases the growth and survival of neurogenic radial glial cells within human neurosphere cultures. Stem Cells. 2008;26:348–355. doi: 10.1634/stemcells.2007-0299. [DOI] [PubMed] [Google Scholar]
- 27.Ostenfeld T, Svendsen CN. Requirement for neurogenesis to proceed through the division of neuronal progenitors following differentiation of epidermal growth factor and fibroblast growth factor-2-responsive human neural stem cells. Stem Cells. 2004;22:798–811. doi: 10.1634/stemcells.22-5-798. [DOI] [PubMed] [Google Scholar]
- 28.Gardiner K, Herault Y, Lott IT, Antonarakis SE, Reeves RH, Dierssen M. Down syndrome: from understanding the neurobiology to therapy. J Neurosci. 2010;30:14943–14945. doi: 10.1523/JNEUROSCI.3728-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Epstein CJ, Epstein LB, Weil J, Cox DR. Trisomy 21: mechanisms and models. Ann N Y Acad Sci. 1982;396:107–118. doi: 10.1111/j.1749-6632.1982.tb26847.x. [DOI] [PubMed] [Google Scholar]
- 30.Hanson JE, Blank M, Valenzuela RA, Garner CC, Madison DV. The functional nature of synaptic circuitry is altered in area CA3 of the hippocampus in a mouse model of Down's syndrome. J Physiol. 2007;579:53–67. doi: 10.1113/jphysiol.2006.114868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanson JE, Madison DV. Presynaptic FMR1 genotype influences the degree of synaptic connectivity in a mosaic mouse model of fragile X syndrome. J Neurosci. 2007;27:4014–4018. doi: 10.1523/JNEUROSCI.4717-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rubinfeld H, Seger R. The ERK cascade: a prototype of MAPK signaling. Mol Biotechnol. 2005;31:151–174. doi: 10.1385/MB:31:2:151. [DOI] [PubMed] [Google Scholar]
- 33.Weeber EJ, Levenson JM, Sweatt JD. Molecular genetics of human cognition. Mol Interv. 2002;2:376–91, 339. doi: 10.1124/mi.2.6.376. [DOI] [PubMed] [Google Scholar]
- 34.Samuels IS, Karlo JC, Faruzzi AN, Pickering K, Herrup K, Sweatt JD, Saitta SC, Landreth GE. Deletion of ERK2 mitogen-activated protein kinase identifies its key roles in cortical neurogenesis and cognitive function. J Neurosci. 2008;28:6983–6995. doi: 10.1523/JNEUROSCI.0679-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Samuels IS, Saitta SC, Landreth GE. MAP'ing CNS development and cognition: an ERKsome process. Neuron. 2009;61:160–167. doi: 10.1016/j.neuron.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cohen S, Greenberg ME. Communication between the synapse and the nucleus in neuronal development, plasticity, and disease. Annu Rev Cell Dev Biol. 2008;24:183–209. doi: 10.1146/annurev.cellbio.24.110707.175235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gardiner K. Predicting pathway perturbations in Down syndrome. J Neural Transm Suppl. 2003;(67):21–37. doi: 10.1007/978-3-7091-6721-2_2. [DOI] [PubMed] [Google Scholar]
- 38.Gardiner K. Transcriptional Dysregulation in Down Syndrome: Predictions for Altered Protein Complex Stoichiometries and Post-translational Modifications, and Consequences for Learning/Behavior Genes ELK, CREB, and the Estrogen and Glucocorticoid Receptors. Behav Genet. 2006;36:439–453. doi: 10.1007/s10519-006-9051-1. [DOI] [PubMed] [Google Scholar]
- 39.Conners FA, Moore MS, Loveall SJ, Merrill EC. Memory profiles of Down, Williams, and fragile X syndromes: implications for reading development. J Dev Behav Pediatr. 2011;32:405–417. doi: 10.1097/DBP.0b013e3182168f95. [DOI] [PubMed] [Google Scholar]
- 40.Scerif G, Steele A. Neurocognitive development of attention across genetic syndromes: inspecting a disorder's dynamics through the lens of another. Prog Brain Res. 2011;189:285–301. doi: 10.1016/B978-0-444-53884-0.00030-0. [DOI] [PubMed] [Google Scholar]
- 41.Wetmore DZ, Garner CC. Emerging pharmacotherapies for neurodevelopmental disorders. J Dev Behav Pediatr. 2010;31:564–581. doi: 10.1097/DBP.0b013e3181ee3833. [DOI] [PMC free article] [PubMed] [Google Scholar]