

Abstract
Apoptosis is defined by specific morphological and biochemical characteristics including cell shrinkage (termed apoptotic volume decrease), a process that results from the regulation of ion channels and plasma membrane transporter activity. The Na+-K+-ATPase is the predominant pump that controls cell volume and plasma membrane potential in cells and alterations in its function have been suggested to be associated with apoptosis. We report here that the Na+-K+-ATPase inhibitor ouabain, potentiates apoptosis in the human lymphoma Jurkat cells exposed to Fas ligand (FasL) or Tumor necrosis factor--related apoptosis-inducing ligand (TRAIL) but not other apoptotic agents such as H2O2, thapsigargin or UV-C implicating a role for the Na+-K+-ATPase in death receptor-induced apoptosis. Interestingly, ouabain also potentiated perturbations in cell Ca2+ homeostasis only in conjunction with the apoptotic inducer FasL but not TRAIL. Ouabain did not affect alterations in the intracellular Ca2+ levels in response to H2O2, thapsigargin or UV-C. FasL-induced alterations in Ca2+ were not abolished in Ca2+-free medium but incubation of cells with BAPTA-AM inhibited both Ca2+ perturbations and the ouabain-induced potentiation of FasL-induced apoptosis. Our data suggest that the impairment of the Na+-K+-ATPase activity during apoptosis is linked to perturbations in cell Ca2+ homeostasis that modulate apoptosis induced by the activation of Fas by FasL.
Keywords: Ouabain, calcium, Na+-pump, Na+-K+-ATPase, apoptosis, Jurkat, cell volume, cell shrinkage, AVD, apoptotic volume decrease, extrinsic pathway, Fas ligand, TRAIL, CD95, Apo-1
INTRODUCTION
Apoptosis is a metabolically active process with importance in organ development and in the regulation and maintenance of cell turnover in various tissues during physiological as well as pathological conditions [1]. This form of programmed cell death is characterized by specific morphological features including cell shrinkage, chromatin condensation, and formation of apoptotic bodies [2]. The signaling cascades that regulate the progression of apoptosis have been extensively studied and characterized. In general both extrinsic and intrinsic pathways have been described for the activation of apoptosis. Induction of apoptosis via the extrinsic pathway is triggered by the activation of death receptors such as those activated by Fas ligand or FasL (Fas (CD95/Apo-1) and by the TNF-related apoptosis-inducing ligand or TRAIL (DR4, DR5). Activation of CD95, DR4 and DR5 leads primarily to the formation of the death-inducing signaling complex (DISC) formed by the recruitment of the (Fas-associated death domain (FADD), caspases 8 (and in some cases caspase 10) and the cellular FLICE-inhibitory protein (FLIP). Initiator caspase 8 is processed and activated and further amplifies the apoptotic cascade by activation of executioner caspases. When upon death receptor activation, cells have lower levels of DISC formation and active caspase-8 (Type II cells), the progression of the cell death program relies on an amplification loop induced by cleavage of the Bcl-2-family protein Bid by caspase 8 and its translocation to the mitochondrial to activate the intrinsic pathway of apoptosis [3, 4].
The intrinsic pathway of apoptosis is also referred to as the mitochondrial pathway. It is activated by a wide variety of stimuli including chemotheraputic and cytotoxic agents, stress (radiation, hyperglycemia, hypoxia, oxidative and osmotic stress) and cytokine withdrawal. Although the mechanisms by which these stimuli trigger apoptosis seems to differ between them, they all convey in the release of proapoptotic proteins from the mitochondria including Cyt C. Cytochrome C release induces the formation of the apoptosome, activation of caspase 9 and the further activation of the execution caspases [5, 6].
During apoptosis, loss of cell volume (apoptotic volume decrease, AVD) has been described as a process that facilitates the breakdown of the cell into smaller apoptotic bodies thus aiding their engulfment and consequently phagocytosis by macrophages [7]. We and others have shown that changes in intracellular ionic homeostasis are associated with the loss of cell volume and that these changes play a critical role in regulating the activity of nucleases and caspases during cell death [8-12]. However, the exact mechanisms by which changes in ionic homeostasis regulate apoptosis remains unclear.
When changes in cell volume occur, they normally result from ionic fluxes across the plasma membrane [13], therefore understanding the molecular basis of these ion changes during apoptosis may aid in our understanding of the cell death process. In general, the cells utilize large amounts of ATP to maintain ionic homeostasis required for normal life. In this respect, the Na+-K+-ATPase (Na+-pump) by consuming a considerable proportion of cellular ATP maintains a high K+ and a low Na+ concentration inside the cell, ensuring osmotic balance, cell volume, cytoplasmic pH, ionic gradients and Na+-coupled transport of nutrients and amino acids into the cells [12-14]. To date there is a excellent understanding of the Na+-pump’s structure, function and regulation in living cells [14, 15]. Ionic gradient imbalance, particularly those of Na+ and K+, is an early hallmark of initial stages of the apoptotic program. Several reports have associated this phenomenon to a reduction in the Na+-K+ ATPase activity during apoptosis [16-25]. Accordingly, inhibition of the Na+-K+ ATPase with cardiac glycosides, has been widely reported to induce cell toxicity and cell death [21, 26-37], or to enhance apoptosis induced by different stimuli in various model systems [17, 18, 38-40]. However, the molecular mechanisms involved in the regulation of apoptosis by impairment of the Na+-K+-ATPase are still unclear.
In the present study, we report that impairment of the Na+-K+-ATPase activity by ouabain potentiates apoptosis in Jurkat cells upon exposure to Fas ligand (FasL) and Tumor necrosis (TNF)-related apoptosis-inducing ligand (TRAIL), but not by other agents that are known to stimulate primarily the intrinsic mitochondrial apoptotic pathway. Additionally we observed that apoptosis by Fas activation and its potentiation by ouabain are associated with Ca2+ release from intracellular stores.
MATERIALS AND METHODS
Cell culture
The Jurkat cell clone J6 (human lymphoma) (empty vector transfected, SFFV-neo), was a kind gift from Dr., Gerald M. Cohen (Medical Research Council Toxicology Unit, University of Leicester, Leicester, United Kingdom) [41] and was cultured in RPMI 1640 supplemented with 10% heat-inactivated fetal calf serum, 2mM glutamine, 31mg/l penicillin, and 50mg/l streptomycin and 0.4mg/ml G418 (GIBCO/Invitrogen Co., Carlsbad, CA) at 37°C, 7% CO2 atmosphere. Jurkat cells (5-7 × 105 cells/ml) were incubated with distinct apoptotic inducers for both the extrinsic (FasL or TRAIL [Kamiya Biomedical, Seattle, WA]) and intrinsic (hydrogen peroxide [H2O2], thapsigargin [Sigma-Aldrich, St. Louis, MO] and 30mJ/cm2 UV-C light [Stratalinker 1800, Stratagene]) pathways of apoptosis over a period of 4h. When indicated, ouabain (Sigma-Aldrich) was added at the concentration indicated at the same time as the apoptotic stimuli and was present throughout the experiment.
Nominally Ca2+-free medium was prepared from powdered RPMI (Hyclone) without Ca2+ nitrate, supplemented as described above, and with addition of 0.5mM ethylene glycol tetraacetic acid, EGTA (Sigma-Aldrich). Intracellular Ca2+ was scavenged by incubation of the cells for 20min with the membrane permeant 1,2-bis-(o-Aminophenoxy)-ethane-N,N,N’,N’-tetraacetic acid, tetraacetoxymethyl ester, BAPTA-AM (25μM; Molecular Probes, Eugene, OR) prior to addition of ouabain and each of the apoptotic stimuli.
Measurement of cell size by flow cytometry
Light scattered in the forward direction is proportional to cell size, while light scattered at a 90° angle (side scatter) is proportional to cell density or granularity [42]. Cell size and changes in the light scattering properties of the cells were determined by flow cytometry as described previously [43] using a BD LSR II flow cytometer equipped with the BD FACSDiva Software (Becton Dickinson, San Jose, CA) for data analysis. For each sample, 10,000 cells were examined after being excited with a 488nm argon laser while their distribution was recorded on a forward-scatter versus side-scatter plot.
Measurement of changes in intracellular Na+, K+ and Ca2+ by flow cytometry
Changes in the intracellular ionic homeostasis were determined as follows. Jurkat cells were preloaded with either SBFI-AM or CoroNa Green (5μM, 1h) for Na+; PBFI-AM (5μM, 1h) for K+; and Fluo-3 AM (1μM; 30min) for Ca2+ (Molecular Probes, Eugene, OR) prior to the end of the incubation period needed to induce apoptosis. Fluorophores were diluted in dimethyl sulfoxide (DMSO) with a final concentration of 0.1%. Ten thousand cells were examined by sequential excitation of the cells containing PBFI, SBFI (340-350nm) or Fluo-3 (488nm) and examining their emissions at 425nm (PBFI/SBFI) or 530nm (Fluo-3).
Measurement of changes in plasma membrane potential
Changes in the plasma membrane potential were measured by flow cytometry using DiBAC4(3) (Molecular Probes). DiBAC4(3) was prepared in DMSO (0.1% final concentration). Thirty minutes prior to each time of examination, DiBAC4(3) was added to a final concentration of 150ng/ml. Ten thousand cells were examined under each condition with excitation performed using a 488nm argon laser, and fluorescent emission was detected at 530nm.
Changes in Plasma Membrane Integrity
To evaluate plasma membrane integrity, propidium iodide (PI) (10μg/ml; Sigma-Aldrich) was added to the cells prior to flow cytometry analysis. PI uptake was evaluated by its excitation with an Argon 488nm laser whereas emission was acquired with a 695/40 filter. High PI fluorescence indicates a loss of membrane integrity.
Analysis of degraded DNA content by flow cytometry
Samples were pelleted and washed in ice-cold PBS. Then, cells were fixed by the addition of 70% cold ethanol to a volume of 1.5ml, adjusted to 5ml with cold 70% ethanol, and stored at -20 °C overnight. Fixed cells were pelleted, washed once in PBS and stained in 1 ml of 20μg/ml PI, 1mg/ml RNase, in PBS for 20 min at RT. Seven thousand five hundred cells were analyzed by flow cytometry using a Becton Dickinson FACSort flow cytometer, and BD Cell Quest Software (Becton Dickinson) for analysis. Region gates were set on a PI area versus width dot plot to exclude cell debris and cell aggregates.
Determination of caspase 3/7-like activity by flow cytometry
Caspase 3/7-like activity was determined using CaspaTag In Situ Apoptosis Detection Kit (Millipore, Billerica MA). Cells were incubated for 1h prior to analysis at 37°C, 7% CO2 atmosphere with FLICA reagent (10μM FAM-DEVD-FMK)), a cell permeable caspase 3/7-like substrate that fluoresces upon cleavage at the DEVD site. Ten thousand cells were excited at 488nm and examined at 530nm for the fluorescence intensity of the FLICA substrate.
Statistical analysis
All calculations were performed using the JMP software (SAS Institute, Cary, NC). Means were compared by one-way analysis of variance followed by a Student’s t-test. A value of P < 0.05 was considered significant.
RESULTS
Death receptor-induced apoptosis is potentiated in the presence of ouabain
We used a flow cytometric-based approach to assess apoptotic volume decrease or cell shrinkage. As a cell shrinks, there is a decrease in the amount of forward-scattered light that is proportional to cell size in coordination with a change in side-scattered light that is relative to cell density or granularity. Figure 1A shows a reduction in the amount of forward-scattered light at each concentration of FasL in the absence (- Ouabain) or presence of ouabain (100μM) (cells outside of the gated area assigned for normal cells). Cell shrinkage or AVD was directly proportional to the concentration of FasL. The addition of ouabain increased the number of apoptotic cells for each concentration of FasL tested (Fig. 1A, Table 1). We next evaluated if ouabain enhanced the occurrence of other parameters of apoptosis. Figure 1B shows that the percentage of cells with degraded DNA content was also enhanced in the presence of ouabain for any given concentration of FasL. Similarly, Figure 1C shows that the percent of FLICA-positive cells indicating active caspase 3/7 like activity was also significantly higher in ouabain-exposed cells when treated with 50 and 100ng/ml FasL. Together, these data demonstrate that impairment of the Na+-K+-ATPase by ouabain potentiates FasL-induced apoptosis, as determined by AVD, DNA degradation and execution caspase activity.
Fig. 1. Effect of ouabain on FasL-induced apoptosis.
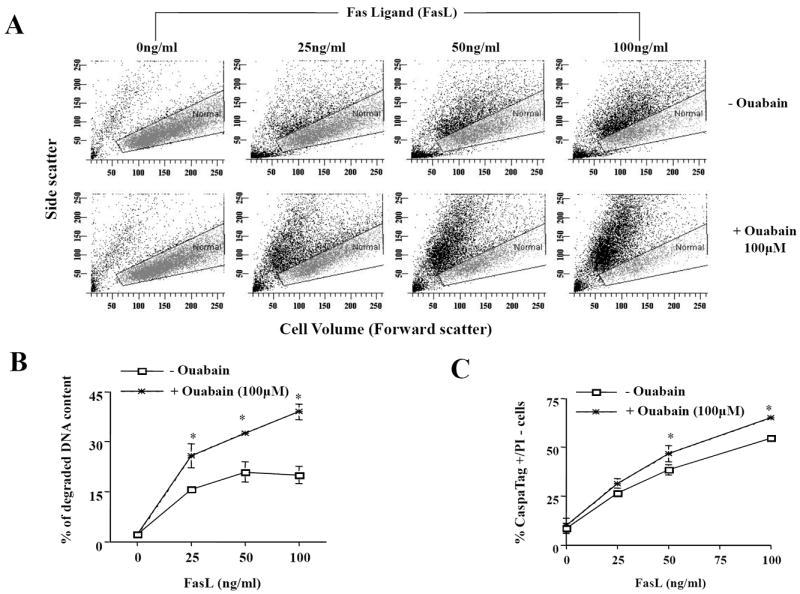
Apoptosis was induced by addition of FasL for 4h in the presence or absence of ouabain at a final concentration of 100μM. Cells were analyzed for changes in cell size on a forward- versus side-scatter plot (A), DNA content (B), and caspase 3/7-like activity (C) as explained in materials and methods. In (A), a gate was drawn to depict cells with normal cell size (grey dots). An increase in the number of apoptotic cells (shrunken cells) is reflected as an increasing number of cells falling outside the gated area assigned for normal cells. Cell shrinkage or AVD is observed as a decrease in the amount of forward-scattered light which is proportional to cell size in coordination with a change in side-scattered light that is proportional to cell density or granularity. In B, data represents the % of cells with subdiploid DNA content In C, data represent the % of cells with increased FLICA (increased activation of execution caspases) and low PI fluorescence (integral plasma membrane integrity). Shown are either averaged values ± SEM from three independent experiments (B and C), or a single representative experiment from five independent experiments (A). Asterisks (*) indicate statistical significance (P<0.05) between the two experimental conditions at each concentration of FasL.
TABLE 1.
Effect of ouabain on FasL-induced apoptosis.
| % “apoptotic” cells
| ||||
|---|---|---|---|---|
| 0ng/ml FasL | 25ng/ml FasL | 50ng/ml FasL | 100ng/ml FasL | |
| - Ouabain | 15.7 ± 1.2 | 31.1 ± 2.2 | 36.0 ± 3.4 | 41.6 ± 2.3 |
| +100μM ouabain | 22.4 ± 1.6 | 41.2 ± 1.7* | 49.3 ± 1.2* | 61.9 ± 1.2* |
Results represent the means ± the standard error of five independent experiments. % of apoptotic cells reflects the percentage of shrunken cells determined as shown in Figure 1A. Asterisks indicate statistical significance (at P<0.05) between mean values of control cells (- Ouabain) and ouabain treated cells.
Ouabain is a specific inhibitor of the Na+-pump, which results in a depletion of intracellular K+ and the accumulation of intracellular Na+. Figure 2A shows the change in intracellular K+ and Na+ upon treatment with 100 uM ouabain, 50 ng/ml FasL, or both, using ionic fluorescent indicators. We have used two independent Na+ indicators due to the low selectivity of Na+ fluorescent dyes. Previously, we demonstrated that FasL treated Jurkat cells results in an increase in intracellular Na+, prior to the loss of both intracellular Na+ and K+ [18, 44]. Here we observed a similar pattern in response to FasL using both the SBFI and CoroNa Green dyes (Figure 2A). As expected, ouabain alone induced a decrease in intracellular K+, along with an increase in intracellular Na+. However, the combined treatment of FasL and ouabain resulted in a greater increase in intracellular Na+ compared to either FasL or ouabain alone. Interestingly, the late stage of both intracellular Na+ and K+ loss was not observed. Additionally, we used the PBFI and CoroNa Green ionic indicators in combination to show that the initial increase in intracellular Na+ occurs prior to the dramatic loss of both Na+ and K+ (Figure 2B).
Fig. 2. Effect of ouabain on intracellular K+ and Na+ content.
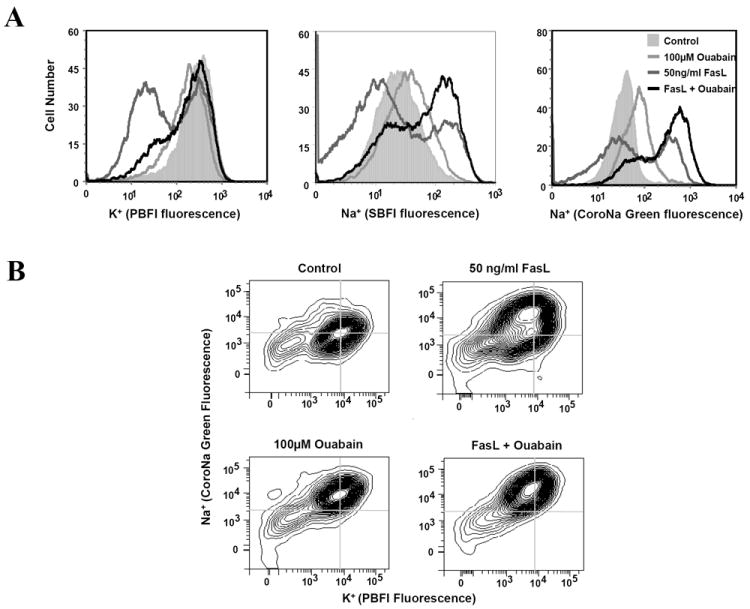
Apoptosis was induced by addition of FasL for 4h in the presence or absence of ouabain. For analysis of K+ and Na+ content, one hour prior to examination, SBFI-AM, CoroNa (Na+) and/or PBFI-AM (K+) dyes were added. Ion content in was analyzed after 4h by flow cytometry using fluorescence histogram plots (A) or contour plots to determine the relationship between alteration in intracellular K+ and Na+ and (B). In C, alterations in plasma membrane potential were determined using DiBAC4(3) incubated 30 minutes prior to flow cytometry analysis. Alterations in plasma membrane potential are presented as changes in mean fluorescence intensity. In D, caspase 3/7-like activity was performed as explained in Figure 1C.
Recently, cell death has been reported to be induced by ouabain independent of alterations in the ionic gradients of Na+ and K+ [45-47]. In Jurkat cells, apoptosis may be induced using concentrations of ouabain 20 times lower than those needed to inhibit the pump [48] suggesting that mechanisms other than ionic transport inhibition may be operative. Potentiation of the death receptor pathway of apoptosis, using lower concentrations of ouabain has also been described in activated lymphocytes [40]. We next decided to determine if ouabain might be activating a Na+/ K+-independent signaling cascade regulating apoptosis. For this we used low concentrations of ouabain where no alteration in ionic gradients is observed. Figure 2C shows that concentrations of 100nm or less of ouabain did not significantly alter the ionic gradients in Jurkat cells reflected by the absence of plasma membrane depolarization. At the same concentrations, ouabain was not able to potentiate FasL-induced apoptosis (Figure 2D). These results suggest that the effects of ouabain on apoptosis are associated to its ability to inhibit the Na+-K+-ATPase and alter cellular ionic homeostasis.
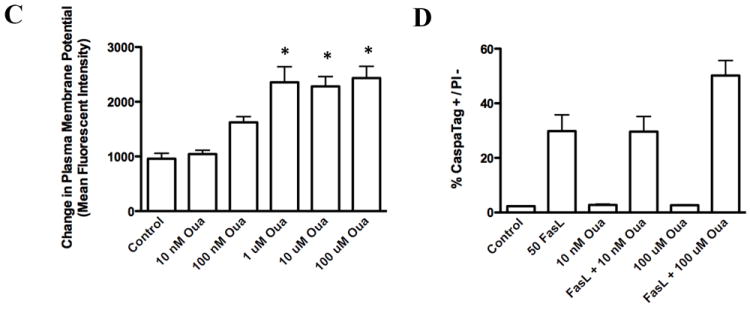
To determine if ouabain-induced potentiation of apoptosis was selective for FasL-induced cell death, we evaluated the effect of ouabain on apoptosis induced by other apoptotic stimuli. Figure 3A shows that the presence of ouabain did not result in a significant increase in apoptosis assessed by the occurrence of cell shrinkage after exposure to either H2O2 (500μM), thapsigargin (5μM) or UV-C (30mJ/cm2) as compared to FasL treatment. However, the presence of ouabain significantly increased the percentage of shrunken cells after exposure to TRAIL when compared to control conditions (Fig. 3B, Table 2) in a manner analogous to FasL induced apoptosis. Together these data suggest that the effect of ouabain in potentiating apoptosis is largely selective to the extrinsic apoptotic pathway activated by the death receptors for FasL and TRAIL, but not to the intrinsic or mitochondrial pathway(s) of apoptosis.
Fig. 3. Effect of ouabain on apoptosis induced by various stimuli.
Apoptosis was induced by addition of H2O2, thapsigargin, UV-C (A) or with increasing concentrations of TRAIL (B) for 4h in the presence or absence of ouabain. Cells were analyzed for changes in their cell size as explained in Figure 1A. Shown are single representative experiments from three independent experiments.
TABLE 2.
Effect of ouabain on TRAIL-induced apoptosis.
| % “apoptotic” cells
| |||||
|---|---|---|---|---|---|
| 0ng/ml TRAIL | 50ng/ml TRAIL | 100ng/ml TRAIL | 250ng/ml TRAIL | 500ng/ml TRAIL | |
| - Ouabain | 16.5 ± 1.5 | 29.1 ± 1.3 | 43.4 ± 1.1 | 48.0 ± 1.9 | 61.5 ± 1.5 |
| + 100μM Ouabain | 22.2 ± 1.6 | 39.9 ± 1.7 | 56.3 ± 1.6* | 59.0 ± 1.9* | 70.5 ± 1.5* |
Results represent the means ± the standard error of three independent experiments. % of apoptotic cells reflects the percentage of shrunken cells determined as shown in Figure 3B. Asterisks indicate statistical significance (at P<0.05) between mean values of control cells (- Ouabain) and ouabain treated cells.
Ouabain-induced potentiation of FasL-induced apoptosis involves perturbations in cell Ca2+ homeostasis
Efficient Na+-K+-ATPAse activity has been shown to be necessary for the maintenance of not only Na+ and K+ homeostasis, but also other cellular ionic gradients including Ca2+ [14]. Thus, we hypothesized that disruption of the ionic homeostasis by ouabain might also result in a secondary alteration in intracellular Ca2+ which is known to regulate several components of apoptosis. Figure 4A shows that FasL induced cell shrinkage was paralleled by an increase in intracellular Ca2+ content as shown by changes in Fluo-3 fluorescence. Additionally, this increase in intracellular Ca2+ by FasL was enhanced in the presence of ouabain. Histograms of the number of cells with an increase in Ca2+ show that FasL induced a dose-dependent increase in intracellular Ca2+, which was significantly higher in the presence of ouabain (Fig. 4B and 4C)
Fig. 4. Effect of ouabain on cell Ca2+ homeostasis in FasL-induced apoptosis.
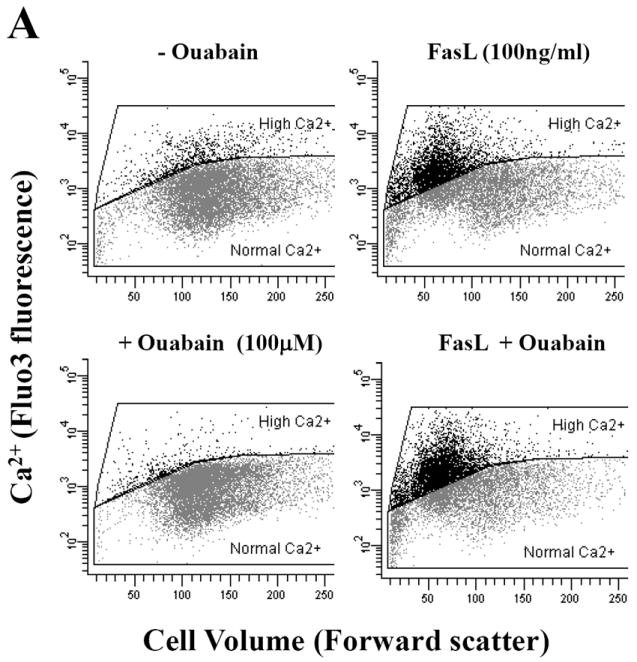
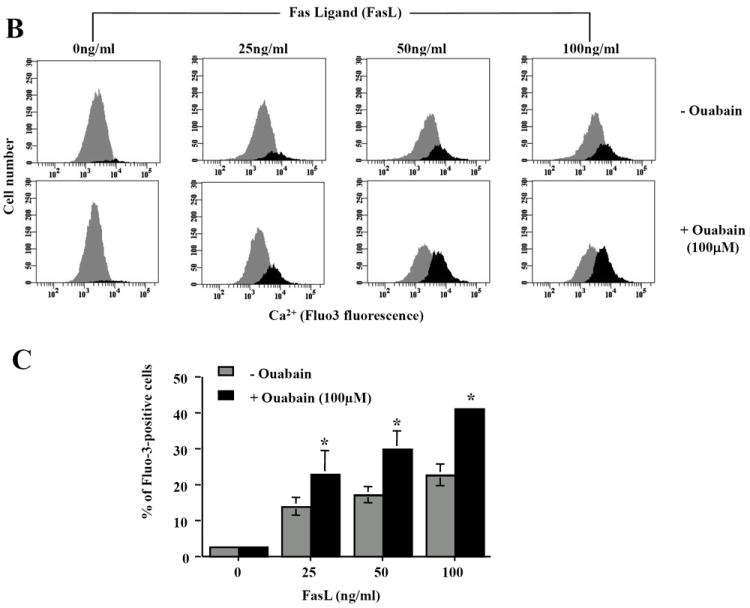
Apoptosis was induced in Jurkat cells by addition FasL for 4h. Cells were loaded with Fluo-3 AM 30min prior to flow cytometry analysis. Intracellular Ca2+ content was analyzed in either Fluo-3 fluorescence versus forward scatter dot plots (A) or Fluo-3 fluorescence histograms (B). Shown are either a single representative experiment from three independent experiments, (A and B), or averaged values ± SEM from three independent experiments (C). Asterisks (*) indicate statistical significance (P<0.05) between the two experimental conditions at each concentration of FasL.
Exposure of cells to ouabain did not result in a significant increase in the number cells with a high intracellular Ca2+ concentration induced in the presence of H2O2 (500μM), thapsigargin (5μM), or UV-C (30mJ/cm2) (Fig. 5, Table 3). Interestingly, although there was a small increase in the intracellular Ca2+ concentration induced by TRAIL (500ng/ml) upon ouabain treatment, this was not statistically significant when averaged (Figure 5, Table 3). Together, these data strongly suggest a specific link between the impairment of the Na+-K+-ATPase activity and changes in the free intracellular Ca2+ during FasL-induced apoptosis but not other apoptotic stimuli. It has been widely reported that thapsigargin rapidly induces a transient Ca2+ release from the endoplasmic reticulum that is immediately counteracted by the activity of the plasma membrane Ca2+ pump or the Na+/Ca2+ exchanger (NCX). This explains why cells treated with thapsigargin after four hours do not show a marked increase in Ca2+ content.
Fig. 5. Effect of ouabain on alterations in intracellular Ca2+ homeostasis induced by various apoptotic stimuli.
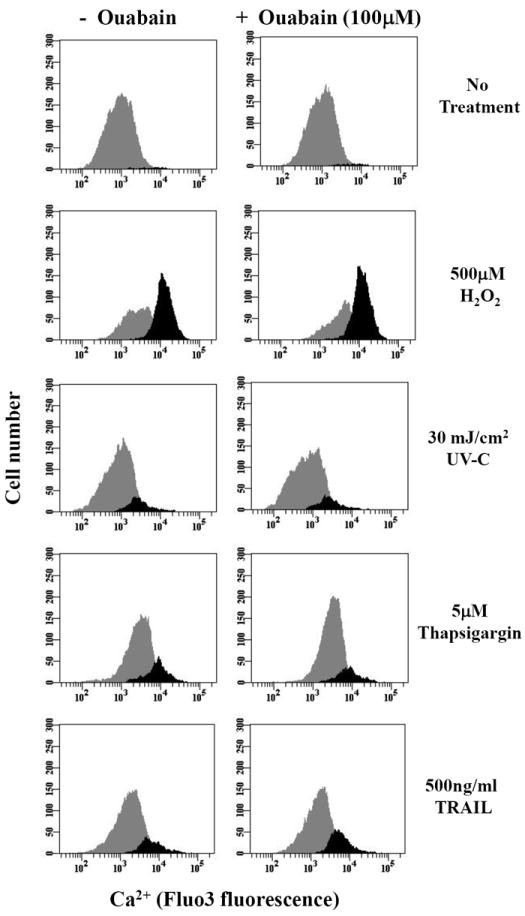
Apoptosis was induced in by addition of H2O2, TRAIL, thapsigargin or UV-C for 4h in the presence or absence of ouabain. Intracellular Ca2+ content was analyzed as explained in Figure 4B. Shown are single representative experiments from three independent experiments.
TABLE 3.
Effect of ouabain on apoptosis-induced Ca2+ increase.
| % of cells with High intracellular Ca2+
| |||||
|---|---|---|---|---|---|
| No treatment | 500μM H2O2 | 5μM Thapsigargin | 30mJ/cm2 UV-C | 500ng/ml TRAIL | |
| - Ouabain | 1.40 +/- 0.23 | 57.0 +/- 6.19 | 2.37 +/- 0.43 | 1.63 +/- 0.37 | 26.0 +/- 6.90 |
| + 100μM Ouabain | 0.97 +/- 0.07 | 58.4 +/- 6.21 | 6.47 +/- 0.78 | 0.70 +/- 0.21 | 31.2 +/- 6.83 |
Results represent the means ± the standard error of three independent experiments. % cells with High intracellular Ca2+ was determined as shown in Figure 4. None of these values are significant (P<0.05) when the individual treatments +/- ouabain are compared.
Ouabain-induced potentiation of FasL-induced apoptosis is associated to changes in cell Ca2+ homeostasis that results from the impairment of the Na+-K+-ATPase activity
Our results suggest a specific link between the impairment of the Na+-K+-ATPase activity and changes in the intracellular Ca2+ homeostasis during FasL-induced apoptosis. To further understand this link, we next determined the source for Ca2+ overload during FasL-induced apoptosis and its potentiation by ouabain. Figure 6A shows that nominally Ca2+-free medium did not prevent FasL-induced intracellular Ca2+ release. These data suggest that extracellular sources do not significantly contribute to the elevation in intracellular Ca2+ content in response to FasL and ouabain. In contrast, preloading cells with 25μM BAPTA-AM, a cell permeable Ca2+ scavenger that is cleaved by cytoplasmic esterases and consequently buffers free Ca2+, completely abolished the rise in intracellular Ca2+ content observed during FasL stimulation either in the presence or absence of ouabain (Fig. 6B).
Fig. 6. Effect of Ca2+-free medium and BAPTA-AM on FasL-induced increase in intracellular Ca2+ homeostasis in the presence or absence of ouabain.
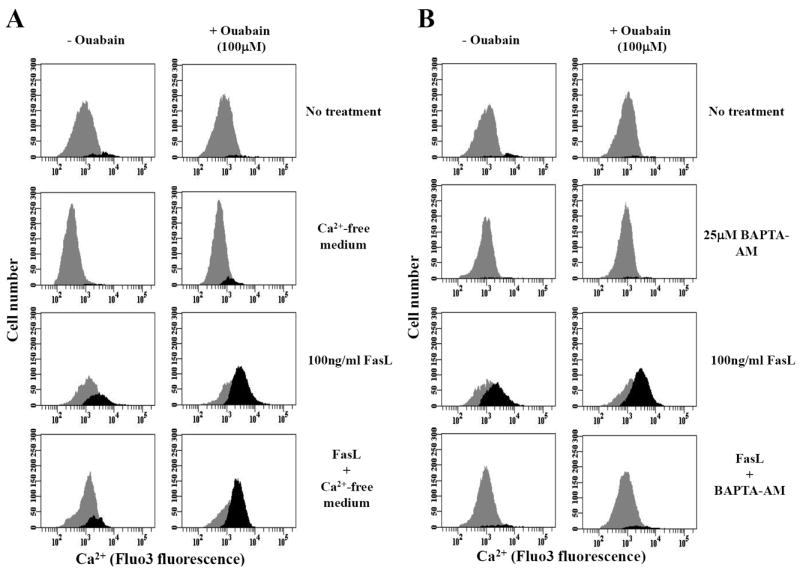
Apoptosis was induced by addition of 100ng/ml FasL for 4h in the presence or absence of ouabain ouabain under experimental conditions involving Ca2+-free medium (A). Alternatively, cells were treated with BAPTA-AM for 30 min prior to addition of FasL (B). Intracellular Ca2+ content was analyzed as explained in Figure 4B. Shown are single representative experiments from three independent experiments.
To elucidate the role of intracellular Ca2+ in FasL-induced apoptosis and on its potentiation by ouabain, cells were pre-incubated for 20min with 25μM BAPTA-AM. The presence of BAPTA-AM significantly reduced apoptosis observed in the presence or absence of ouabain (Fig. 7, Table 4). Moreover, the percentage of apoptotic cells in the presence of BAPTA-AM and BAPTA-AM plus ouabain was not significantly different suggesting that ouabain induced potentiation of apoptosis is mediated by stimulation of intracellular Ca2+ overload. Additionally, BAPTA-AM also significantly reduced the percentage of cells with degraded DNA induced by Fas activation and potentiated by ouabain (not shown). These data suggest that FasL-induced apoptosis and its potentiation by ouabain involve a rise in intracellular Ca2+. Thus, the regulation of FasL-induced apoptosis by the Na+-K+-ATPase activity seems to be specifically regulated by alterations in intracellular Ca2+ homeostasis.
Fig. 7. Effect of BAPTA-AM on FasL-induced apoptosis in the presence or absence of ouabain.
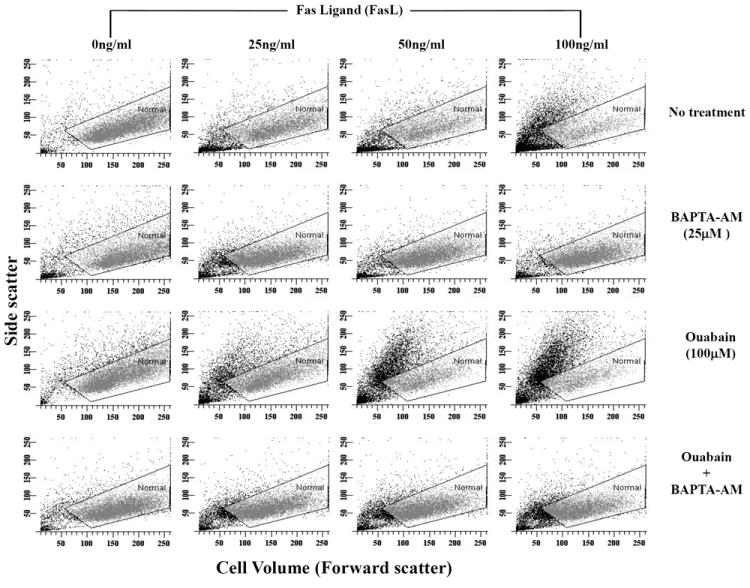
Apoptosis was induced in Jurkat cells by addition of FasL for 4h in the presence or absence of ouabain at a final concentration of 100μM. Thirty minutes prior to addition of FasL, cells were treated with BAPTA-AM. Cells were analyzed for changes in their cell size as explained in Figure 1A. Shown are single representative experiments from four independent experiments.
TABLE 4.
Effect of BAPTA-AM on the potentiation of FasL-induced apoptosis by ouabain.
| % “apoptotic” cells
| ||||
|---|---|---|---|---|
| 0ng/ml FasL | 25ng/ml FasL | 50ng/ml FasL | 100ng/ml FasL | |
| No treatment | 24.5 ± 1.6 | 48.5 ± 0.8 | 54.2 ± 2.8 | 57.7 ± 4.4 |
| BAPTA-AM | 32.5 ± 1.9 | 37.8 ± 3.0# | 38.7 ± 3.7# | 40.5 ± 3.7# |
| Ouabain | 28.5 ± 2.1 | 52.1 ± 3.0 | 63.6 ± 1.8 | 68.5 ± 2.2 |
| BAPTA-AM + Ouabain | 32.8 ± 1.2 | 37.7 ± 1.0* | 42.8 ± 2.2* | 46.5 ± 4.4* |
Results represent the means ± the standard error of four independent experiments. % of apoptotic cells reflects the percentage of shrunken cells determined as shown in Figure 7. * and # indicate statistical significance (at P<0.05) between mean values of cells treated with FasL (*) ± Ouabain (#) and cells preloaded with the intracellular Ca2+ scavenger BAPTA-AM.
DISCUSSION
Apoptosis is a physiological mode of cell death by which unwanted cells are removed in response to selective stimuli. This mode of cell death has been the subject of extensive research because of its involvement in human diseases [1]. Cell death by apoptosis is a process well characterized and defined by many distinct morphological and biochemical characteristics. Among these parameters is cell shrinkage or AVD that is independent of cell type and stimulus employed to induce cell death. AVD has been shown to be driven by the movement of ions across membranes. Such alterations in ionic homeostasis are important because they have also been shown to modify many biological processes including metabolism and gene expression [7, 10, 11, 13].
We and others have previously demonstrated that apoptosis can impair the activity of the Na+-K+-ATPase [17, 18, 21, 22, 49, 50]. The present study now examined the role of the Na+-K+-ATPase with various apoptotic stimuli by its inhibition with ouabain. Ouabain potentiation of apoptosis in Jurkat cells was restricted to the death receptor pathway (FasL and TRAIL) since it had no effect when other stimuli (H2O2, thapsigargin and UV-C) were used to activate the cell death process. Although these results are in agreement with other studies showing that ouabain and other cardiac glycosides potentiate apoptosis [17, 18, 38-40], our data demonstrates for the first time that the effect of ouabain is restricted to the extrinsic death receptor pathway in lymphoid cells. Additionally, we showed that in FasL-induced apoptosis, impairment of the Na+-K+-ATPase results in an increase in Ca2+ release from intracellular stores that specifically regulates the progression of the cell death program. Recent reports have shown that degradation of the Na+-K+ ATPase occurs during apoptosis induced by death receptor activation [18] glucocorticoids [25] and staurosporine [51]. On the other hand apoptotic resistant phenotypes of bcl-2 overexpressing cells [52, 53] have been associated with an elevated Na+-K+ ATPase activity. However, the mechanisms by which apoptosis modulates ATPase activity are still unclear. Recent reports suggest the role of caspases [25, 51], protein kinases [24, 54], and reactive oxygen/nitrogen species [22, 50, 55-57] on the modulation of Na+-K+-ATPase expression and activity during apoptosis.
The Na+-K+-ATPase exports three Na+ ions and imports two K+ ions (at the expense of one ATP molecule) and thus serves to maintain high K+ and low Na+ intracellular concentrations. Failure or inhibition of the pump results in intracellular depletion of K+ and consequently accumulation of Na+ both of which lead to plasma membrane depolarization [12, 14, 58]. Because the Na+ and K+ gradients maintained by the Na+-K+-ATPase have been shown necessary for the maintenance of the overall ionic homeostasis of the cell [12, 14, 15, 58], we analyzed the relationship between the activity of the Na+-K+-ATPase and changes in intracellular Ca2+ levels. Interestingly, ouabain only potentiated intracellular Ca2+ release induced by FasL but not by other apoptotic stimuli. This result was surprising when we take into account that ouabain potentiated cell shrinkage or AVD in Jurkat cells treated with TRAIL. Furthermore, it has been stipulated that activation of the Fas receptor and DR4 and DR5 (TRAIL receptors) lead to common signaling apoptotic pathways including the formation of DISC formed by the recruitment of the FADD) caspases 8 and FLIP. Previous reports have suggested that FasL- and TRAIL-induced apoptosis might be distinctively regulated by Bcl-2 proteins [59-61]. Thus, it is possible that although ouabain potentiates apoptosis and AVD induced by activation of death receptors for TRAIL and FasL, it does this by distinct signaling events.
The data described above suggest that ouabain has a specific effect on FasL-induced apoptosis through the regulation of changes in intracellular Ca2+ content. We observed that the presence of Ca2+-free medium did not abolish the rise in intracellular Ca2+ induced by FasL. In contrast, the intracellular Ca2+ scavenger BAPTA-AM prevented FasL-induced Ca2+ increase, which demonstrates that Ca2+ release from intracellular stores rather than its influx from the extracellular medium are involved in the Ca2+ overload during apoptosis induced by activation of Fas and its potentiation by ouabain. This finding also argues against the possibility that the intracellular Ca2+ rise might result from the reversal of the NCX (where intracellular Na+ is exchanged for extracellular Ca2+) since it would require extracellular Ca2+ [62]. However, it is important to mention that inhibition of the Na+-K+-ATPse by ouabain might lead to the impairment of NCX activity by inversion of the Na+/K+ gradient. This might in turn contribute to further accumulation of released Ca2+ because the NCX is one of the main pathways for intracellular Ca2+ clearance [63]. It has been widely reported that the effect of cardiac glycosides on Ca2+ signaling and overload is linked to the activity of NCX [64-71].
Calcium is known to regulate both extrinsic and intrinsic pathways of apoptosis at the level of the mitochondria [72]. Jurkat cells are considered type II cells which means that the activation of death receptors leads to a limited amount of DISC formation and activation of caspase 8. Thus type II cells depend on a positive feedback loop triggered by the release of mitochondrial cytochrome c and the activation of caspase-9 for the efficient execution of the cell death program [73]. Thus, the regulatory role of ouabain on FasL-induced Ca2+ perturbations might occur prior to the activation of the mitochondrial pathway of apoptosis. We and others have previously shown that changes in intracellular Ca2+ are necessary for DNA degradation during late stages of apoptosis induced by activation of Fas [74, 75]. Recently, Wozniak et al., reported that apoptosis induced by FasL is regulated by early oscillatory Ca2+ signals prior to the release of cytochrome C [76]. In this work, the authors demonstrated that Fas-induced apoptosis required an early endoplasmic reticulum (ER)-mediated Ca2+ release induced by phospholipase C-gamma1 (PLC-gamma1) phosphorylation and Ca2+ release from inositol 1,4,5-trisphosphate receptor (IP3R) channels [76]. Indeed previous reports have demonstrated that T cells deficient in IP3R are resistant to Fas-induced apoptosis [77]. The mechanism by which Fas activation mediates PLC-induced Ca2+ release is still unknown, however, it has been previously reported that T-cells deficient of FADD have impaired Ca2+ release from intracellular stores [78].
Similarly to Fas, ouabain has been reported to induce Ca2+ oscillation by a direct interaction of the Na+-K+-ATPase with the IP3R [66, 79, 80]. Moreover, ouabain has also been shown to induce the Src-dependent phosphorylation of PLC-gamma1 [81] and induce/potentiate Ca2+ release from the ER. We report here that elimination of ouabain-induced Ca2+ overload from intracellular stores through the use of BAPTA-AM abolishes the potentiation of FasL-induced apoptosis observed via direct inhibition of the Na+-K+-ATPase using ouabain. Thus it is plausible that ouabain might be regulating apoptosis upon Fas stimulation by acting on this early PLC-mediated Ca2+ release from the ER prior to the activation of the mitochondria feedback loop, which will explain why ouabain does not potentiate apoptosis induced by intrinsic pathways. In fact, a recent report demonstrates that apoptosis induced by activation of the mitochondrial (intrinsic) pathway does not depend on Ca2+ release IP3R receptors [82].
Ouabain and other Na+-K+-ATPase inhibitors seem to have tissue-specific effects on the modulation of cell death (by either necrosis or apoptosis) and cell survival. Inhibition of the Na+-K+-ATPase with cardiac glycosides, has been widely reported to induce or enhance apoptosis induced in various model systems including lymphocytes [17, 18, 30, 40], neural cells [27, 34, 37, 39] and a variety of other cell types [21, 26, 28, 29, 32, 33, 38]. In contrast, other studies have reported that chronic inhibition of the Na+-K+-ATPase activity does not affect survival of vascular smooth muscle cells (VSMC) [83, 84], lymphocytes [85, 86] and astrocytes [87]. We have previously reported the potentiation of Fas-induced apoptosis in Jurkat cells by ouabain [18]. Previous studies have demonstrated that chronic inhibition of the Na+-K+-ATPase with nM concentrations of cardiac glycosides induces apoptosis in Jurkat cells [48, 88]. Interestingly, contradictory results have been reported by Orlov and collaborators [84]. In this study, Orlov et al. described that Jurkat cells preincubated 2h with high concentrations of ouabain (1mM) did not observe an increase in apoptosis after stimulation with FasL [84]. In contrast, in our studies, ouabain was added in parallel to the induction of apoptosis. Pretreatment of ouabain seems to protect against apoptosis induced by different stimuli [83, 84, 89-92]. It is likely that prolonged alterations in the intracellular ionic homeostasis lead to altered conditions for apoptosis signaling. In fact it is known that prolonged ouabain treatment triggers the activation of PKC-, PI3K-, MAPK-survival pathways and the induction up-regulation of anti-apoptotic proteins such as Bcl-2 and mortalin [86, 89-95], which might occur independently from alterations in the Na+ gradient [15, 96]. We hypothesize then that activation of these signaling cascades alters the cellular environment prior to the induction of apoptosis impairing death receptor signaling. These observations might explain these contradictory observations.
Recently, it was demonstrated that ouabain binding to the Na+-K+-ATPase induces cell signaling events independent from Na+ gradient/transport [45, 65, 66, 97]. Orlov et al., demonstrated that ouabain binding to the Na+-K+-ATPase induces cell death by Na+/K+-independent activation of p38 MAPK. In this work, however, cells were exposed to chronic incubations with ouabain which were shown to induce necrosis rather than apoptosis [98]. Non-apoptotic cell death is also induced by chronic treatment of MDCK cells [47, 99] and porcine aorta endothelial cells [88]. These results clearly indicate that ouabain might also regulate other distinct types of cell death by different signaling pathways in a variety of cell types which might depend as well on the incubation time of the cardiac glycoside.
Ihenetu and coworkers recently demonstrated that chronic inhibition of the Na+-K+-ATPase with cardiac glycosides, at concentrations 20 times lower than that needed to inhibit the ATPase induces apoptosis in Jurkat cells [48]. These results suggested that mechanisms other than ionic transport inhibition may be operative. However, in this study, the IC50 required for inhibition of Na+-K+-ATPase was determined in porcine cerebral cortex tissue but not in Jurkat cells. In fact, Nobel and coworkers previously determined that at concentrations below 0.5μM ouabain does not inhibit the Na+-K+-ATPase activity measured by the ability of the cells to take up radiolabeled 86Rb+ as a tracer for K+, instead, ouabain was reported to stimulate the activity of the Na+-pump [17]. This phenomenon has been reported in other cell types [100]. Similar to these results, in this work we have demonstrated that ouabain, at concentrations that do not induce any alteration in the ionic gradients, reflected by the lack of plasma membrane depolarization, had no effect on AVD or apoptosis.
As reviewed elsewhere [13], cells contain impermeable, polyvalent anionic macromolecules and are constantly threatened by colloid osmotic cell swelling due to the entrance of diffusible ions and water. According to the “pump and leak” concept, cell swelling is avoided by low Na+ permeability and active Na+ extrusion via the Na+-K+-ATPase which renders the plasma membrane impermeable to Na+. The Na+-K+-ATPase builds up the outwardly directed gradient for K+ and in combination with the high K+ permeability of the plasma membrane, generates an inside negative membrane potential (Em), which drives Cl- out of the cell, allowing essential organic molecules within the cytoplasm [101-104]. It is expected then, that inhibition of the Na+-K+-ATPase by ouabain will induce cell swelling in Jurkat cells as it has been reported in other cell types where ouabain induces cell death by necrosis concomitant with cell swelling [47, 88, 99, 105] (see above). After pump inhibition, the fall in the K+ gradient should collapse the Em, which would result in the influx of Cl- and Na+ leading to cell swelling. However, in Jurkat cells, ouabain by itself does not induce apoptosis or cell shrinkage/AVD. AVD has been shown to be mediated by the activation of volume regulated K+ and Cl- channels which should counteract influx of Cl- triggered by inhibition of the Na+-K+-ATPase [8, 106-109]. We have previously demonstrated that during apoptosis, intracellular Na+ increase (mediated by impairment of the Na+-K+-ATPase activity and activation of Na+ channels) is followed by a secondary loss of intracellular Na+ which counteracts Na+ overload [8, 18, 44]. Although it is possible that the absence of cell swelling in ouabain treated Jurkat cells is related to tissue specific effects of ouabain, it is more likely that cell swelling is counteracted by the AVD process induced by either FasL (when ouabain treatment is done acutely) or by chronic treatment with ouabain. In fact, it has been previously demonstrated that in those cell types where ouabain induces/potentiates apoptosis, treatment with the cardiac glycoside triggers a transient cell swelling followed by AVD and cell shrinkage [37, 39].
We have recently demonstrated that AVD occurs in two stages characterized by an initial reduction in cell volume associated with the influx of Na+ ions followed by a secondary shrinkage linked to the loss of both K+ and Na+ [8, 18, 44]. Cell shrinkage is a hallmark of apoptotic cell death, but it is as yet unclear whether a reduction in cell volume is a primary signal for the activation of apoptosis or just a consequence of the alterations in ionic gradients [110-112]. We have previously demonstrated that sodium influx but not cell shrinkage or AVD is required for the progression of apoptosis [44]. In our study ouabain treatment reduced K+ content. However, ouabain-induced K+ loss was not sufficient to induce apoptosis. This result might be explained by the fact that under the experimental conditions tested here (4h incubation with ouabain), ouabain does not decrease K+ to the same extent as it occurs during apoptosis triggered by stimuli such as FasL (Figure 2). Indeed, we have observed that chronic treatment of Jurkat cells for18h with 100μM ouabain induces caspase-dependent apoptosis (Razik and Cidlowski data not shown).
In conclusion, we report here that ouabain selectively potentiates apoptosis in Jurkat cells induced by activation of death receptors which suggests a role for the Na+-K+-ATPase during apoptosis. Interesintgly, our results also suggest that apoptosis by Fas activation and its potentiation by ouabain are linked to Ca2+ release from intracellular stores. In contrast, the regulation of TRAIL-induced apoptotis and AVD by ouabain was shown to involve a different mechanism independent from alterations in intracellular Ca2+. Our data demonstrates a link between the impairment of the Na+-K+-ATPase during apoptosis and changes in cell Ca2+ homeostasis that are exclusively related to the FasL-induced apoptosis.
Acknowledgments
We acknowledge Dr. Elizabeth Murphy and Dr. Gary St. J. Bird for the internal review and comments on this manuscript. We appreciate MS. Maria Sifre, for technical support in the flow cytometry studies. The research was supported by the Intramural Research Program of the NIH/ NIEHS.
References
- 1.Fadeel B, Orrenius S. Apoptosis: a basic biological phenomenon with wide-ranging implications in human disease. J Intern Med. 2005;258:479–517. doi: 10.1111/j.1365-2796.2005.01570.x. [DOI] [PubMed] [Google Scholar]
- 2.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 3.Khosravi-Far R, Esposti MD. Death receptor signals to mitochondria. Cancer Biol Ther. 2004;3:1051–1057. doi: 10.4161/cbt.3.11.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barnhart BC, Alappat EC, Peter ME. The CD95 type I/type II model. Semin Immunol. 2003;15:185–193. doi: 10.1016/s1044-5323(03)00031-9. [DOI] [PubMed] [Google Scholar]
- 5.Ravagnan L, Roumier T, Kroemer G. Mitochondria, the killer organelles and their weapons. J Cell Physiol. 2002;192:131–137. doi: 10.1002/jcp.10111. [DOI] [PubMed] [Google Scholar]
- 6.Bras M, Queenan B, Susin SA. Programmed cell death via mitochondria: different modes of dying. Biochemistry (Mosc) 2005;70:231–239. doi: 10.1007/s10541-005-0105-4. [DOI] [PubMed] [Google Scholar]
- 7.Bortner CD, Cidlowski JA. Apoptotic volume decrease and the incredible shrinking cell. Cell Death Differ. 2002;9:1307–1310. doi: 10.1038/sj.cdd.4401126. [DOI] [PubMed] [Google Scholar]
- 8.Bortner CD, Hughes FM, Jr, Cidlowski JA. A primary role for K+ and Na+ efflux in the activation of apoptosis. J Biol Chem. 1997;272:32436–32442. doi: 10.1074/jbc.272.51.32436. [DOI] [PubMed] [Google Scholar]
- 9.Hughes FM, Jr, Bortner CD, Purdy GD, Cidlowski JA. Intracellular K+ suppresses the activation of apoptosis in lymphocytes. J Biol Chem. 1997;272:30567–30576. doi: 10.1074/jbc.272.48.30567. [DOI] [PubMed] [Google Scholar]
- 10.Franco R, Bortner CD, Cidlowski JA. Potential roles of electrogenic ion transport and plasma membrane depolarization in apoptosis. J Membr Biol. 2006;209:43–58. doi: 10.1007/s00232-005-0837-5. [DOI] [PubMed] [Google Scholar]
- 11.Lang F, Foller M, Lang K, et al. Cell volume regulatory ion channels in cell proliferation and cell death. Methods Enzymol. 2007;428:209–225. doi: 10.1016/S0076-6879(07)28011-5. [DOI] [PubMed] [Google Scholar]
- 12.Panayiotidis MI, Bortner CD, Cidlowski JA. On the mechanism of ionic regulation of apoptosis: would the Na+/K+-ATPase please stand up? Acta Physiol (Oxf) 2006;187:205–215. doi: 10.1111/j.1748-1716.2006.01562.x. [DOI] [PubMed] [Google Scholar]
- 13.Hoffmann EK, Lambert IH, Pedersen SF. Physiology of cell volume regulation in vertebrates. Physiol Rev. 2009;89:193–277. doi: 10.1152/physrev.00037.2007. [DOI] [PubMed] [Google Scholar]
- 14.Lingrel JB, Kuntzweiler T. Na+,K(+)-ATPase. J Biol Chem. 1994;269:19659–19662. [PubMed] [Google Scholar]
- 15.Pierre SV, Xie Z. The Na,K-ATPase receptor complex: its organization and membership. Cell Biochem Biophys. 2006;46:303–316. doi: 10.1385/cbb:46:3:303. [DOI] [PubMed] [Google Scholar]
- 16.Arrebola F, Zabiti S, Canizares FJ, Cubero MA, Crespo PV, Fernandez-Segura E. Changes in intracellular sodium, chlorine, and potassium concentrations in staurosporine-induced apoptosis. J Cell Physiol. 2005;204:500–507. doi: 10.1002/jcp.20306. [DOI] [PubMed] [Google Scholar]
- 17.Nobel CS, Aronson JK, van den Dobbelsteen DJ, Slater AF. Inhibition of Na+/K(+)-ATPase may be one mechanism contributing to potassium efflux and cell shrinkage in CD95-induced apoptosis. Apoptosis. 2000;5:153–163. doi: 10.1023/a:1009684713784. [DOI] [PubMed] [Google Scholar]
- 18.Bortner CD, Gomez-Angelats M, Cidlowski JA. Plasma membrane depolarization without repolarization is an early molecular event in anti-Fas-induced apoptosis. J Biol Chem. 2001;276:4304–4314. doi: 10.1074/jbc.M005171200. [DOI] [PubMed] [Google Scholar]
- 19.Tang MJ, Cheng YR, Lin HH. Role of apoptosis in growth and differentiation of proximal tubule cells in primary cultures. Biochem Biophys Res Commun. 1996;218:658–664. doi: 10.1006/bbrc.1996.0118. [DOI] [PubMed] [Google Scholar]
- 20.Orlov SN, Pchejetski DV, Sarkissian SD, et al. [3H]-thymidine labelling of DNA triggers apoptosis potentiated by E1A-adenoviral protein. Apoptosis. 2003;8:199–208. doi: 10.1023/a:1022931028235. [DOI] [PubMed] [Google Scholar]
- 21.Yu SP. Na(+), K(+)-ATPase: the new face of an old player in pathogenesis and apoptotic/hybrid cell death. Biochem Pharmacol. 2003;66:1601–1609. doi: 10.1016/s0006-2952(03)00531-8. [DOI] [PubMed] [Google Scholar]
- 22.Wang XQ, Xiao AY, Sheline C, et al. Apoptotic insults impair Na+, K+-ATPase activity as a mechanism of neuronal death mediated by concurrent ATP deficiency and oxidant stress. J Cell Sci. 2003;116:2099–2110. doi: 10.1242/jcs.00420. [DOI] [PubMed] [Google Scholar]
- 23.Wang XQ, Xiao AY, Yang A, LaRose L, Wei L, Yu SP. Block of Na+,K+-ATPase and induction of hybrid death by 4-aminopyridine in cultured cortical neurons. J Pharmacol Exp Ther. 2003;305:502–506. doi: 10.1124/jpet.102.045013. [DOI] [PubMed] [Google Scholar]
- 24.Nowak G. Protein kinase C-alpha and ERK1/2 mediate mitochondrial dysfunction, decreases in active Na+ transport, and cisplatin-induced apoptosis in renal cells. J Biol Chem. 2002;277:43377–43388. doi: 10.1074/jbc.M206373200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mann CL, Bortner CD, Jewell CM, Cidlowski JA. Glucocorticoid-induced plasma membrane depolarization during thymocyte apoptosis: association with cell shrinkage and degradation of the Na(+)/K(+)-adenosine triphosphatase. Endocrinology. 2001;142:5059–5068. doi: 10.1210/endo.142.12.8516. [DOI] [PubMed] [Google Scholar]
- 26.Chueh SC, Guh JH, Chen J, Lai MK, Teng CM. Dual effects of ouabain on the regulation of proliferation and apoptosis in human prostatic smooth muscle cells. J Urol. 2001;166:347–353. [PubMed] [Google Scholar]
- 27.Hennion JP, el-Masri MA, Huff MO, el-Mailakh RS. Evaluation of neuroprotection by lithium and valproic acid against ouabain-induced cell damage. Bipolar Disord. 2002;4:201–206. doi: 10.1034/j.1399-5618.2002.01162.x. [DOI] [PubMed] [Google Scholar]
- 28.Kurosawa M, Tani Y, Nishimura S, Numazawa S, Yoshida T. Distinct PKC isozymes regulate bufalin-induced differentiation and apoptosis in human monocytic cells. Am J Physiol Cell Physiol. 2001;280:C459–464. doi: 10.1152/ajpcell.2001.280.3.C459. [DOI] [PubMed] [Google Scholar]
- 29.McConkey DJ, Lin Y, Nutt LK, Ozel HZ, Newman RA. Cardiac glycosides stimulate Ca2+ increases and apoptosis in androgen-independent, metastatic human prostate adenocarcinoma cells. Cancer Res. 2000;60:3807–3812. [PubMed] [Google Scholar]
- 30.Olej B, dos Santos NF, Leal L, Rumjanek VM. Ouabain induces apoptosis on PHA-activated lymphocytes. Biosci Rep. 1998;18:1–7. doi: 10.1023/a:1022259832207. [DOI] [PubMed] [Google Scholar]
- 31.Stelmashook EV, Weih M, Zorov D, Victorov I, Dirnagl U, Isaev N. Short-term block of Na+/K+-ATPase in neuro-glial cell cultures of cerebellum induces glutamate dependent damage of granule cells. FEBS Lett. 1999;456:41–44. doi: 10.1016/s0014-5793(99)00922-9. [DOI] [PubMed] [Google Scholar]
- 32.Watabe M, Masuda Y, Nakajo S, Yoshida T, Kuroiwa Y, Nakaya K. The cooperative interaction of two different signaling pathways in response to bufalin induces apoptosis in human leukemia U937 cells. J Biol Chem. 1996;271:14067–14072. doi: 10.1074/jbc.271.24.14067. [DOI] [PubMed] [Google Scholar]
- 33.Kawazoe N, Aiuchi T, Masuda Y, Nakajo S, Nakaya K. Induction of apoptosis by bufalin in human tumor cells is associated with a change of intracellular concentration of Na+ ions. J Biochem (Tokyo) 1999;126:278–286. doi: 10.1093/oxfordjournals.jbchem.a022446. [DOI] [PubMed] [Google Scholar]
- 34.Lang H, Schulte BA, Schmiedt RA. Ouabain induces apoptotic cell death in type I spiral ganglion neurons, but not type II neurons. J Assoc Res Otolaryngol. 2005;6:63–74. doi: 10.1007/s10162-004-5021-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang YT, Chueh SC, Teng CM, Guh JH. Investigation of ouabain-induced anticancer effect in human androgen-independent prostate cancer PC-3 cells. Biochem Pharmacol. 2004;67:727–733. doi: 10.1016/j.bcp.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 36.Schmiedt RA, Okamura HO, Lang H, Schulte BA. Ouabain application to the round window of the gerbil cochlea: a model of auditory neuropathy and apoptosis. J Assoc Res Otolaryngol. 2002;3:223–233. doi: 10.1007/s1016200220017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xiao AY, Wei L, Xia S, Rothman S, Yu SP. Ionic mechanism of ouabain-induced concurrent apoptosis and necrosis in individual cultured cortical neurons. J Neurosci. 2002;22:1350–1362. doi: 10.1523/JNEUROSCI.22-04-01350.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Penning LC, Denecker G, Vercammen D, Declercq W, Schipper RG, Vandenabeele P. A role for potassium in TNF-induced apoptosis and gene-induction in human and rodent tumour cell lines. Cytokine. 2000;12:747–750. doi: 10.1006/cyto.1999.0626. [DOI] [PubMed] [Google Scholar]
- 39.Xiao AY, Wang XQ, Yang A, Yu SP. Slight impairment of Na+,K+-ATPase synergistically aggravates ceramide- and beta-amyloid-induced apoptosis in cortical neurons. Brain Res. 2002;955:253–259. doi: 10.1016/s0006-8993(02)03472-8. [DOI] [PubMed] [Google Scholar]
- 40.Esteves MB, Marques-Santos LF, Affonso-Mitidieri OR, Rumjanek VM. Ouabain exacerbates activation-induced cell death in human peripheral blood lymphocytes. An Acad Bras Cienc. 2005;77:281–292. doi: 10.1590/s0001-37652005000200008. [DOI] [PubMed] [Google Scholar]
- 41.Sun XM, Bratton SB, Butterworth M, MacFarlane M, Cohen GM. Bcl-2 and Bcl-xL inhibit CD95-mediated apoptosis by preventing mitochondrial release of Smac/DIABLO and subsequent inactivation of X-linked inhibitor-of-apoptosis protein. J Biol Chem. 2002;277:11345–11351. doi: 10.1074/jbc.M109893200. [DOI] [PubMed] [Google Scholar]
- 42.Shiffer Z, Zurgil N, Shafran Y, Deutsch M. Analysis of laser scattering pattern as an early measure of apoptosis. Biochem Biophys Res Commun. 2001;289:1320–1327. doi: 10.1006/bbrc.2001.6127. [DOI] [PubMed] [Google Scholar]
- 43.Bortner CD, Cidlowski JA. Absence of volume regulatory mechanisms contributes to the rapid activation of apoptosis in thymocytes. Am J Physiol. 1996;271:C950–961. doi: 10.1152/ajpcell.1996.271.3.C950. [DOI] [PubMed] [Google Scholar]
- 44.Bortner CD, Cidlowski JA. Uncoupling cell shrinkage from apoptosis reveals that Na+ influx is required for volume loss during programmed cell death. J Biol Chem. 2003;278:39176–39184. doi: 10.1074/jbc.M303516200. [DOI] [PubMed] [Google Scholar]
- 45.Akimova OA, Hamet P, Orlov SN. [Na+]i/[K+]i -independent death of ouabain-treated renal epithelial cells is not mediated by Na+,K+ -ATPase internalization and de novo gene expression. Pflugers Arch. 2008;455:711–719. doi: 10.1007/s00424-007-0283-6. [DOI] [PubMed] [Google Scholar]
- 46.Orlov SN, Hamet P. The death of cardiotonic steroid-treated cells: evidence of Na+i,K+i-independent H+i-sensitive signalling. Acta Physiol (Oxf) 2006;187:231–240. doi: 10.1111/j.1748-1716.2006.01546.x. [DOI] [PubMed] [Google Scholar]
- 47.Pchejetski D, Taurin S, Der Sarkissian S, et al. Inhibition of Na+,K+-ATPase by ouabain triggers epithelial cell death independently of inversion of the [Na+]i/[K+]i ratio. Biochem Biophys Res Commun. 2003;301:735–744. doi: 10.1016/s0006-291x(02)03002-4. [DOI] [PubMed] [Google Scholar]
- 48.Ihenetu K, Qazzaz HM, Crespo F, Fernandez-Botran R, Valdes R., Jr Digoxin-like immunoreactive factors induce apoptosis in human acute T-cell lymphoblastic leukemia. Clin Chem. 2007;53:1315–1322. doi: 10.1373/clinchem.2006.082081. [DOI] [PubMed] [Google Scholar]
- 49.Dussmann H, Rehm M, Kogel D, Prehn JH. Outer mitochondrial membrane permeabilization during apoptosis triggers caspase-independent mitochondrial and caspase-dependent plasma membrane potential depolarization: a single-cell analysis. J Cell Sci. 2003;116:525–536. doi: 10.1242/jcs.00236. [DOI] [PubMed] [Google Scholar]
- 50.Sen N, Das BB, Ganguly A, Mukherjee T, Bandyopadhyay S, Majumder HK. Camptothecin-induced imbalance in intracellular cation homeostasis regulates programmed cell death in unicellular hemoflagellate Leishmania donovani. J Biol Chem. 2004;279:52366–52375. doi: 10.1074/jbc.M406705200. [DOI] [PubMed] [Google Scholar]
- 51.Dussmann H, Kogel D, Rehm M, Prehn JH. Mitochondrial membrane permeabilization and superoxide production during apoptosis. A single-cell analysis. J Biol Chem. 2003;278:12645–12649. doi: 10.1074/jbc.M210826200. [DOI] [PubMed] [Google Scholar]
- 52.Gilbert M, Knox S. Influence of Bcl-2 overexpression on Na+/K(+)-ATPase pump activity: correlation with radiation-induced programmed cell death. J Cell Physiol. 1997;171:299–304. doi: 10.1002/(SICI)1097-4652(199706)171:3<299::AID-JCP8>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 53.Gilbert MS, Saad AH, Rupnow BA, Knox SJ. Association of BCL-2 with membrane hyperpolarization and radioresistance. J Cell Physiol. 1996;168:114–122. doi: 10.1002/(SICI)1097-4652(199607)168:1<114::AID-JCP14>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 54.Wang XQ, Yu SP. Novel regulation of Na, K-ATPase by Src tyrosine kinases in cortical neurons. J Neurochem. 2005;93:1515–1523. doi: 10.1111/j.1471-4159.2005.03147.x. [DOI] [PubMed] [Google Scholar]
- 55.Thevenod F, Friedmann JM. Cadmium-mediated oxidative stress in kidney proximal tubule cells induces degradation of Na+/K(+)-ATPase through proteasomal and endo-/lysosomal proteolytic pathways. Faseb J. 1999;13:1751–1761. doi: 10.1096/fasebj.13.13.1751. [DOI] [PubMed] [Google Scholar]
- 56.Cimen B, Turkozkan N, Seven I, Unlu A, Karasu C. Impaired Na+,K+-ATPase activity as a mechanism of reactive nitrogen species-induced cytotoxicity in guinea pig liver exposed to lipopolysaccharides. Mol Cell Biochem. 2004;259:53–57. doi: 10.1023/b:mcbi.0000021344.64317.a2. [DOI] [PubMed] [Google Scholar]
- 57.Yin W, Cheng W, Shen W, et al. Impairment of Na(+),K(+)-ATPase in CD95(APO-1)-induced human T-cell leukemia cell apoptosis mediated by glutathione depletion and generation of hydrogen peroxide. Leukemia. 2007;21:1669–1678. doi: 10.1038/sj.leu.2404791. [DOI] [PubMed] [Google Scholar]
- 58.Therien AG, Blostein R. Mechanisms of sodium pump regulation. Am J Physiol Cell Physiol. 2000;279:C541–566. doi: 10.1152/ajpcell.2000.279.3.C541. [DOI] [PubMed] [Google Scholar]
- 59.Lee Y, Shacter E. Fas aggregation does not correlate with Fas-mediated apoptosis. J Immunol. 2001;167:82–89. doi: 10.4049/jimmunol.167.1.82. [DOI] [PubMed] [Google Scholar]
- 60.Velthuis JH, Rouschop KM, De Bont HJ, Mulder GJ, Nagelkerke JF. Distinct intracellular signaling in tumor necrosis factor-related apoptosis-inducing ligand- and CD95 ligand-mediated apoptosis. J Biol Chem. 2002;277:24631–24637. doi: 10.1074/jbc.M111572200. [DOI] [PubMed] [Google Scholar]
- 61.Werner AB, de Vries E, Tait SW, Bontjer I, Borst J. TRAIL receptor and CD95 signal to mitochondria via FADD, caspase-8/10, Bid, and Bax but differentially regulate events downstream from truncated Bid. J Biol Chem. 2002;277:40760–40767. doi: 10.1074/jbc.M204351200. [DOI] [PubMed] [Google Scholar]
- 62.Balasubramanyam M, Rohowsky-Kochan C, Reeves JP, Gardner JP. Na+/Ca2+ exchange-mediated calcium entry in human lymphocytes. J Clin Invest. 1994;94:2002–2008. doi: 10.1172/JCI117553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 64.Golovina VA, Song H, James PF, Lingrel JB, Blaustein MP. Na+ pump alpha 2-subunit expression modulates Ca2+ signaling. Am J Physiol Cell Physiol. 2003;284:C475–486. doi: 10.1152/ajpcell.00383.2002. [DOI] [PubMed] [Google Scholar]
- 65.Zhang L, Zhang Z, Guo H, Wang Y. Na+/K+-ATPase-mediated signal transduction and Na+/K+-ATPase regulation. Fundam Clin Pharmacol. 2008;22:615–621. doi: 10.1111/j.1472-8206.2008.00620.x. [DOI] [PubMed] [Google Scholar]
- 66.Tian J, Xie ZJ. The Na-K-ATPase and calcium-signaling microdomains. Physiology (Bethesda) 2008;23:205–211. doi: 10.1152/physiol.00008.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Reuter H, Henderson SA, Han T, Ross RS, Goldhaber JI, Philipson KD. The Na+-Ca2+ exchanger is essential for the action of cardiac glycosides. Circ Res. 2002;90:305–308. doi: 10.1161/hh0302.104562. [DOI] [PubMed] [Google Scholar]
- 68.Echevarria-Lima J, de Araujo EG, de Meis L, Rumjanek VM. Ca2+ mobilization induced by ouabain in thymocytes involves intracellular and extracellular Ca2+ pools. Hypertension. 2003;41:1386–1392. doi: 10.1161/01.HYP.0000072801.90600.C2. [DOI] [PubMed] [Google Scholar]
- 69.Swift F, Birkeland JA, Tovsrud N, et al. Altered Na+/Ca2+-exchanger activity due to downregulation of Na+/K+-ATPase alpha2-isoform in heart failure. Cardiovasc Res. 2008;78:71–78. doi: 10.1093/cvr/cvn013. [DOI] [PubMed] [Google Scholar]
- 70.Lynch RM, Weber CS, Nullmeyer KD, Moore ED, Paul RJ. Clearance of store-released Ca2+ by the Na+-Ca2+ exchanger is diminished in aortic smooth muscle from Na+-K+-ATPase alpha 2-isoform gene-ablated mice. Am J Physiol Heart Circ Physiol. 2008;294:H1407–1416. doi: 10.1152/ajpheart.00855.2007. [DOI] [PubMed] [Google Scholar]
- 71.Altamirano J, Li Y, DeSantiago J, Piacentino V, 3rd, Houser SR, Bers DM. The inotropic effect of cardioactive glycosides in ventricular myocytes requires Na+-Ca2+ exchanger function. J Physiol. 2006;575:845–854. doi: 10.1113/jphysiol.2006.111252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rizzuto R, Pinton P, Ferrari D, et al. Calcium and apoptosis: facts and hypotheses. Oncogene. 2003;22:8619–8627. doi: 10.1038/sj.onc.1207105. [DOI] [PubMed] [Google Scholar]
- 73.Samraj AK, Keil E, Ueffing N, Schulze-Osthoff K, Schmitz I. Loss of caspase-9 provides genetic evidence for the type I/II concept of CD95-mediated apoptosis. J Biol Chem. 2006;281:29652–29659. doi: 10.1074/jbc.M603487200. [DOI] [PubMed] [Google Scholar]
- 74.Scoltock AB, Bortner CD, St JBG, Putney JW, Jr, Cidlowski JA. A selective requirement for elevated calcium in DNA degradation, but not early events in anti-Fas-induced apoptosis. J Biol Chem. 2000;275:30586–30596. doi: 10.1074/jbc.M004058200. [DOI] [PubMed] [Google Scholar]
- 75.Oshimi Y, Miyazaki S. Fas antigen-mediated DNA fragmentation and apoptotic morphologic changes are regulated by elevated cytosolic Ca2+ level. J Immunol. 1995;154:599–609. [PubMed] [Google Scholar]
- 76.Wozniak AL, Wang X, Stieren ES, Scarbrough SG, Elferink CJ, Boehning D. Requirement of biphasic calcium release from the endoplasmic reticulum for Fas-mediated apoptosis. J Cell Biol. 2006;175:709–714. doi: 10.1083/jcb.200608035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jayaraman T, Marks AR. T cells deficient in inositol 1,4,5-trisphosphate receptor are resistant to apoptosis. Mol Cell Biol. 1997;17:3005–3012. doi: 10.1128/mcb.17.6.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hueber AO, Zornig M, Bernard AM, Chautan M, Evan G. A dominant negative Fas-associated death domain protein mutant inhibits proliferation and leads to impaired calcium mobilization in both T-cells and fibroblasts. J Biol Chem. 2000;275:10453–10462. doi: 10.1074/jbc.275.14.10453. [DOI] [PubMed] [Google Scholar]
- 79.Aizman O, Uhlen P, Lal M, Brismar H, Aperia A. Ouabain, a steroid hormone that signals with slow calcium oscillations. Proc Natl Acad Sci U S A. 2001;98:13420–13424. doi: 10.1073/pnas.221315298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Miyakawa-Naito A, Uhlen P, Lal M, et al. Cell signaling microdomain with Na,K-ATPase and inositol 1,4,5-trisphosphate receptor generates calcium oscillations. J Biol Chem. 2003;278:50355–50361. doi: 10.1074/jbc.M305378200. [DOI] [PubMed] [Google Scholar]
- 81.Yuan Z, Cai T, Tian J, Ivanov AV, Giovannucci DR, Xie Z. Na/K-ATPase tethers phospholipase C and IP3 receptor into a calcium-regulatory complex. Mol Biol Cell. 2005;16:4034–4045. doi: 10.1091/mbc.E05-04-0295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Khan MT, Bhanumathy CD, Schug ZT, Joseph SK. Role of inositol 1,4,5-trisphosphate receptors in apoptosis in DT40 lymphocytes. J Biol Chem. 2007;282:32983–32990. doi: 10.1074/jbc.M705183200. [DOI] [PubMed] [Google Scholar]
- 83.Orlov SN, Taurin S, Thorin-Trescases N, Dulin NO, Tremblay J, Hamet P. Inversion of the intracellular Na(+)/K(+) ratio blocks apoptosis in vascular smooth muscle cells by induction of RNA synthesis. Hypertension. 2000;35:1062–1068. doi: 10.1161/01.hyp.35.5.1062. [DOI] [PubMed] [Google Scholar]
- 84.Orlov SN, Thorin-Trescases N, Kotelevtsev SV, Tremblay J, Hamet P. Inversion of the intracellular Na+/K+ ratio blocks apoptosis in vascular smooth muscle at a site upstream of caspase-3. J Biol Chem. 1999;274:16545–16552. doi: 10.1074/jbc.274.23.16545. [DOI] [PubMed] [Google Scholar]
- 85.Falciola J, Volet B, Anner RM, Moosmayer M, Lacotte D, Anner BM. Role of cell membrane Na,K-ATPase for survival of human lymphocytes in vitro. Biosci Rep. 1994;14:189–204. doi: 10.1007/BF01200248. [DOI] [PubMed] [Google Scholar]
- 86.Xu JW, Jin RM, Li EQ, Wang YR, Bai Y. Signal pathways in ouabain-induced proliferation of leukemia cells. World J Pediatr. 2009;5:140–145. doi: 10.1007/s12519-009-0028-z. [DOI] [PubMed] [Google Scholar]
- 87.Akimova OA, Mongin AA, Hamet P, Orlov SN. The rapid decline of MTT reduction is not a marker of death signaling in ouabain-treated cells. Cell Mol Biol (Noisy-le-grand) 2006;52:71–77. [PubMed] [Google Scholar]
- 88.Orlov SN, Thorin-Trescases N, Pchejetski D, et al. Na+/K+ pump and endothelial cell survival: [Na+]i/[K+]i-independent necrosis triggered by ouabain, and protection against apoptosis mediated by elevation of [Na+]i. Pflugers Arch. 2004;448:335–345. doi: 10.1007/s00424-004-1262-9. [DOI] [PubMed] [Google Scholar]
- 89.Trevisi L, Visentin B, Cusinato F, Pighin I, Luciani S. Antiapoptotic effect of ouabain on human umbilical vein endothelial cells. Biochem Biophys Res Commun. 2004;321:716–721. doi: 10.1016/j.bbrc.2004.07.027. [DOI] [PubMed] [Google Scholar]
- 90.Isaev NK, Stelmashook EV, Halle A, et al. Inhibition of Na(+),K(+)-ATPase activity in cultured rat cerebellar granule cells prevents the onset of apoptosis induced by low potassium. Neurosci Lett. 2000;283:41–44. doi: 10.1016/s0304-3940(00)00903-4. [DOI] [PubMed] [Google Scholar]
- 91.Taurin S, Seyrantepe V, Orlov SN, et al. Proteome analysis and functional expression identify mortalin as an antiapoptotic gene induced by elevation of [Na+]i/[K+]i ratio in cultured vascular smooth muscle cells. Circ Res. 2002;91:915–922. doi: 10.1161/01.res.0000043020.45534.3e. [DOI] [PubMed] [Google Scholar]
- 92.Zhou X, Jiang G, Zhao A, Bondeva T, Hirszel P, Balla T. Inhibition of Na,K-ATPase activates PI3 kinase and inhibits apoptosis in LLC-PK1 cells. Biochem Biophys Res Commun. 2001;285:46–51. doi: 10.1006/bbrc.2001.5126. [DOI] [PubMed] [Google Scholar]
- 93.de Rezende Correa G, Araujo do Santos A, Frederico Leite Fontes C, Giestal de Araujo E. Ouabain induces an increase of retinal ganglion cell survival in vitro: the involvement of protein kinase C. Brain Res. 2005;1049:89–94. doi: 10.1016/j.brainres.2005.04.082. [DOI] [PubMed] [Google Scholar]
- 94.Golden WC, Martin LJ. Low-dose ouabain protects against excitotoxic apoptosis and up-regulates nuclear Bcl-2 in vivo. Neuroscience. 2006;137:133–144. doi: 10.1016/j.neuroscience.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 95.Pshezhetsky AV. Proteomic analysis of vascular smooth muscle cells treated with ouabain. Methods Mol Biol. 2007;357:253–269. doi: 10.1385/1-59745-214-9:253. [DOI] [PubMed] [Google Scholar]
- 96.Liu J, Tian J, Haas M, Shapiro JI, Askari A, Xie Z. Ouabain interaction with cardiac Na+/K+-ATPase initiates signal cascades independent of changes in intracellular Na+ and Ca2+ concentrations. J Biol Chem. 2000;275:27838–27844. doi: 10.1074/jbc.M002950200. [DOI] [PubMed] [Google Scholar]
- 97.Akimova OA, Bagrov AY, Lopina OD, et al. Cardiotonic steroids differentially affect intracellular Na+ and [Na+]i/[K+]i-independent signaling in C7-MDCK cells. J Biol Chem. 2005;280:832–839. doi: 10.1074/jbc.M411011200. [DOI] [PubMed] [Google Scholar]
- 98.Akimova OA, Lopina OD, Rubtsov AM, et al. Death of ouabain-treated renal epithelial cells: evidence for p38 MAPK-mediated Na (i) (+) /K (i) (+) -independent signaling. Apoptosis. 2009;14:1266–1273. doi: 10.1007/s10495-009-0404-0. [DOI] [PubMed] [Google Scholar]
- 99.Contreras RG, Shoshani L, Flores-Maldonado C, Lazaro A, Cereijido M. Relationship between Na(+),K(+)-ATPase and cell attachment. J Cell Sci. 1999;112(Pt 23):4223–4232. doi: 10.1242/jcs.112.23.4223. [DOI] [PubMed] [Google Scholar]
- 100.Giunta C, Cavaletto M, Pergola L, Pessione E, Bracchino P. Modulation of Na+/K+ pump in intact erythrocytes by cardioglycosides, steroid hormones and ouabain-like compounds. Gen Pharmacol. 1992;23:683–687. doi: 10.1016/0306-3623(92)90148-d. [DOI] [PubMed] [Google Scholar]
- 101.Alvarez-Leefmans FJ, Cruzblanca H, Gamino SM, Altamirano J, Nani A, Reuss L. Transmembrane ion movements elicited by sodium pump inhibition in Helix aspersa neurons. J Neurophysiol. 1994;71:1787–1796. doi: 10.1152/jn.1994.71.5.1787. [DOI] [PubMed] [Google Scholar]
- 102.Alvarez-Leefmans FJ, Gamino SM, Reuss L. Cell volume changes upon sodium pump inhibition in Helix aspersa neurones. J Physiol. 1992;458:603–619. doi: 10.1113/jphysiol.1992.sp019436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Armstrong CM. The Na/K pump, Cl ion, and osmotic stabilization of cells. Proc Natl Acad Sci U S A. 2003;100:6257–6262. doi: 10.1073/pnas.0931278100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dierkes PW, Wusten HJ, Klees G, Muller A, Hochstrate P. Ionic mechanism of ouabain-induced swelling of leech Retzius neurons. Pflugers Arch. 2006;452:25–35. doi: 10.1007/s00424-005-0009-6. [DOI] [PubMed] [Google Scholar]
- 105.Akimova OA, Lopina OD, Rubtsov AM, et al. Death of ouabain-treated renal epithelial cells: evidence for p38 MAPK-mediated Na (i) (+) /K (i) (+) -independent signaling. Apoptosis. 2009 doi: 10.1007/s10495-009-0404-0. [DOI] [PubMed] [Google Scholar]
- 106.Okada Y, Shimizu T, Maeno E, Tanabe S, Wang X, Takahashi N. Volume-sensitive chloride channels involved in apoptotic volume decrease and cell death. J Membr Biol. 2006;209:21–29. doi: 10.1007/s00232-005-0836-6. [DOI] [PubMed] [Google Scholar]
- 107.Szabo I, Lepple-Wienhues A, Kaba KN, Zoratti M, Gulbins E, Lang F. Tyrosine kinase-dependent activation of a chloride channel in CD95-induced apoptosis in T lymphocytes. Proc Natl Acad Sci U S A. 1998;95:6169–6174. doi: 10.1073/pnas.95.11.6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Franco R, DeHaven WI, Sifre MI, Bortner CD, Cidlowski JA. Glutathione depletion and disruption of intracellular ionic homeostasis regulate lymphoid cell apoptosis. J Biol Chem. 2008;283:36071–36087. doi: 10.1074/jbc.M807061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Storey NM, Gomez-Angelats M, Bortner CD, Armstrong DL, Cidlowski JA. Stimulation of Kv1.3 potassium channels by death receptors during apoptosis in Jurkat T lymphocytes. J Biol Chem. 2003;278:33319–33326. doi: 10.1074/jbc.M300443200. [DOI] [PubMed] [Google Scholar]
- 110.Friis MB, Friborg CR, Schneider L, et al. Cell shrinkage as a signal to apoptosis in NIH 3T3 fibroblasts. J Physiol. 2005;567:427–443. doi: 10.1113/jphysiol.2005.087130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Orlov SN, Pchejetski D, Taurin S, et al. Apoptosis in serum-deprived vascular smooth muscle cells: evidence for cell volume-independent mechanism. Apoptosis. 2004;9:55–66. doi: 10.1023/B:APPT.0000012122.47197.03. [DOI] [PubMed] [Google Scholar]
- 112.Fumarola C, Zerbini A, Guidotti GG. Glutamine deprivation-mediated cell shrinkage induces ligand-independent CD95 receptor signaling and apoptosis. Cell Death Differ. 2001;8:1004–1013. doi: 10.1038/sj.cdd.4400902. [DOI] [PubMed] [Google Scholar]