

Abstract
Live cell imaging is an important technique applied to a number of Drosophila tissues used as models to investigate topics such as axis specification, cell differentiation and organogenesis 1. Correct preparation of the experimental samples is a crucial, often neglected, step. The goal of preparation is to ensure physiological relevance and to establish optimal imaging conditions. To maintain tissue viability, it is critical to avoid dehydration, hypoxia, overheating or medium deterioration 2.
The Drosophila egg chamber is a well established system for examining questions relating, but not limited, to body patterning, mRNA localization and cytoskeletal organization 3,4. For early- and mid-stage egg chambers, mounting in halocarbon oil is good for survival in that it allows free diffusion of oxygen, prevents dehydration and hypoxia and has superb optical properties for microscopy. Imaging of fluorescent proteins is possible through the introduction of transgenes into the egg chamber or physical injection of labeled RNA, protein or antibodies 5-7. For example, addition of MS2 constructs to the genome of animals enables real time observation of mRNAs in the oocyte 8. These constructs allow for in vivo labeling of mRNA through utilization of the MS2 bacteriophage RNA stem loop interaction with its coat protein 9.
Here, we present a protocol for the extraction of ovaries as well as isolating individual ovarioles and egg chambers from the female Drosophila. For a detailed description of Drosophila oogenesis see Allan C. Spradling (1993, reprinted 2009) 10.
Keywords: Molecular Biology, Issue 60, Drosophila oocytes, wide-field microscopy, cytoskeleton, RNA injection, mRNA localization
Protocol
1. Drosophila preparation prior for dissection (According to E. Gavis, Princeton University)
Discard the adults from a sparsely seeded bottle and transfer the bottle to 25 °C in advance of your experiment. Once new adults have begun to emerge, wait two days.
Take a fly vial containing approximately 10ml of solid food and add 0.5-0.7 grams of dry yeast (see table) on a 45 degree angle. Do not cover the whole surface of the fly food with yeast.
Add a few drops of water to the yeast, sufficient to hydrate (Figure 1A). Place the vial at 45 degrees and allow about 3 minutes for the yeast to soak up the water (Figure 1B).
Note: Alternatively, premix a yeast paste in a separate container and add the paste to the vial with a spatula.
Anesthetize the flies in the vial by injecting CO2 gas. When they stop moving, tip the unconscious flies onto the CO2 fly pad for sorting (Figure 1C).
Isolate 10-15 females and 5 males of the desired genotype under a dissecting scope with a fine tipped paint brush (Figure 1D). Males are obvious by the presence of claspers, a dark protruding structure, at their posterior.
Add selected flies to the yeasted vial (Figure 1E). Place at 25 °C for 24-60 hours. Incubate for shorter time periods for a larger percent of young to mid stage egg chambers and longer time periods for late stage egg chambers.
2. Drosophila ovary dissection
Place a small drop of halocarbon oil on a No. 1.5, 22 x 50 mm cover slip (Figure 2A). Set the prepared cover slip to the side where it will not be in the way.
Anesthetize the flies in the yeasted vile by injecting CO2 gas and tip onto a CO2 pad.
Use sharp fine-nosed forceps to grasp an individual fattened female at the anterior of the abdomen (not between the abdomen and thorax) (Figure 2B-C).
Hold the female with the forceps and view under a dissecting microscope, roughly 2-3 cm above the prepared coverslip.
Still holding at the anterior, use a second set of forceps to remove the tissue at the extreme posterior of the female (Figure 2D). Note: Gut tissue will usually pull out with the posterior end. Wipe the removed tissue from the forceps.
While still holding the female at the anterior, use the wiped forceps to gently squeeze or massage the abdomen, working from the anterior towards the posterior. Massage the abdomen in the same fashion as removing toothpaste from the end of a tube.
Two large opaque structures will be pushed from the abdomen and stick on the end of the forceps (Figure 2E-F). These are the paired ovaries.
Touch the ovaries onto the halocarbon oil on the slide below.
Repeat the dissection 1-2 times to give a total of 4-6 ovaries in the oil on the coverslip (Figure 2G).
3. Isolating ovarioles
Note: Each ovary contains about 16 ovarioles made up of 6 to 10 egg chambers of different stages arranged like beads on a string10.
Note: Prior to isolating the ovarioles, adjust the light source on the dissecting microscope so that the illumination strikes at a shallow angle to the sample. This gives contrast to the sample and allows visualization of the young stage egg chambers.
In the oil, separate the two ovaries by removing the connecting tissue at the posterior end (old stages) with closed forceps (Figure 3A).
Orient an individual ovary with the oldest stages to the left and youngest stage to the right. The young stage egg chambers in the ovary are transparent and form the more pointed end of the ovary.
With a pair of forceps in the dominant hand, gently grasp the posterior of an ovary, preferably by the remnants of the connective tissue. While holding the ovary in place with the forceps, use a dissecting probe (or tungsten needle) to tease out individual ovarioles (Figure 3B-D).
Note: Individual ovarioles will break between the young stages (germarium to stage 10) as the older stages are too large to be separated from the full ovary.
Note: Puncturing a late stage oocyte with the dissecting probe will result in the cytoplasm leaking into the oil and make extraction of individual ovarioles very challenging. If a late stage oocyte is punctured, move to a fresh ovary.
Once the ovariole is free of the ovary, use the dissecting probe to drag it to the center of the oil drop and orient in the desired orientation for the experiment. Note: Ovarioles in oil will settle and adhere to the glass.
Repeat this process (3.3-3.4) until there are 15-25 individual ovarioles. Note: This may require the dissection of multiple ovaries. The whole process should take less than 15 minutes.
Note: It is advisable to dissect one ovary each from several flies rather than dissecting both ovaries from fewer flies to increase the n number of flies for the experiment.
Note: Rough handling of the ovariole will result in unhealthy oocytes and can lead to artifacts in the experiment.
Note: Oocytes will begin to display phenotypic changes due to stress 40 minutes after being dissected from the ovary (compare Figure 7E to F).
Once ovarioles are in an adequate orientation (Figure 3E), use the dissecting probe and closed forceps to move excess ovarian tissue out of the oil. This material can be wiped from the forceps onto a paper towel.
4. Isolating individual late stage egg chambers (According to E. Gavis, Princeton University)
With a pair of forceps in the dominant hand, grasp the posterior of an isolated ovary, preferably by the remnants of the connective tissue. While holding the ovary in place with the forceps, introduce the dissecting probe into the posterior end of the ovary near the forceps (Figure 4A). Avoid puncturing any egg chambers by inserting the probe between late stage egg chambers.
Pull the dissecting needle through the ovary until it exits at the early stage egg chambers (Figure 4B). When done correctly, the probe will remove the connective tissue holding the ovarioles and late stage egg chambers in place.
Repeat step 4.2 until the ovarioles are spread out on the coverslip. Note: When egg chambers are no longer stacked on top of each other, this step is complete.
Using the dissecting probe, separate the late oocytes to allow for easy imaging (Figure 4C).
Note: Puncturing an oocyte while dissecting late stage egg chambers is not as condemning as with dissecting individual ovarioles for younger stages.
Note: For stage 14 egg chambers, use the forceps to grasp the dorsal appendages for orientation.
5. Injection preparation
Note: For the injection of mid stage oocytes, orient the ovarioles perpendicularly to the long axis of the cover slip.
Turn on the imaging microscope [in our case a DeltaVision Core from Applied Precision] and load the appropriate imaging settings (Figure 5A-B).
Turn on the injection apparatus (Figure 5C).
Load the injection needle with a loading tip (Figure 5D-E). Solutions to be injected should be centrifuged briefly at high speed to avoid deposits that might block the injection needle.
Attach the loaded injection needle to the injector. Take care to avoid breaking the needle (Figure 5F).
To assist with unclogging needles, place a small piece of glass 2x10 mm from a cut or broken coverslip at one side of the oil drop containing the ovarioles. Make sure this glass fragment is covered in oil. This glass will serve to ram the needle tip against for breaking or unclogging the needle.
6. Injection of fluorescent RNA
Before you begin: Set up the injection apparatus and micro-manipulator controls such that the injection needle is positioned over the center of the field of view.
Mount the sample to be injected on the microscope stage.
Select the 20x objective. Higher or lower magnification objectives can also be used.
Note: If using an automated stage with point visiting capability it is advisable to mark all the stage 8-9 oocytes for injection before beginning. This allows for easy visiting and injection of selected oocytes.
Lower the injection needle until it touches the oil. Center the needle tip over the objective lens.
Turn off the room lights and turn on the bright-field light for the microscope. Select the first point (oocyte) on the marked points list. While looking through the oculars, focus on the oocyte (Figure 6A).
Lower the needle manually until it is in the same plane of focus as the oocyte and visible next to the oocyte.
Using the manipulator controls, move the point of the injection needle until it is inside the oocyte. A fast stabbing movement is often more successful than a slow advance. Once inside, eject a small volume from the needle and visually assess the effects, repeat as required. If the injection is successful, the cytoplasm inside the oocyte will be displaced, if this is not apparent, repeat. It is not necessary to image in fluorescence to determine if material has been injected. After injection, remove the needle from inside the oocyte (Figure 6B-G).
The precise amount of fluid injected is difficult to quantitate. To increase the injected volume, adjust the pressure or time of the injection pulse, break the needle tip to increase the opening slightly or use repeated injection pulses.
Note: The whole injection process should take less than 10 seconds when executed properly. Extensive stabbing or lingering with the needle inside to oocyte will result in severe damage to the egg chamber.
Before selecting the next marked oocyte, raise the injection needle high enough in the oil so as not to contact other oocytes in the travel path.
At the new oocyte, repeat step 6.6. Carry on in this way until all the required oocytes are injected.
Note: If a mistake is made while injecting, move to the next marked oocyte. Do not waste time on damaged oocytes.
Note: The needle may become clogged during an injection session. In this case, move the needle adjacent to the broken piece of glass in the oil on the coverslip. With the glass and needle in the same focal plane, gently ram the needle into the glass (Figure 6H). Test that the needle is unclogged by pushing the injection button and seeing if any fluid exits the tip of the needle.
When all the oocytes have been injected, manually move the needle up, out of the oil and orient it out of the beam path for the microscope. Make sure that it is in a safe orientation, to avoid eye damage.
Note: If an extended period of time has elapsed between egg chamber isolation and oocyte injection, oocytes will be difficult to inject and exhibit phenotypic changes associated with stress. Similar issues can arise with rough handling of the oocyte or injection of excess volumes (compare Figures 6I, J, K).
7. Live imaging experimental design
Select the first injected oocyte from the list. Test that the injection was inside the oocyte by imaging one frame in fluorescence (as in Figure 6G).
Configure the experimental parameters, including: excitation wavelength, emission path, excitation time, Z-stack and time between collections. Note: For more information on this, see Parton R., et al. (2010) 2.
Gather the data set.
8. Representative Results
In vitro synthesized Alexa-546 grk RNA injected into an Me31B::GFP egg chamber (Figure 7A). The RNA localizes to the dorsal anterior of the oocyte and forms a cap around the nucleus (Figure 7B). For more examples of RNA localization see MacDougall N., et al. (2003) 11.
Mid and late stage oocytes expressing fluorescent labeled protein (Tau-GFP, Figure 7C) or RNA tagging systems (grk*mCherry, Figure 7D) can be imaged without injection.
 Figure 1.
Figure 1.
 Figure 2.
Figure 2.
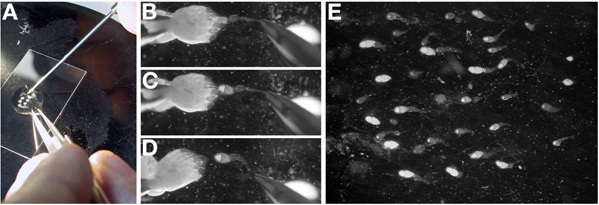 Figure 3.
Figure 3.
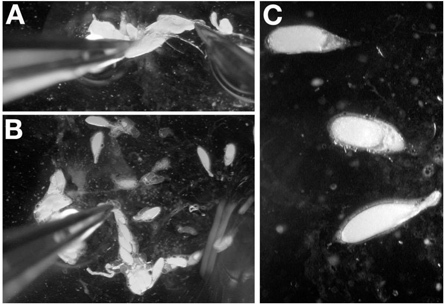 Figure 4.
Figure 4.
 Figure 5.
Figure 5.
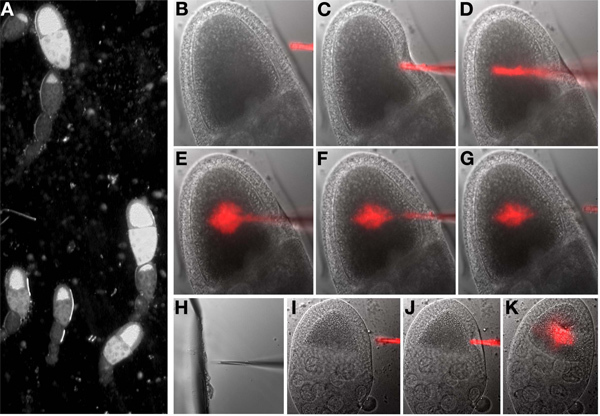 Figure 6.
Figure 6.
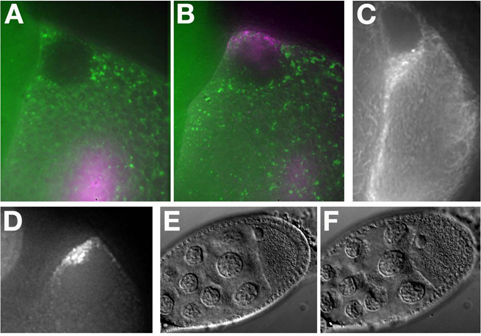 Figure 7.
Figure 7.
Discussion
Live cell imaging is a powerful assay for examining cellular processes in real time. In addition to simple bright field observation, the addition of fluorescent labels to proteins and RNAs of interest has lead to many breakthroughs. Here we have outlined a protocol for imaging individual living oocytes that can be utilized in combination with genetic and biochemical assays.
In this protocol, we also explain how to experimentally manipulate living oocytes by injection. There are many possibilities for material to inject, including in vitro synthesized fluorescent labeled RNA to assay the ability of a secondary structure to direct RNA localization (Ball and Davis, unpublished) and antibodies that inhibit the function of proteins 6. Future work will likely see the introduction of other labeling components and cellular machinery to living oocytes enabling molecular mechanisms to be tested.
Preserving the viability and health of the tissue is essential when working with live cells. In this protocol, we point out a number of steps that can lead to stress of the egg chamber. For example, despite oil being superior for imaging, extended culture in the oil can lead to stress on the egg chamber. This can be easily monitored under bright field exposure by examining the nuclear morphology and position, oocyte membrane that will distort and bleb under stress (compare Figure 7E (unstressed) to Figure7F (stressed)). Stage 9 egg chambers show RNA localization and border cell migration 12 over multiple hours. However, a stage 7/8 egg chamber will begin to exhibit ill effects in halo carbon oil after approximately 40 minutes of the ovary being removed from the female. Late stage egg chambers, stage 11 to 14, can develop normally in either oil or aqueous media because of the secretion of the egg shell from the follicle cells at these stages 13. It has been reported that the addition of insulin to aqueous insect medium can maintain stage 9 oocytes for up to 6 hours 14 and germaria up to 14 hours 15. In all cases, aggressive maneuvering of the egg chamber should be avoided, as it stresses the oocytes and reduces its viability. Imaging fewer egg chambers and taking care to treat each gently is the best way to ensure optimal viability.
Disclosures
We have nothing to disclose.
Acknowledgments
This work was supported by a Wellcome Trust Senior Research Fellowship to I. Davis.
References
- Arias AM. Drosophila melanogaster and the development of biology in the 20th century. Methods Mol. Biol. 2008;420:1–25. doi: 10.1007/978-1-59745-583-1_1. [DOI] [PubMed] [Google Scholar]
- Parton RM, Valles A-M, Dobbie ID, Davis I. Pushing the Limits of Live Cell Imaging in Drosophila. In: Goldman RD, Swedlow JR, Spector DL, editors. Live Cell Imaging: A Laboratory. Cold Spring Harbor Laboratory Press; 2010. pp. 387–418. [Google Scholar]
- Johnston D. Moving messages: the intracellular localization of mRNAs. Nat. Rev. Mol. Cell Biol. 2005;6(5):363–375. doi: 10.1038/nrm1643. [DOI] [PubMed] [Google Scholar]
- Jansen R. mRNA Localization: Message on the Move. Nat. Rev. Mol. Cell Biol. 2001;2:247–256. doi: 10.1038/35067016. [DOI] [PubMed] [Google Scholar]
- Van De Bor V, Hartswood E, Jones C, Finnegan D, Davis I. gurken and the I factor retrotransposon RNAs share common localization signals and machinery. Dev. Cell. 2005;9(1):51–62. doi: 10.1016/j.devcel.2005.04.012. [DOI] [PubMed] [Google Scholar]
- Delanoue R, Herpers B, Soetaert J, Davis I, Rabouille C. Drosophila Squid/hnRNP helps Dynein switch from a gurken mRNA transport motor to an ultrastructural static anchor in sponge bodies. Dev. Cell. 2007;13(4):523–538. doi: 10.1016/j.devcel.2007.08.022. [DOI] [PubMed] [Google Scholar]
- Jaramillo AM, Weil TT, Goodhouse J, Gavis ER, Schupbach T. The dynamics of fluorescently labeled endogenous gurken mRNA in Drosophila. J. Cell. Sci. 2008;121(Pt. 6):887–894. doi: 10.1242/jcs.019091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest KM, Gavis ER. Live imaging of endogenous mRNA reveals a diffusion and entrapment mechanism for nanos mRNA localization in Drosophila. Curr. Biol. 2003;13:1159–1168. doi: 10.1016/s0960-9822(03)00451-2. [DOI] [PubMed] [Google Scholar]
- Bertrand E. Localization of ASH1 mRNA particles in living yeast. Mol. Cell. 1998;2(4):437–445. doi: 10.1016/s1097-2765(00)80143-4. [DOI] [PubMed] [Google Scholar]
- Spradling AC. Developmental genetics of oogenesis. In: Bate M, Martinez-Arias A, editors. The Development of Drosophila melanogaster. Vol. 1. Cold Spring Harbor: Cold Spring Harbor Press; 1993. pp. 1–70. [Google Scholar]
- MacDougall N, Clark A, MacDougall E, Davis I. Drosophila gurken (TGFalpha) mRNA localizes as particles that move within the oocyte in two dynein-dependent steps. Dev. Cell. 2003;4(3):307–319. doi: 10.1016/s1534-5807(03)00058-3. [DOI] [PubMed] [Google Scholar]
- Tekotte H, Tollervey D, Davis I. Imaging the migrating border cell cluster in living Drosophila egg chambers. Dev. Dyn. 2007;236(10):2818–2824. doi: 10.1002/dvdy.21305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weil TT, Parton R, Davis I, Gavis ER. Changes in bicoid mRNA anchoring highlight conserved mechanisms during the oocyte-to-embryo transition. Curr. Biol. 2008;18:1055–1061. doi: 10.1016/j.cub.2008.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad M, Jang AC, Starz-Gaiano M, Melani M, Montell DJ. A protocol for culturing Drosophila melanogaster stage 9 egg chambers for live imaging. Nat. Protoc. 2007;2(10):2467–2473. doi: 10.1038/nprot.2007.363. [DOI] [PubMed] [Google Scholar]
- Morris LX, Spradling AC. Long-term live imaging provides new insight into stem cell regulation and germline-soma coordination in the Drosophila ovary. Development. 2011;138(11):2207–2215. doi: 10.1242/dev.065508. [DOI] [PMC free article] [PubMed] [Google Scholar]