

Abstract
The American Cancer Society’s 2009 statistics estimate that 1 out of every 4 deaths is cancer related. Genomic instability is a common feature of cancerous states, and an increase in genomic instability is the diagnostic feature of Bloom Syndrome. Bloom Syndrome, a rare disorder characterized by a predisposition to cancer, is caused by mutations of the BLM gene. This study focuses on the partnerships of BLM protein to RAD51, a Homologous Recombination repair protein essential for survival. A systematic set of BLM deletion fragments were generated to refine the protein binding domains of BLM to RAD51 and determine interacting regions of BLM and ssDNA. Results show that RAD51 and ssDNA interact in overlapping regions; BLM100–214 and BLM1317–1367. The overlapping nature of these regions suggests a preferential binding for one partner that could function to regulate homologous recombination and therefore helps to clarify the role of BLM in maintaining genomic stability.
Keywords: Bloom syndrome, Protein–protein interaction, RAD51, Homologous recombination, Cancer
1 Introduction
Maintenance of an accurate genome is critical to cellular survival. In fact, increased genomic instability is thought to be essential to the multi-step process through which cells accumulate mutational characteristics and transition to a tumorigenic state [11]. Therefore, cells have developed complex repair systems to address the vast array of DNA lesions caused by both endogenous and environmental genotoxins. Homologous recombination repair (HR) is responsible for the repair of lethal DNA double-strand breaks (DSB) and the restoration of blocked or collapsed replication forks. Additionally, HR plays critical roles in homologous chromosome segregation, meiotic shuffling and telomere maintenance [1, 9, 10, 19, 22, 23].
HR is initiated by the MRN complex [Mre11, Rad50, NBN (formerly NBS1)] resecting the DSB ends to generate 3′ overhangs flanking the break site. The resulting single-stranded DNA (ssDNA) ends are first protected with RPA before eventually being coated with RAD51 protein to form the critical nucleoprotein filament necessary for accurate repair. This filament searches the genome for an appropriately homologous sequence to catalyze the strand exchange and form a D-loop HR intermediate [20]. The 3′DNA ends are then elongated using the homologous sequence as a template for repair. If only one end of the DSB is used as a template, the D-loop can dissociate with repair being completed via two analogous processes: Synthesis-Dependent Strand Annealing (SDSA) or Break Induced Replication (BIR) [13]. Alternatively, if both ends of the DSB are elongated, a double Holliday junction (DHJ) is formed that must be resolved in order to complete repair [13]. DHJ resolution to obtain the minimum amount of crossed DNA sequence is accomplished with the BLM-TopIIIalpha-RmiI complex [14]. Inappropriate or misaligned homologous recombination events can lead to large-scale chromosomal insertions, deletions and/or translocations. Thus, HR regulation is critical to global genomic stability.
Bloom syndrome (BS) is an autosomal recessive disorder that results from a mutation of the BLM gene, a RecQ DNA helicase family member. BS patients exhibit extraordinarily high levels of sister chromatid exchange (SCE) events, a marker of genomic instability. The RecQ family of DNA helicases, named for similarities to the E. coli helicase recQ, plays a major role in the maintenance of genomic stability (reviewed in [2, 16]). RecQ proteins are characterized as ATP- and Mg2+-dependent helicases able to unwind a variety of duplex DNA structures in the 3′ → 5′ direction. Human cells contain five RecQ-like helicases, encoded by the BLM, WRN, RECQL/RECQ1, RECQL4 and RECQL5 genes. Defects in BLM, WRN, and RECQL4 result in the human autosomal recessive disorders Bloom syndrome, Werner syndrome and Rothmund-Thomson syndrome, respectively. Unlike other DNA helicases, the RecQ family, and in particular BLM shows a preference for non-Watson–Crick DNA structures such as Holliday Junctions, Y-form DNA (that mimic replication forks), and D-loops. Spontaneous chromosomal breakage, increased genomic instability, and significantly elevated levels of cancer susceptibility characterize these syndromes, providing a direct link between RecQ helicase function and the onset of genomic instability.
The catalytic core of BLM houses a helicase that is suggested to stabilize stalled replication forks and suppress genomic instability. Since blocked replication forks are a universal type of DNA damage irrespective of the specific genotoxic agent, a thorough knowledge of BLM-mediated fork stability is essential to the broad understanding of cellular responses to genotoxic agents. Additionally, a recent study demonstrated that depletion of BLM undermines cellular survival in response to chemotherapeutic agents; providing a rationale for developing BLM as a biomarker for chemotherapeutic responsiveness [15].
BLM facilitates its replication-fork stabilizing function through multiple protein–protein interactions. BLM was identified in a large DNA damage surveillance complex with BRCA1 and the MRN complex [21] and has been shown to associate with many HR proteins. For example, BLM partners with TopIIIα and RmiI to form the RTR complex that is required for DHJ resolution [14]. An essential BLM interacting partner is RAD51, a protein central to the HR pathway [21, 24]. BLM physically and functionally interacts with RAD51 and has been reported to displace RAD51 from the nucleo-protein filament that is responsible for homology searching and strand invasion [3, 4]. The studies described herein refine the amino acid interaction domain between BLM and RAD51 as well as define an adjacent BLM domain that interacts with DNA. Together these data provide a better understanding of the conditions necessary to modulate this critical interaction and help clarify the role of HR in maintaining a stable genome.
2 Materials and Methods
2.1 Plasmid Construction
BLM protein fragment DNA was PCR amplified from full length BLM cDNA (kind gift from Dr. Nathan A. Ellis, University of Illinois, Chicago, IL) without a start codon but containing a C-terminal epitope FLAG tag. Amplified DNA was agarose gel-purified and cloned into the Gateway entry vector pENTR-D (Invitrogen Corp., Carlsbad, CA). Accurate sequence was confirmed on an ABI 3130xl genetic analyzer with KB™ Basecaller software (Genomic Sequencing Center, Kingston, RI) then recombined into the pDEST-17 destination vector to incorporate an N-terminal 6x-HIS epitope tag (Invitrogen Corp., Carlsbad, CA) using DH5α as E. coli host. The final expression plasmid was verified by restriction endonuclease digestion and transformed into either Rosetta-2 competent cells (Novagen, Gibbstown, NJ) or C41 (DE3) competent cells (Lucingen Corp., Middleton, WI). The final BLM fragments contain both an N-terminal 6x-HIS and a C-terminal FLAG epitope tag.
2.2 Protein Expression and Purification
E. coli strain C41(DE3) was grown in YT Broth while Rosetta-2 strains were grown in Luria–Bertani (LB). Both strains were supplemented with 100 ug/mL amplicillin for positive selection. Bacterial overnight cultures (50 mL) were inoculated into 1 L fresh broth and cultured for 2–2.5 h to log phase (OD600 of 0.5–0.7). Protein expression was initiated with addition of 1 mM IPTG. Cultures were incubated for an additional 2 h with gentle shaking at RT for C41 cells and 37 °C for Rosetta cells. An aliquot was removed for verification while the remaining culture was centrifuged, supernatant discarded and stored at −20 °C. Recombinant BLM protein fragments were purified on either Ni2+-NTA Agarose (Qaigen Corp, Valencia, CA) or Rapid S cation exchange resin (Bio-Rad Laboratories, Hercules, CA). Cell pellets were lysed in the corresponding manufacturers’ recommended buffer supplemented with benzonase, lysozyme and protease inhibitors. Cellular debris was removed via centrifugation and the remaining supernatant was incubated with 1 mL of the appropriate resin for 30 min with end-over-end rotation. The resin was then transferred to a column support, drained, washed with 10 column volumes of buffer, and eluted with two column volumes of buffer containing either 50 mM NaH2PO4, 300 mM NaCl and 500 mM Imidazole for Nickel or 250 mM NaCl for Rapid S resin. Elutions were desalted on Zeba columns (Pierce Biotechnology, Rockford, IL) into 60 mM Tris–HCl pH 7.5, 100 mM NaCl, 1 mM EDTA, 1 mM 2-mercaptoethanol and stored at −80 °C in 25% glycerol.
2.3 Conventional Western Blotting
BLM fragments were resolved by SDS–PAGE and transferred to nitrocellulose membranes (BioRad, Inc, Hercules, CA) for further processing. Blots were blocked in TBST containing 5% milk powder for 1 h at RT before being incubated overnight at 4 °C in TBST/5% milk powder supplemented with primary antibody (anti-FLAG M2, Sigma–Aldrich, Inc, St. Louis, MO—1:1,000 dilution). Blots were washed 8 times in TBST before being incubated with anti-Mouse IgG conjugated horseradish peroxidase (1:10,000, from Sigma–Aldrich, Inc, St. Louis, MO) for 1 h at RT. Signal was detected using Supersignal Pico (Pierce Biotechnologies, Rockford, IL) following manufacturers’ instructions and imaged on an SRX-101A developer from Healthcare Technologies, Inc (Marietta, GA).
2.4 Far Western Immunoblotting
BLM N-terminal fragments were resolved by SDS–PAGE and transferred to nitrocellulose membranes (BioRad, Inc., Hercules, CA) for further processing. All subsequent steps were performed at 4 °C, unless stated otherwise. Blots were immersed in denaturation buffer (6 M guanidine HCl in PBS) for 10 min and then incubated 6 times in serial dilutions (1:1) of denaturation buffer in PBS for 10 min. Membranes were blocked for 30 min in PBS containing 10% milk powder supplemented with 0.1% Tween-20 then incubated overnight with fresh RAD51 cell lysate in PBS 0.25% milk powder and 0.1% Tween-20 supplemented with Halt protease inhibitor (Pierce Biotechologies, Rockford, IL) per manufacturers’ instruction. Blots were washed 4 times for 10 min in PBS with 0.25% milk powder and 0.1% Tween-20. The second wash contained 0.0001% glutaraldehyde. Conventional western blotting was then performed to detect the presence of RAD51 protein using anti-RAD51, 1:1,000 dilution (EMD Chemicals, Inc, Gibbstown, NJ) and anti-Mouse IgG conjugated horseradish peroxidase, 1:10,000 dilution. Signal was detected using Supersignal Pico (Pierce Biotechnologies, Rockford, IL) following manufacturers’ instructions and imaged on an SRX-101A developer from Healthcare Technologies, Inc (Marietta, GA). Signal intensity was quantified using Quantity One software from BioRad, Inc. Stastistical significance was determined using a one-way ANOVA calculation with a Dunnett’s Multiple comparison test.
2.5 Southwestern Analysis
Fragments of the BLM termini proteins were handled similar to the Far western protocol with the following exceptions. Blots were incubated with 0.8 μM of biotin labeled ssDNA (sequence: BIOTIN- CGGGTCAACGT GGGCAAAGATGTCCTAGCAA) in 0.25% milk, 1 × PBS, 0.1% Tween 20 for 24 h. Blots were washed then crosslinked with 0.0001% glutaraldehyde in 0.25% milk, 1 × PBS, 0.1% Tween 20 at 4 °C and probed with anti-Avidin conjugated to HRP (Sigma–Aldrich, Inc, St. Louis, MO) (in 0.5% milk, 1 × TBS, 0.1% Tween 20 at 4 °C for 18 h. Signal was detected using Supersignal Pico (Pierce Biotechnologies, Rockford, IL) following manufacturers’ instructions and imaged on a SRX-101A developer from Healthcare Technologies, Inc (Marietta, GA). Signal intensity was quantified using Quantity One software from BioRad, Inc. Statistical significance was determined using a one-way ANOVA calculation with a Dunnett’s multiple comparison test.
3 Results
3.1 Construction of the BLM Expression Clone, Protein Expression and Purification
Wu et al. reported two independent interaction domains between RAD51 and BLM [24]. Specifically, BLM amino acids 1–213 on the N-terminus and 1,317–1,417 of the C-terminus physically interact with RAD51 (Fig. 1a). Although the termini of BLM are likely to be responsible for its unique affects on genomic stability, little information is available regarding these regions. No crystal structure of BLM termini exists. Thus, a refinement of the interaction domains is vital to understanding this critical interaction. To that end, we generated a set of BLM protein fragments to study the association of BLM with RAD51. The set includes the two fragments known to interact with RAD51 (BLM1–214 and BLM1317–1417) as well as a systematic set of deletion mutants within these domains: BLM1–50, BLM1–100, BLM1–150, BLM1367–1417 (Fig. 1b). An additional fragment, BLM125–360, was generated to complete the set (Fig. 1b).
Fig. 1.
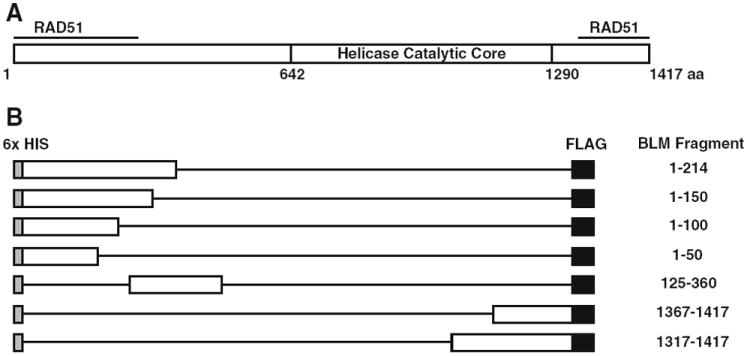
Schematic of BLM fragments generated. a Full length BLM protein with regions that interact with RAD51 marked. b All fragments generated include an N-terminal 6× Histidine epitope tag and a C-terminal FLAG epitope tag. Exact amino acids in each fragment are listed on the right
To express and purify the human BLM protein fragments in E. coli, cDNA of the corresponding fragment sequence was PCR amplified with a FLAG-epitope tag that was engineered into the C-terminus (Fig. 1b). Amplified DNA was purified and inserted into the pENTR-D vector (Gateway, Invitrogen). DNA sequencing of the BLM ORF verified each entry vector prior to recombination into an E. coli protein expression vector containing a hexahistidine epitope tag fused to the N-terminus. Final destination vectors were verified by restriction endonuclease analysis and transformed into Rosetta-2 (Novagen, Gibbstown, NJ) chemically competent cells.
Initial induction experiments showed low level expression of the BLM fragments when expressed in the E. coli strain Rosetta-2 but with evidence of rapid proteolysis (not shown), suggesting elevated expression of BLM fragments in this strain might be toxic. To diminish this effect, the BLM protein fragments were expressed following transformation into the E. coli strain C41 (DE3) (Lucigen Corp., Middleton, WI), a strain developed with genetic mutations phenotypically selected for conferring toxicity tolerance. The optimized expression of BLM protein fragments was determined following induction and activation of the T7 promoter with IPTG addition to the culture medium. BLM expression was verified via immunoblotting using the anti-FLAG M2 antibody. Calculated masses for each fragment were compared to protein electrophoresis migration of molecular weight protein markers. The calculated mass of BLM1–214 is 28.3 kDa but its apparent molecular mass appears slightly greater, migrating to just under the 37 kDa molecular marker. Repeated electrophoresis consistently identified the location of this fragment at 35 kDa (Fig. 2a left panel). Each BLM fragment migrated slightly higher than the calculated mass: BLM1–50 to 15 kDa, BLM1–100 to 19 kDa, BLM1–150 to 28 kDa and BLM1367–1417 to 19 kDa (Fig. 2a). This slightly increased kDa pattern is consistent with numerous other 6x-HIS epitope-tagged proteins purified in our lab (unpublished observation).
Fig. 2.
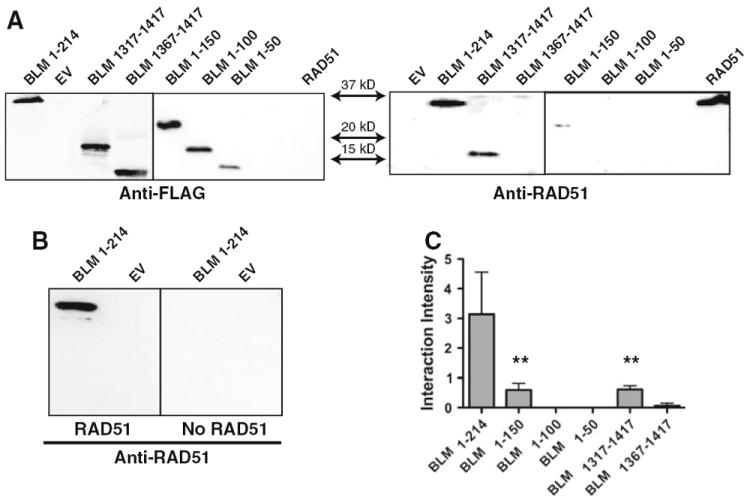
Far western Immunoblotting of BLM termini. a Left panel Conventional western analysis of the BLM fragments using anti-FLAG M2 antibody. Right panel Far western analysis of BLM fragments interaction with RAD51 using anti-RAD51 antibody. b Left panel Far western of BLM fragments incubated with RAD51 cell lysate. Right panel Far western of identical blot incubated with empty vector control cell lysate. c Quantification of data represented in (a). RAD51 far western interaction bands were normalized for BLM fragment loading controls. Graph represents the mean of three independent trials with standard deviations indicated. One-way ANOVA analysis was completed for all fragments with Dunnett’s multiple comparison test. Statistical significance of p < 0.01 is indicated with double asterisks
The N-terminal BLM fragments were enriched using nickel-chelate affinity chromatography. Imidazole elution (500 mM) removed the hexahistidine fragments from the resin to enrich the eluant to 60–99% pure BLM fragment. Impurities remaining included endogenous E. coli proteins that were used as protein normalizing controls when comparing to the empty vector control lysates. C-terminal fragments were unresponsive to Nickel affinity column purification presumably due to the N-terminal epitope tag being masked by the tertiary structure of the native protein fragment. These fragments were purified by cation exchange chromatography and were also studied as protein lysates.
3.2 BLM-RAD51 Interacting Domains
The protein–protein interaction domains between BLM and RAD51 were refined using a Far western analysis protocol. Results of the N-terminal BLM fragments confirmed the BLM1–214 fragment partners with the RAD51 protein [24]. BLM1–150 also interacts with RAD51 although the interaction is significantly weaker, as compared to the interaction between RAD51 and the BLM1–214 fragment. The BLM1–50 and BLM1–100 fragments showed no observable partnership with RAD51 (Fig. 2). This clearly establishes that the preponderance of the BLM N-terminus interacting domain for RAD51 is housed between amino acids 100 and 214 although amino acids 150–214 may be responsible for the majority of the interaction.
C-terminal screening of BLM1317–1417 for interaction with RAD51 lysate confirmed the independent interaction of the C-Terminus amino acids 1,317–1,417, while the truncated, BLM1367–1417, yielded no significant interaction. Therefore, the primary BLM interaction domain with RAD51 is between amino acid residues 100–214 and 1,317–1,367, with half of the interacting strength housed within the 150–214 amino acid region (Fig. 2b). One-tailed ANOVA analysis of the interaction normalized to fragment loading revealed a statistically significant difference in BLM1–150 and BLM1317–1417 as compared to BLM1–214 binding (p < 0.01).
3.3 BLM-DNA Interaction
This BLM region may influence genomic stability by regulating the interaction between RAD51 and DNA. To investigate the interaction of these BLM protein fragments with DNA directly, we utilized a southwestern analysis of the BLM fragments with ssDNA. This analysis revealed a BLM-DNA interacting domain similar to the BLM-RAD51 interacting domain. Fragments 1–214, 1–150 and 1,317–1,417 all yielded positive DNA interaction results (Fig. 3a). In this case however, the majority of the interaction is housed in the 1,317–1,367 domain. Fragments BLM1–214 and BLM1–150 interacted with equal intensity to the ssDNA (Fig. 3b), suggesting that the interaction domain is present in both fragments. However, BLM125–360 showed no association with DNA (Fig. 3). This further refines the primary ssDNA interacting domain to the 25 amino acids encompassed by BLM100–125. Interestingly, the pI for this interacting BLM sequence (residues 100–125) is calculated as 8.7 while the calculated pI for the non-interacting BLM domain (residues 125–214) is 9.1, suggesting that the interaction is not likely the result of electrostatic attraction to the DNA backbone.
Fig. 3.
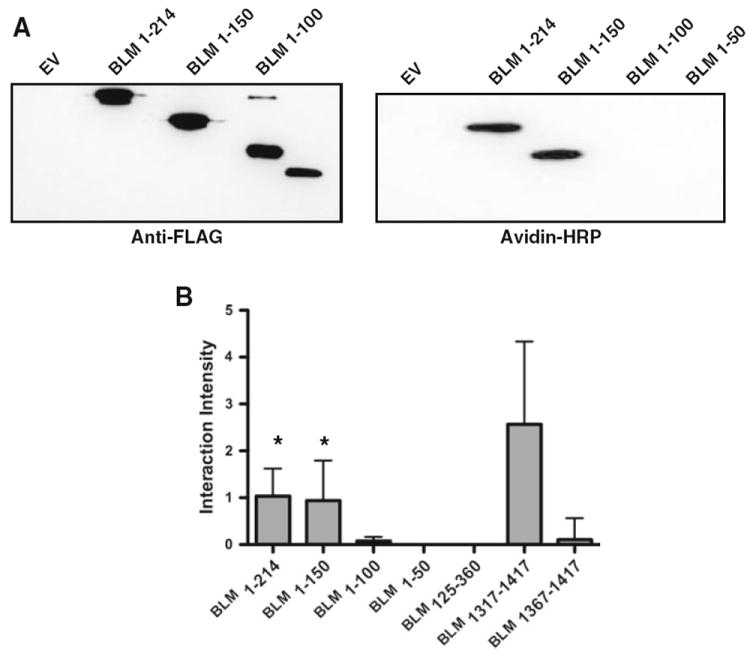
Southwestern analysis. a Representative data from one of three independent trials. The left panel shows the BLM fragments probed with anti-FLAG (M2) antibody. The right panel shows the same fragments incubated with biotin labeled ssDNA and probed with HRP-conjugated avidin. b Quantification of southwestern analysis of BLM fragments shown in (a). Graph represents the mean of three independent trials with standard deviations indicated. One-way ANOVA analysis was completed for all fragments with Dunnett’s multiple comparison test with BLM1317–1417 as control. Statistical significance of p < 0.05 is indicated with an asterisk
4 Discussion
Bloom syndrome is characterized by a predisposition to early-onset cancer that is unique among RecQ helicases. Although the catalytic core of the 5 RecQ family human paralogs is highly conserved, the termini are significantly different and thus could contribute to the variation of the resulting human disease states. BLM protein interacts with numerous DNA replication and repair proteins at its temini, for example topoisomerase IIIα [25], Flap endonuclease 1 [18] DNA mismatch repair protein MLH1 [12, 17] and RAD51 [24]. Additionally, all of these proteins interact with DNA. Currently, each of these proteins is thought to bind in common regions of BLM termini, specifically, the first 214 amino acids and the last 100 amino acids are frequent sites of binding. A refinement of these interaction domains will increase our understanding of the nature of these interactions (Fig. 4).
Fig. 4.
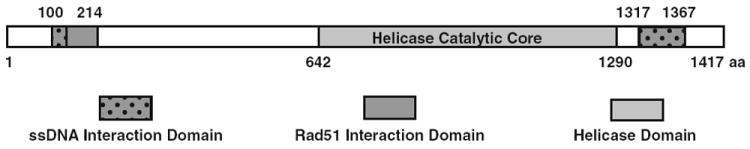
Representation of BLM interaction domains with RAD51 and ssDNA
BLM demonstrates anti-recombinagenic activity in a variety of proposed pathways [3] two of which involve the RAD51-ssDNA filament that is essential for homology searching and strand invasion. BLM has been shown to regulate the RAD51-ssDNA filament formation by displacing RAD51 from the DNA whereby suppressing HR initiation. Additionally, BLM can suppress genome-destabilizing crossover events by promoting the use of the SDSA pathway. In this case, the RAD51 filament has invaded the homologous DNA strand forming a D-loop. BLM, in the presence of RPA, can then stimulate DNA extension via polymerase η [4]. This extended D-loop could then be disrupted, allowing the newly extended strand to anneal to its original complementary strand. Although the mechanism of this displacement has yet to be determined, it could occur through protein–protein interaction with RAD51 to remove the RAD51 from the ssDNA. Alternatively, BLM may interact with RAD51 to dislodge it from the ssDNA then complete the process by binding to the ssDNA whereby liberating the RAD51 molecule. The data reported here supports this stepwise model as the interacting domains for both RAD51 and ssDNA are overlapping, however further studies are clearly required to identify a mechanism of activity.
The results herein also support the recent model proposed by Yodh et al. [26] that hypothesized a DNA binding domain within BLM that is independent of the helicase catalytic core domain. The study by Yodh and colleagues suggests that BLM unwinds then reanneals the DNA in a repetitive manner. BLM rapidly unwinds a number of duplexed base pairs then switches strands and translocates along the ssDNA to reanneal the same sequence. In this model, BLM642–1290 interacts with dsDNA at the junction between single-stranded and duplexed DNA. When the maximum length of DNA has been unwound, BLM642–1290 releases the duplex but remains attached to the ssDNA via alternate BLM domains. This study reinforces the Yodh model of BLM activity in that BLM can interact with ssDNA between amino acids 100–125 and 1,317–1,367 independent of the helicase catalytic activity. The ssDNA binding domains reported herein overlap the single-strand annealing domains of BLM. Cheok et al. reported the initial single-strand annealing function of BLM [6] between amino acids 1,290 and 1,350, while Chen et al. recently reported a DNA strand annealing activity in the N-terminus of BLM (amino acids 1–294) [5]. Given that the ssDNA annealing domains reported above align with the ssDNA binding domains reported herein, it is likely that the two functions are connected. However, this remains to be determined.
BLM is also implicated in the DNA damage-signaling pathway. In response to DNA damage during S-phase, BLM forms foci with γ-H2AX then together recruit BRCA1 and NBN to sites of DNA damage [7]. BLM deficient cells exhibit a delayed assembly of DNA repair complexes, specifically complexes involving BRCA1 and NBN [7]. Furthermore, BLM is required for the optimal activation of ATM in response to replication damage and recruits 53BP1, the p53 binding protein, to sites of damage independent of its helicase activity [8]. Taken together these data suggest that BLM is an early responder to DNA damage formed during replication and that the DNA helicase activity of BLM may be independent of the DNA signaling regions.
To clarify the function of the BLM termini with respect to RAD51 interaction, we have generated and expressed overlapping deletion fragments of the N- and C-termini of BLM. Each protein fragment was purified and assessed for its ability to bind either RAD51 or ssDNA. Our results show that the domains responsible for RAD51 partnership coincide with the domains responsible for the binding of ssDNA, BLM100–214 and BLM1317–1367. This suggests that the ability of BLM to suppress HR may involve both the displacement of RAD51 from the D-loop HR intermediate and the binding of BLM to the newly liberated ssDNA to reinstate the duplex and completely impede genetic exchange through homologous recombination.
Acknowledgments
The authors thank Drs. Susan A. Gerbi and Robert W. Sobol, for their helpful discussions and critical reading of this manuscript. We also thank Dr. Nathan Ellis for his kind gift of BLM cDNA. This work was supported by the RI-INBRE Grant #P20 RR16457 from National Center for Research Resources/National Institutes of Health to KHA.
Abbreviations
- HR
Homologous recombination
- DSB
DNA double-strand breaks
- SDSA
Synthesis-dependent strand annealing
- BIR
Break induced replication
- DHJ
Double holliday junction
- BS
Bloom syndrome
- SCE
Sister chromatid exchange
References
- 1.Agarwal S, Tafel AA, Kanaar R. DNA double-strand break repair and chromosome translocations. DNA Repair (Amst) 2006;5:1075–1081. doi: 10.1016/j.dnarep.2006.05.029. [DOI] [PubMed] [Google Scholar]
- 2.Bachrati CZ, Hickson ID. RecQ helicases: suppressors of tumorigenesis and premature aging. Biochem J. 2003;374:577–606. doi: 10.1042/BJ20030491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Branzei D, Foiani M. RecQ helicases queuing with Srs2 to disrupt Rad51 filaments and suppress recombination. Genes Dev. 2007;21:3019–3026. doi: 10.1101/gad.1624707. [DOI] [PubMed] [Google Scholar]
- 4.Bugreev DV, Yu X, Egelman EH, Mazin AV. Novel pro- and anti-recombination activities of the Bloom’s syndrome helicase. Genes Dev. 2007;21:3085–3094. doi: 10.1101/gad.1609007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen CF, Brill SJ. An essential DNA strand-exchange activity is conserved in the divergent N-termini of BLM orthologs. EMBO J. 2010;29:1713–1725. doi: 10.1038/emboj.2010.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheok CF, Bachrati CZ, Chan KL, Ralf C, Wu L, Hickson ID. Roles of the Bloom’s syndrome helicase in the maintenance of genome stability. Biochem Soc Trans. 2005;33:1456–1459. doi: 10.1042/BST0331456. [DOI] [PubMed] [Google Scholar]
- 7.Davalos AR, Campisi J. Bloom syndrome cells undergo p53-dependent apoptosis and delayed assembly of BRCA1 and NBS1 repair complexes at stalled replication forks. J Cell Biol. 2003;162:1197–1209. doi: 10.1083/jcb.200304016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davalos AR, Kaminker P, Hansen RK, Campisi J. ATR and ATM-dependent movement of BLM helicase during replication stress ensures optimal ATM activation and 53BP1 focus formation. Cell Cycle. 2004;3:1579–1586. doi: 10.4161/cc.3.12.1286. [DOI] [PubMed] [Google Scholar]
- 9.Helleday T, Lo J, van Gent DC, Engelward BP. DNA double-strand break repair: from mechanistic understanding to cancer treatment. DNA Repair (Amst) 2007;6:923–935. doi: 10.1016/j.dnarep.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 10.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 11.Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361:1475–1485. doi: 10.1056/NEJMra0804615. [DOI] [PubMed] [Google Scholar]
- 12.Langland G, Kordich J, Creaney J, Goss KH, Lillard-Wetherell K, Bebenek K, Kunkel TA, Groden J. The Bloom’s syndrome protein (BLM) interacts with MLH1 but is not required for DNA mismatch repair. J Biol Chem. 2001;276:30031–30035. doi: 10.1074/jbc.M009664200. [DOI] [PubMed] [Google Scholar]
- 13.Li X, Heyer WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008;18:99–113. doi: 10.1038/cr.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mankouri HW, Hickson ID. The RecQ helicase-topoisomerase III-Rmi1 complex: a DNA structure-specific ‘dissolvasome’? Trends Biochem Sci. 2007;32:538–546. doi: 10.1016/j.tibs.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 15.Mao FJ, Sidorova JM, Lauper JM, Emond MJ, Monnat RJ. The human WRN and BLM RecQ helicases differentially regulate cell proliferation and survival after chemotherapeutic DNA damage. Cancer Res. 2010;70:6548–6555. doi: 10.1158/0008-5472.CAN-10-0475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakayama H. RecQ family helicases: roles as tumor suppressor proteins. Oncogene. 2002;21:9008–9021. doi: 10.1038/sj.onc.1205959. [DOI] [PubMed] [Google Scholar]
- 17.Pedrazzi G, Perrera C, Blaser H, Kuster P, Marra G, Davies SL, Ryu GH, Freire R, Hiskson ID, Jiricny J, Stagljar I. Direct association of Bloom’s syndrome gene product with the human mismatch repair protein MLH1. Nucleic Acids Res. 2001;29:4378–4386. doi: 10.1093/nar/29.21.4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharma S, Sommers JA, Wu L, Bohr VA, Hickson ID, Brosh RM., Jr Stimulation of flap endonuclease-1 by the Bloom’s syndrome protein. J Biol Chem. 2004;279:9847–9856. doi: 10.1074/jbc.M309898200. [DOI] [PubMed] [Google Scholar]
- 19.Sung P, Klein H. Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol. 2006;7:739–750. doi: 10.1038/nrm2008. [DOI] [PubMed] [Google Scholar]
- 20.Sung P, Krejci L, Van Komen S, Sehorn MG. Rad51 recombinase and recombination mediators. J Biol Chem. 2003;278:42729–42732. doi: 10.1074/jbc.R300027200. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000;14:927–939. [PMC free article] [PubMed] [Google Scholar]
- 22.West SC. Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol. 2003;4:435–445. doi: 10.1038/nrm1127. [DOI] [PubMed] [Google Scholar]
- 23.Whitby MC. Making crossovers during meiosis. Biochem Soc Trans. 2005;33:1451–1455. doi: 10.1042/BST0331451. [DOI] [PubMed] [Google Scholar]
- 24.Wu L, Davies SL, Levitt NC, Hickson ID. Potential role for the BLM helicase in recombinational repair via a conserved interaction with RAD51. J Biol Chem. 2001;276:19375–19381. doi: 10.1074/jbc.M009471200. [DOI] [PubMed] [Google Scholar]
- 25.Wu L, Davies SL, North PS, Goulaouic H, Riou JF, Turley H, Gatter KC, Hickson ID. The Bloom’s syndrome gene product interacts with topoisomerase III. J Biol Chem. 2000;275:9636–9644. doi: 10.1074/jbc.275.13.9636. [DOI] [PubMed] [Google Scholar]
- 26.Yodh JG, Stevens BC, Kanagaraj R, Janscak P, Ha T. BLM helicase measures DNA unwound before switching strands and hRPA promotes unwinding reinitiation. EMBO J. 2009;28:405–416. doi: 10.1038/emboj.2008.298. [DOI] [PMC free article] [PubMed] [Google Scholar]