

Abstract
Metabolic adaptation to stress is a crucial yet poorly understood phenomenon, particularly in the central nervous system (CNS). The ability to identify essential metabolic events which predict neuronal fate in response to injury is critical to developing predictive markers of outcome, for interpreting CNS spectroscopic imaging, and for providing a richer understanding of the relevance of clinical indices of stress which are routinely collected. In this work, real-time multianalyte microphysiometry was used to dynamically assess multiple markers of aerobic and anaerobic respiration through simultaneous electrochemical measurement of extracellular glucose, lactate, oxygen, and acid. Pure neuronal cultures and mixed cultures of neurons and glia were compared following a 90 min exposure to aglycemia. This stress was cytotoxic to neurons yet resulted in no appreciable increase in cell death in age-matched mixed cultures. The metabolic profile of the cultures was similar in that aglycemia resulted in decreases in extracellular acidification and lactate release in both pure neurons and mixed cultures. However, oxygen consumption was only diminished in the neuron enriched cultures. The differences became more pronounced when cells were returned to glucose-containing media upon which extracellular acidification and oxygen consumption never returned to baseline in cells fated to die. Taken together, these data suggest that lactate release is not predictive of neuronal survival. Moreover, they reveal a previously unappreciated relationship of astrocytes in maintaining oxygen uptake and a correlation between metabolic recovery of neurons and extracellular acidification.
Keywords: Glycemic stress, metabolism, multianalyte microphysiometry, acidosis, real-time, stroke, electrochemical
The brain is a highly aerobic organ consuming 20% of all ATP generated by the body.1−3 This supply is threatened during events such as stroke, diabetic neuropathy, and myocardial infarction, when glucose becomes limiting, depriving cells of vital nutrients. In the mature central nervous system (CNS), neurons are the most vulnerable population of cells to nutrient deprivation and, in concert with glia, they can evoke powerful protective programs in response to such environments.4
Induction of metabolic dysfunction in the CNS has been shown to be a powerful inducer of preconditioning (PC), a cytoprotective program which is evoked when exposure to mild stress activates cellular defensive programs. PC is highly conserved across a number of tissues both in the CNS and periphery and leads to a limited period of increased resistance to subsequent stresses, which is mediated by chaperones, reactive oxygen species, alterations in mitochondrial biochemistry, and other molecules.5−7 At the molecular level, the CNS responds to ATP depletion by rapidly altering metabolism, employing nonpreferred substrates such as glycogen and lactate to generate ATP, and by evoking the transcription of genes associated with mitochondrial biosynthesis, oxygen and glucose uptake, and metabolism.7−9 One of the earliest consequences of limiting glucose is that cells rely more heavily on glycolysis, where the anaerobic biosynthesis of lactate results in the extrusion of lactate and protons. This process activates proton sensitive ion channels which impacts both neuronal membrane potential and cell signaling, both of which have been linked to PC protection.10
In spite of a wealth of information about the molecular underpinnings of PC protection,11 we have yet to identify the central metabolic mediators of the neuronal stress response. These data are essential to being able to capitalize on metabolic adaptation to potentially provide neuroprotection in the clinical settings where glucose is limiting such as stroke, diabetic stress, or acute CNS injury. This gap in our understanding can be largely attributed to the paucity of analytical tools capable of dynamic measurements of stress. (12)
The multianalyte microphysiometer (MAMP) provides real-time simultaneous electrochemical detection of key metabolites in a microfluidic chamber. These metabolites include extracellular glucose, oxygen, lactate, and acid. The MAMP has provided new insights into the immediate metabolic effects of protein toxins,13,14 cancer metabolism,15 and responses of murine islets to nutrient stimulation.16 A previous study from our group in which pure neuronal cultures were exposed to either mild (5 min) or lethal (90 min) oxygen and glucose deprivation (OGD) found that neurons that are capable of withstanding mild stress rapidly recover oxygen consumption and lactate generation. Indeed, there is a significant increase in oxygen consumption in preconditioned neurons and a rise in total ATP production. In addition, this paper assisted in the validation of a new model of PC protection provided by an in vitro environment that recapitulates transient ischemic attack as seen in vivo.5
Metabolic flux in vivo, however, is highly dependent upon glia, which vastly outnumber neurons in the CNS. In this work, we assesed multiple markers of biological stress including lipid and protein oxidation as well as utilized the MAMP in order to observe rapid metabolic changes in neurons and glia suffering from extended nutrient deprivation in vitro, providing the first study utilizing these real-time dynamic measures to identify essential events that mediate injury. We found a dramatic increase in neuronal lipid oxidation in mixed cultures even under conditions that were nontoxic, suggesting that increased lipid oxidation is not the most immediately sensitive indicator of cellular fate. Moreover, markers of protein oxidation, another event thought to be indicative of cell fate, did not correlate with neuronal cell death. The use of real-time microphysiometry, however, revealed that the greatest single predictor of neuronal survival was extracellular acidification, as neurons fated to die experienced far greater and more rapid acid generation than protected cells. Moreover, lactate levels, currently a key metric in the clinic, were poorly correlated with neuronal cell fate.
Results
Real-Time Analysis of Metabolic Flux of Aglycemic Neurons and Mixed Cultures Using MAMP
The MAMP allows for direct comparison of the real-time metabolic responses of primary pure neuronal cultures (98% neurons) and mixed cultures composed of 20% neurons and 80% glia exposed to 90 min glucose deprivation (GD) within the cell chamber (Figure 1) by simultaneously measuring changes in extracellular glucose, lactate, oxygen, and acid. Our previous experiments had revealed that 90 min OGD was sufficient to lead to the death of pure neurons within 24 h.5 Therefore, we hypothesized that neurons would be uniquely vulnerable to the aglycemic conditions used in this study. To test this, we compared the responses of pure neuronal cultures to mixed cultures exposed to either 60 or 90 min GD without cocurrent oxygen deprivation.
Figure 1.
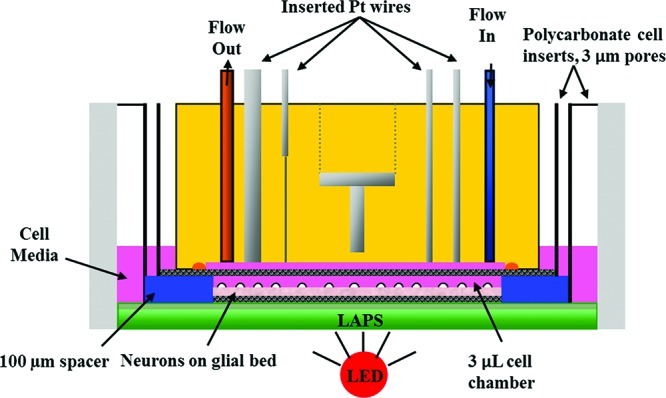
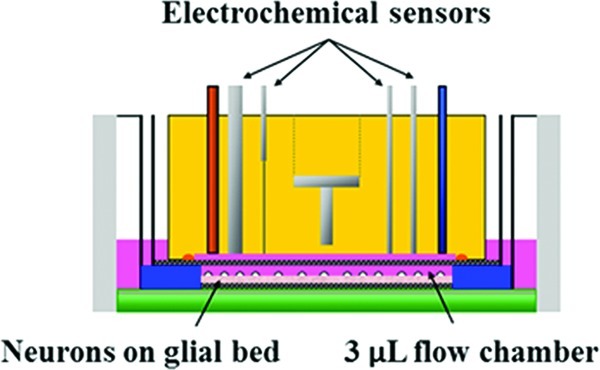
Cross-view of microphysiometry cell chamber. A polycarbonate insert containing neurons cultured as described with and without glia were placed in the sensor cup. A spacer and additional insert are placed on top of the cells, and the sensor head lowered over the assembly. The sensor head features inlet and outlet tubes to allow for 100 μL/min flow through the 3 μL chamber, as well as the Pt wires for amperometric detection of glucose, lactate, and oxygen, and a stainless steel counter electrode for the acid-sensitive LAPS electrode. A Ag/AgCl (2 M KCl) reference electrode is placed downstream of the chamber.
After a 30 min perfusion of media containing 5 mM glucose, cultures were exposed to glucose-free media for 90 min. After deprivation, 5 mM glucose was returned for 1 h. Metabolite concentrations and acidification rates of each replicate chamber were graphed as percent change from the basal rate (see Methods) to allow comparison between replicate cell chambers, which consisted of cultures from several dissections. The real-time data was then normalized to the control chambers and grouped to find average levels of each analyte during deprivation and recovery (Figure 2). The overall decrease in signal observed for all analytes is due to a combination of electrochemical phenomena and changes in neuronal metabolism that occur in response to removal from a humidified environment, exchanges of cellular media, and the introduction of flow. These phenomena cannot be separated, making the use of control chambers critical for interpretation of data collected when using this method.
Figure 2.
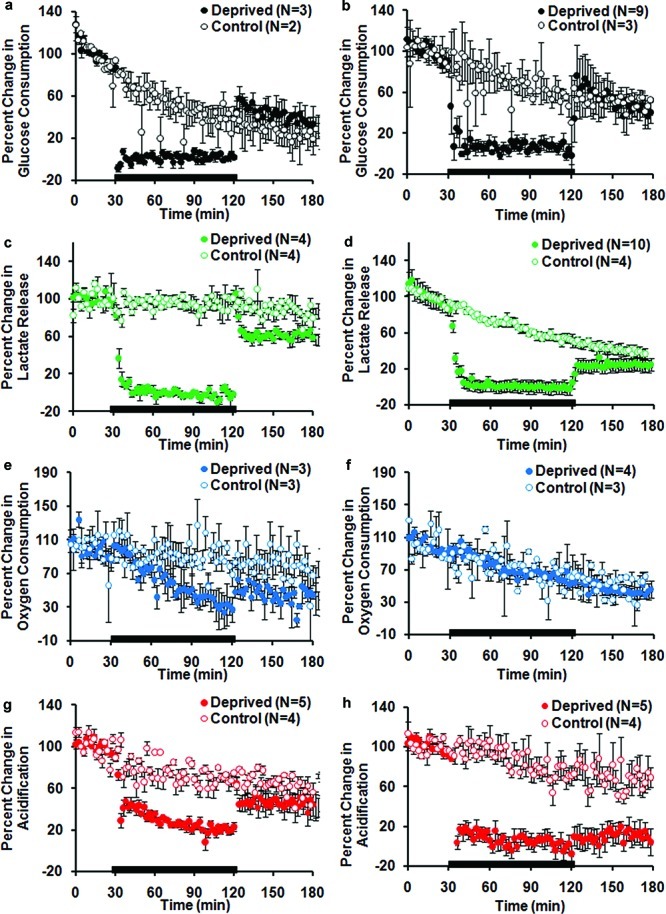
Percent change in pure neuronal and mixed culture metabolites during 90 min GD. Each data point is the average rate of consumption or production at each stop-flow period (every 2 min). Average of replicate for deprived and control chambers ± SEM. The black bar represents the GD time course. (a) Glucose consumption, neurons. (b) Glucose consumption, mixed. (c) Lactate production, neurons. (d) Lactate production, mixed. (e) Oxygen consumption, neurons. (f) Oxygen consumption, mixed. (g) Acidification, neurons. (g) Acidification, mixed.
The loss of extracellular glucose caused by altering culture media composition was detected immediately by the MAMP in both neuron enriched cultures (Figure 2a) as well as in mixed cultures (Figure 2b). Both systems avidly utilized glucose from the culture media when it was reintroduced, recovering to levels of glucose consumption similar to those observed at baseline. This data was consistent with visual assessment of the cultures which demonstrated that both pure neuronal and mixed cultures had intact processes and phase bright somas indicative of normal cellular electrochemical coupling immediately following energetic challenge.
In the absence of glucose, cells can use lactate to fuel aerobic respiration as long as oxygen is not limited. Lactate is present in both neurons and glia in a free form and can also be liberated from glycogen stores in glia. While the enzymatic conversion of lactate to pyruvate by lactate dehydrogenase runs in both directions, lactate generation has often been used as a measure of glycogen utilization in vivo and in vitro.17 The presence of lactate was detected within the cell chamber for both cell groups, with no other possible source but the cells themselves, demonstrating that anaerobic metabolism of glucose or liberation from glycogen stores must be occurring. These levels are, however, significantly lower in neurons than has been observed in immortalized cell lines.13,14
In both pure neurons (Figure 2c) and mixed cultures (Figure 2d), lactate levels quickly dropped off in the absence of glucose, suggesting that lactate extrusion from anaerobic respiration and astrocytic glycogen and pyruvate is minimal under these conditions. When normalized to respective control chambers, both mixed and neuronal cultures were shown to be able to generate lactate within 1 h after the termination of the GD stress (Figure 3a). Figure 3a demonstrates a comparable increase in lactate where mixed cultures recovered lactate release to 0.62 ± 0.20 of control chambers (p < 0.01, Student’s t test, n = 10) and neuronal cultures recovered to 0.77 ± 0.06 of control chambers (p < 0.004, Student’s t test, n = 4).
Figure 3.

Metabolic recovery of pure neuronal and mixed cultures can be compared by normalizing to control chambers. (a) Lactate release, (b) oxygen consumption, and (c) acidification were measured continuously following 90 min GD using microphysiometry. Replicate chambers were grouped into two groups: 0′ denotes measurements during GD and 60′ denotes the average metabolic levels 60 min after GD. Data were normalized to control cell measurements at the corresponding time point and represent the mean from at least three independent experiments. Asterisk (*) denotes statistical significance as compared to control cells (p < 0.05). Triangle (△) denotes statistically significant difference between mixed and neuronal cultures in the specified group.
The oxygen consumption of mixed cultures was not impacted by aglycemia, whereas that of pure neuronal cultures was irreparably altered (Figure 2e and f). When compared to control chambers (Figure 3b), the oxygen consumption of pure neuronal cultures was significantly different immediately following 90 min GD where a reduction to 0.63 ± 0.10 of control chambers was observed (p < 0.01, Student’s t test, n = 3). In addition, after 1 h of recovery, oxygen consumption of neuronal cultures further decreased to 0.48 ± 0.14 of control chambers (p < 0.01, Student’s t test, n = 3).
Extracellular acid release during 90 min GD was also strikingly different in pure neuronal and mixed cultures. Acidification decreased for both neuronal (Figure 2g) and mixed cultures (Figure 2h) immediately following GD, but only in the mixed cultures did acidification drop toward the lower limit of detection of the MAMP. When normalized to control chambers, mixed cultures (Figure 3c) exhibited lower extracellular acidification, 0.09 ± 0.07 of control levels (p < 0.0001, Student’s t test, n = 5), while the acidification of neuronal cultures decreased to only 0.36 ± 0.03 of control levels (p < 0.0002, Student’s t test, n = 5). After reintroduction of glucose, mixed cultures exhibited less acidification compared to pure neuronal cultures which may be indicative of less acid generation/extrusion. Mixed culture acidification was 0.24 ± 0.18 of control levels 1 h after GD (p < 0.02, Student’s t test, n = 5). Neuronal cultures generated much more acid returning to 0.73 ± 0.18 of control levels 1 h after GD (p < 0.03, Student’s t test, n = 5). Taken together, these data support a model whereby mixed cultures have a far longer period of altered pH following aglycemia than neurons.
Validation of Neuronal Sensitivity to Glucose Deprivation
Based on our previous publications, we know that the combination of oxygen and GD is not well tolerated by pure neurons but can produce only mild changes in survival of mixed cultures.5,6 Similarly, both 60 and 90 min GD resulted in increased cell death in our neuronal cultures as evidenced by the increase in lactate dehydrogenase (LDH) release measured 24 h after GD (Figure 4a). Strikingly, neither 60 nor 90 min GD resulted in increased cell death in mixed cultures (Figure 4b) or substantially changed neuronal morphology (data not shown).
Figure 4.

Mixed cultures survive nutrient deprivation which is lethal in pure neuronal cultures. LDH release assay showing viability of (a) neurons and (b) mixed cultures 24 h after 60 min and 90 min GD. (c) OxyBlot of total protein oxidation in mixed cultures detected 24 h after various GD durations. (d) Neuroprostane levels measured at 24 h revealed significant oxidation of DHA-enriched lipids in neurons in response to GD. Data were normalized to control cell measurements at the corresponding time point and represent the mean from at least three independent experiments. Asterisk (*) denotes statistical significance as compared to control cells (p < 0.05).
The stark contrast in survival of these two different neuronal cultures coupled with the real-time, multiparameter measurements acquired during and after the insult provides us with the opportunity to correlate survival with other biochemical features associated with cell stress which are also used as surrogates of injury, including lipid and protein oxidation, as well as compare their predictive validity to the metabolic changes.
Oxidative modification of proteins and lipids has been used as a common indicator of cell injury following stress and has been used in some settings to assess cell injury. We have previously demonstrated that formation of carbonyl groups on proteins and nonenzymatic oxidation of docosahexaenoic acid (DHA) enriched in neuronal membranes are increased in post mortem tissue from individuals who had suffered from stroke or transient ischemic attacks as well as in mixed culture models of OGD.18,19 OxyBlot analysis performed 24 h after GD in mixed cultures revealed that levels of protein oxidation were similar between control and 60 min GD cells (Figure 4c), whereas longer periods of GD (90 min) resulted in less protein oxidation due to either fewer proteins being synthesized/oxidized or a more robust removal of oxidized proteins via autophagy and/or proteasomal degradation.
Injury to lipids caused by reactive oxygen species can be assessed by the detection of DHA enriched lipid peroxidation which results in the formation of F4-Neuroprostane (NeuroP). This method allows us to specifically assess neuronal lipid stress. NeuroP formation was significantly increased in response to GD.5,6,19 NeuroP formation was both time and stress dependent with longer periods of GD resulting in higher oxidation of DHA although the levels were slightly less than half of those observed following 90 min exposure to NMDA, which induces 100% neuronal cell death by 24 h (Figure 4d). Taken together, these data suggest that protein oxidation measured 24 h after stress does not correlate with the length of GD, whereas NeuroP formation does. Moreover, these data demonstrate that GD causes the generation of appreciable amounts of radicals in neurons but this oxidative stress is indeed survivable if glial support is present.
Real-Time Analysis of Metabolic Flux of Mildly Aglycemic Mixed Cultures Using MAMP
Given the physiological relevance of our mixed culture population and their ability to induce metabolic and molecular adaptation following 90 min aglycemia, we were eager to determine if shorter periods of aglycemia could be used to reveal differences in metabolic recovery as GD becomes more protracted using the MAMP. Figure 5 compares the real-time percent change in metabolic consumption of all four analytes in mixed cultures in response to 60 min GD. The real-time data was normalized to control chambers and grouped to find average levels of the analytes at different time points during the deprivation and recovery (Figure 6). The shift in glucose consumption observed following 60 min GD (Figure 5a) appears to be due to a change in baseline current at the electrode as glucose is removed and then returned to the sensing chamber (raw data not shown). It is possible that an increase in glucose consumption occurred as a means to increase energetic status following aglycemia, but this signal could not be separated from the changes in current measured at the glucose electrodes.
Figure 5.
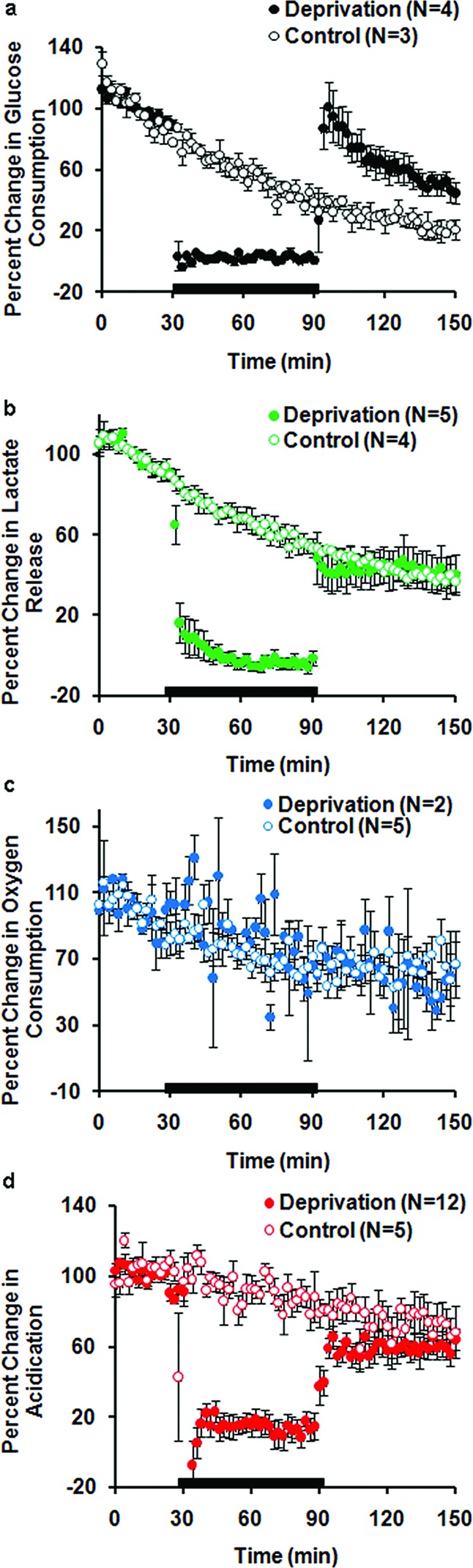
Percent change in mixed cultures metabolites during 60 min GD. Each data point is the average rate of consumption or production at each stop-flow period (every 2 min). Average of replicates for deprived and control chambers ± SEM. The black bar represents the GD duration. (a) Glucose consumption, 60 min GD. (b) Lactate production, 60 min GD. (c) Oxygen consumption, 60 min GD. (d) Acidification, 60 min GD.
Figure 6.

Normalizing to control chambers reveals extracellular acid is reduced during recovery of mixed cultures from mild GD. (a) Lactate release, (b) oxygen consumption, and (c) acidification were measured using microphysiometry. Replicate chambers were grouped into two groups: 0′ denotes measurements during GD, and 60′ denotes the average metabolic levels 60 min after GD. Asterisk (*) denotes statistical significance as compared to control cells (p < 0.05).
Based on our previous data, we speculated that the most robust and reproducible indices of injury would be extracellular acidification and suspected that there would be a stronger parallel between lactate and extracelluar acidification given that lactate extrusion during anaerobic respiration requires proton expulsion. Notably, while lactate levels in mixed cultures receiving mild deprivation returned to control levels, these cultures did not fully recover extracellular acidification following 60 min GD (Figure 5d), reaching 0.81 ± 0.13 of control (p < 0.002, Student’s t test, n = 12) (Figure 6c). These data would suggest either that the glycogen pools of astrocytes can be maximally driven by 60 min GD or that anaerobic respiration is playing a greater role in mixed cultures with mild GD. As previously discussed, recovery of acid levels after 90 min GD was also incomplete (Figure 3c), reaching only 0.24 ± 0.18 of control levels (p < 0.01, Student’s t test, n = 5).
We also noted that oxygen consumption was not impaired in response to 60 min GD, suggesting that cells were continuing to uptake oxygen with both periods of aglycemia (Figures 2f and 3b) and the notable lack of increased oxygen consumption would suggest that aerobic respiration is not upregulated in response to GD.
Discussion
Diabetes in the United States has reached epidemic levels and patients with diabetes have a higher incidence of stroke and worse outcome following stroke. We remain, however, severely limited in identifying accurate measures of cellular stress in the period after acute injuries that could predict the fate of injured cells in the clinical settings and in preclinical trials. Mild metabolic dysfunction can evoke short-term neuroprotection but thus far we have been unable to identify a metabolic profile which predicts the fate of cells exposed to GD. We have made great strides in developing technologies for in vivo measurement of oxygen saturation, acidification, glucose consumption, and lactate generation by magnetic resonance spectroscopy (MRS) which have informed our understanding of regional specification of activity in the brain and how metabolism is impacted by injury. These measurements, are, however, complicated by the speed with which these biochemical events must be captured as well as complications arising from the unique cellular and neurochemical composition of different parts of the CNS. The MAMP provides an invaluable opportunity to bridge the in vivo and in vitro data on metabolism and dissect cellular interactions in a systematic way to develop a stronger sense of the outcomes and effectiveness of potential neuroprotective therapies.
By coupling our novel MAMP method with measures of cytoplasmic rupture, we found that lactate release drops off precipitously when glucose is limiting, but quickly recovers in neurons that ultimately survive the stress as well as those that are fated to die, thus demonstrating that lactate release is not predictive of cell survival. This is in stark contrast to early clinical practices in which lactate levels were monitored as a metric of edema, or brain swelling, which is correlated with poor outcomes.20 As new proteins associated with cell lysis and blood-brain barrier disruption were identified, markers such as sperimidine, S-100 protein, and adenylyl cyclase were all studied as surrogates of injury with varying degrees of success. Given the powerful imaging technologies which now exist enabling in vivo analysis of lactate, glucose, oxygenation, and acidification, there is a pressing need to understand the fundamental aspects of metabolism as they relate to the complex interplay of cells within the CNS.21
The continuous measurement of oxygen consumption and extracellular acid levels proved to be superior predictors of neuronal survival in comparison to lactate accumulation following prolonged energetic stress. Oxygen consumption appears to be an immediate predictor of neuronal survival, as neurons fated to die within 24 h exhibited an immediate decrease in oxygen consumption at the onset of the 90 min GD. While both neurons and glia rely on glucose to fuel the Kreb's cycle and feed oxidative phosphorylation, mixed, but not pure, neuronal cultures continue to consume oxygen, which may be correlated with a more robust ability to convert lactate to pyruvate and recapture this energetic substrate as this process is oxygen-dependent.
Additionally, we observed that extracellular acid levels are significantly lower in mixed cultures after GD, which, unlike pure neurons, survived the 90 min GD. This highlights our key finding that extracellular acid levels are the best metabolic index of neuronal survival in response to stress. We would predict that guided technologies to assess acidification in areas of infarction following stroke or trauma would be more valuable than current tools which measure CNS and plasma lactate levels. It is also likely that metabolic adaptation can occur in glial cells in order to reduce extracellular acid during and after stress, thereby contributing to biologically significant events that impact neuronal injury such as activation of proton sensitive, acid-sensing ion channels (ASICs). These proteins alter neuronal membrane potential and cell signaling, allowing for intake of extracellular Ca2+ and Na+, and in turn leading to neuronal excitotoxicity and cell death.10,22,23 Further investigation of neurons using ASIC inhibitors in the MAMP combined with traditional measures of cellular stress could further illustrate the importance of reduced acid levels in mixed cultures. From a biological standpoint, the observation that acid levels were significantly lower in mixed cultures receiving a mild, 60 min, energetic challenge (whereas no other metabolites were permanently altered) has two clear implications: (1) that acidification occurs incredibly rapidly in the absence of glia when neurons are returned to normal glycemic conditions, and (2) that the active removal of acid by glia is essential for neuronal survival.
From this work, it is clear that real-time measurement of metabolites during and following insults has important implications for the study of neuroprotection. Additionally, the design of the MAMP may be easily modified to allow for sensing of other analytes in the extracellular space. Future work could add sensors for Ca2+ and K+ flux, which would enable correlation of metabolic and ionic flux during times of stress. The MAMP is essential to further our understanding of the involvement of metabolism in neuronal stress and survival, thus improving knowledge of neuroprotection under conditions of energetic stress.
Methods
User-friendly versions of standard toxicological protocols and procedures can be found on our Web site at http://www.mc.vanderbilt.edu/root/vumc.php?site=mclaughlinlab&doc=17838.
Materials and Reagents
LDH Toxicology Assay Kit (Tox7), potassium cyanide, glucose oxidase (GOx, Type IIS from Aspergillus niger), bovine serum albumin (BSA, fraction V, 96%), and glutaraldehyde (25 wt % solution in water) were purchased from Sigma (St. Louis, MO). OxyBlot Protein Oxidation Detection Kit was provided by Millipore (Temecula, CA). Stabilized lactate oxidase (LOx) was purchased from Applied Enzyme Technology (Pontypool, U.K.). Nafion (perfluorosulfonic acid-PTFE copolymer, 5% w/w solution in ethanol) and platinum wire were purchased from Alfa Aesar (Ward Hill, MA). Sterile 20% glucose solution was purchased from Teknova (Hollister, CA). Sterile (l)-lactic acid was purchased from Fisher Scientific (Pittsburgh, PA). Lyophilized alamethicin was obtained from A.G. Scientific, Inc. (San Diego, CA) and reconstituted with 1 mL absolute ethanol. All media and media supplements were from Invitrogen (Carlsbad, CA) except for the microphysiometry experiments which used custom RPMI (1 mM phosphate buffer, glucose and bicarbonate-free) from Mediatech, Inc. (Manassas, VA). Transwell polyester permeable supports with 3 μm pores were purchased from Corning Life Sciences (Lowell, MA). All MAMP consumables were obtained from Molecular Devices Corp. (Sunnyvale, CA).
Cell Culture
Near pure neuronal (98% neurons) cortical cultures were prepared from embryonic day 18 Sprague–Dawley rats as previously described.18 Briefly, cortices were digested in trypsin and dissociated. The resultant cell suspension was adjusted to 335 000 cells/mL, and cells were plated in 6-well tissue culture plates containing poly-l-ornithine-treated coverslips in growth media (80% Dulbecco’s modified Eagle's medium (DMEM), 10% Ham’s F12-nutrients, 10% fetal bovine serum (Hyclone) with 24 U/mL penicillin, 24 μg/mL streptomycin, and 2 mM l-glutamine). On day in vitro (DIV) 2, cytosine arabinoside (1 μM) was added to inhibit glial cell proliferation, after which cells were maintained in Neurobasal media (Invitrogen) containing 2× N2 supplement, 50× B27 supplement, 50× NS21 supplement,24 penicillin, and streptomycin. All experiments were conducted between DIV21 and DIV25, when excitotoxicity is fully expressed.
Mixed cultures (80% glial/20% cortical neuron cultures) were prepared as described above for pure neuronal cultures with minor changes. Cells were maintained in growth media without inhibition of glial cells until DIV13, at which point cytosine arabinoside was added. After glial inhibition, cells were maintained in D2C media (94% DMEM, l-glutamine, 2% FBS, 0.025 M HEPES, 0.0125 mM l-glutamine, 24 U/mL penicillin, 24 μg/mL streptomycin) for up to 1 month (DIV25–29). Alternatively, both cell types were grown as stated but at twice the density and on polyester cell inserts used for microphysiometry.
Measurement of F4-Neuroprostanes
Neuronal lipid peroxidation was assessed through quantification of F4-neuroprostanes (F4-NeuroPs), F2-IsoP-like compounds generated from the free radical-mediated peroxidation of docosahexaenoic acid,25−28 a polyunsaturated fatty acid enriched in neurons 10-fold over astrocytes.29 F4-NeuroPs were measured using gas chromatography–mass spectrometry as previously described.30 Briefly, cells were harvested and 500 μL of the lysate mixed with methanol containing 0.05% butylated hydroxytoluene (BHT) to prevent auto-oxidation. The remaining lysate was saved for protein assay and normalized for protein concentrations. F4-NeuroPs esterified to phospholipids were hydrolyzed by chemical saponification, after which total F4-NeuroPs were extracted using C-18 and silica Sep-Pak cartridges, purified by thin-layer chromatography, converted to pentafluorobenzyl ester trimethylsilyl ether derivatives, and quantified by stable isotope dilution techniques using gas chromatography/negative ion chemical ionization mass spectrometry using [2H4]-8-iso-PGF2α (m/z 573) as an internal standard. F4-NeuroPs are detected at m/z 593.
Detection of Oxidized Proteins
Twenty-four hours following either 60 or 90 min GD exposure, cells were harvested into 200 μL of TNEB (50 mM Tris, 2 mM EDTA, 150 mM NaCl, 8 mM β-glycerophosphate, 100 μM orthovanadate, 1% Triton X-100 (1%), 1:1000 protease inhibitor) and total oxidized proteins were determined using the OxyBlot Protein Oxidation Detection Kit. Following the cell harvest, 100 μL of the lysate was used for protein assay and the other 100 μL was immediately treated with 50 mM DTT to prevent protein oxidation. The DTT treated lysate was split into two separate 50 μL aliquots, one for the derivatization reaction containing 2,4-dinitrophenylhydrazine and the other for the negative control containing derivatization control solution. Samples were stored at 4 °C for no longer than 7 days after derivatization. Equal protein concentrations were analyzed by Western blot using antibodies specific for the detection of oxidized proteins provided by the manufacturer. Data represent results from at least three independent experiments. Statistical significance was determined by two-tailed paired t test with p < 0.05.
LDH Toxicity Assays
Twenty four hours following each evaluated insult, 40 μL of cell media was removed from each well and used to assess cell viability using a lactate dehydrogenase (LDH)-based in vitro toxicity kit as previously described.31 In order to account for variation in total LDH content, raw LDH values were normalized to the toxicity caused by 100 μM NMDA plus 10 μM glycine, which is known to cause 100% cell death in this system.6 All experiments were performed with a minimum “n” of 3 using cells derived from at least three independent primary dissections.
Analysis and Statistics
Except where otherwise noted, data were summarized and are represented as mean ±
SEM. The statistical significance of differences between means was assessed using one-way ANOVA at the 95% confidence level (p < 0.05), followed by Tukey multiple-comparison tests using GraphPad Prism software.
Neuronal and Mixed Culture Glucose Deprivation
Mature neuronal cultures were perfused with 5 mM glucose RPMI in the microphysiometer for 90 min. Following the initial 90 min perfusion, RPMI containing no glucose was perfused for 90 min, after which the 5 mM glucose RPMI was returned and perfused for an additional 60 min. Control experiments in which no GD occurred were performed simultaneously.
Mixed cultures were perfused with 5 mM glucose RPMI in the microphysiometer for 90 min. Following the initial 90 min perfusion, the media was replaced with RPMI containing no glucose and perfused for either 60 or 90 min, after which the 5 mM glucose RPMI was returned to the cells and perfused for an additional 60 min. Control experiments in which no GD occurred were performed simultaneously.
Instrumentation
A modified sensor head was prepared by adding four platinum wires to the Cytosensor sensor head designed by Molecular Devices. Four 0.6 mm paths were drilled through the sensor head with the hole for the counter electrode widened on the surface to ∼2 mm. The counter electrode was formed by melting a 0.5 mm platinum electrode to form a 1.5 mm ball at the end. Two 0.5 mm platinum wires were inserted for glucose and lactate measurements. A 127 μm platinum wire was wrapped multiple times around a 0.5 mm platinum wire for added mechanical stability, and silver epoxy was used to aid the electrical contact. Each wire was embedded in the sensor head with white epoxy, ground down flush to the face of the sensor head using silicon carbide grinding paper, and polished with 3 μm diamond polish. The wires extending from the sensor head were reinforced with copper socket pins to add mechanical strength and reinforced with white epoxy. The resulting sensor contains two 0.5 mm diameter disk electrodes for glucose and lactate detection, one 127 μm diameter disk electrode for oxygen detection, and a 1.5 mm diameter disk electrode as the counter electrode. The surface of the sensor head was cleaned by scrubbing with water and ethanol, or by polishing with 3 μm diamond polish from Buehler (Lake Bluff, IL).
A four channel microphysiometer and Cytosoft program (Molecular Devices) were used to control pump cycles and hold the temperature at 37 °C. A 120 s stop-flow cycle was used with 80 s at a flow rate of 100 μL/min and a stop period of 40 s. Constant flow replenished glucose and oxygen consumed by the neurons within the chamber, as well as washed away byproducts released from the neurons, such as lactate and acid. Periodically stopping the flow allows for the measurable consumption and accumulation of metabolites, increasing sensitivity and enabling detection of small changes in metabolism.
Glucose and lactate-sensing electrode films were prepared similarly to those previously described.5,32,33 Briefly, 800 μL of phosphate buffer (50 mM PB, pH 7.00) was added to 50 mg of bovine serum albumin (BSA). For glucose-sensing films, 2 mg of GOx was dissolved in 300 μL of a BSA-buffer solution then vortexed with 3 μL of 25% glutaraldehyde for 5 s. For lactate-sensing films, 1.8 mg of LOx was dissolved in 100 μL of a BSA-buffer solution and then vortexed with 0.8 μL of 25% glutaraldehyde for 5 s. Electrode films were cast by allowing a droplet of the enzyme solution to dry on the platinum electrode surface of the sensor head. New solutions were prepared for each experiment. A droplet of the 5% Nafion solution was also applied to the oxygen electrode to reduce biofouling as shown in the literature.15,32,34 Although heat-curing of the Nafion film at 200 °C is typical for electrochemical sensors for direct detection of dopamine and other electroactive compounds at a carbon fiber electrode, it is not suitable for enzymatic electrodes, and thus, the common practice is to allow the films to dry in ambient air.35,36
Glucose, lactate, and oxygen measurements were performed with a multichamber bipotentiostat built by the Vanderbilt Institute for Integrative Biosystems Research and Education (VIIBRE). Data was collected using LabVIEW software, which when coupled with the potentiostat enabled monitoring of multiple analytes in four chambers simultaneously. The glucose and lactate sensing electrodes were held at a potential of +0.6 V to oxidize H2O2 produced within enzyme films, while the oxygen electrode was held at −0.45 V to reduce dissolved oxygen. All potentials were set versus the Cytosensor Ag/AgCl (2 M KCl) reference electrode in the effluent stream.
Acidification, or the decrease in solution pH due to the release of cellular byproduct, was measured using the light-addressable potentiometric sensor (LAPS), the original acid-sensing component of the Cytosensor microphysiometer.
Microphysiometry Analysis
Cytosensor spacers and inserts were placed in prepared cell inserts containing pure neuronal or mixed cultures. This assembly was then placed in the sensing cup and modified sensor heads were secured in the chamber to create the 3 μL cell chamber. Modified RPMI (5 mM glucose, 1 mM PO43-) media was perfused through the chamber at 100 μL/min using Cytosoft to maintain a pump-on/pump-off cycle (80 s pump-on, 40 s pump-off), allowing for measurable consumption of oxygen and glucose as well as accumulation of lactate and acidic byproducts. When replacements were available, cell inserts and sensor heads were exchanged after preliminary measurements if cell or electrode performance was not optimal. Additionally, primary cell lines were only used if the extracellular acidification rate was found to be between 20 and 100 μV/s. This selection process is one way that biological variation is controlled for in the MAMP.
Glucose, lactate, oxygen, and acid signals were sampled by the potentiostat and Cytosensor once per second for the entirety of the experiment. At the completion of each experiment, the neurons were perfused with 15 μM alamethicin in RPMI, which forms pores in the cellular membrane and causes cellular death. This allows determination of sensor response during zero metabolic activity and calibration of the glucose and lactate sensors.
In all experiments, the sensors were calibrated with modified RPMI media with no glucose and no lactate, with 0.025 mM lactate and 1 mM glucose, with 0.05 mM lactate and 2 mM glucose, with 0.1 mM lactate and 3 mM glucose, and with 0.2 mM lactate and 5 mM glucose.
After signal collection, raw amperometric signals were filtered using a program with a Fourier transform low pass filter written in LabVIEW to remove the peristaltic pump noise present in the baseline. Successful removal of the pump noise reduces the noise in the baseline current by a factor of 4. As this value is required in the calculation of metabolic rates from raw signals, filtration is essential for removal of instrumental pump noise.
Amperometric signals were analyzed by comparing the stop-flow peaks for live and dead cells. The current response of a given stop flow period, ip, is defined as the difference between the baseline current and the current at the end of the stop flow. The current response due to cellular activity, Δip, is obtained by subtracting the response during zero metabolic activity from the response with live cells for each stop flow period. It should be noted that the apparent negative values observed in the glucose and lactate signals during glucose deprivation in Figures 2 and 5 are an artifact of subtracting the sensor response of zero metabolic activity from sensor response when levels of glucose and lactate are at null levels within the chamber.
Molar glucose consumption and lactate release per stop-flow period were calculated by comparing Δip to calibrations at the end of each experiment. Molar oxygen was calculated by assuming the oxygen baseline of dead cells to be the concentration of dissolved oxygen, 0.24 mM, and was calculated as described previously.37 Acidification rates at the LAPS were automatically calculated by Cytosoft as the slope of the change in potential during each stop flow, as previously described.38 While all four analytes were measured in each chamber for each experiment, successful and physiologically relevant data was not always collected at all four electrodes in the same chamber, leading to the disparity in number of replicates for each analyte. This was due to either the inability to replace a faulty sensor head at the beginning of the experiment, or the decay of the electrochemical signal throughout the experiment, yielding largely negative and nonphysiologically relevant results, requiring their removal from further consideration.
As each chamber of cells differs slightly in neuronal density and therefore metabolic activity, all signals were normalized to 100% of the average metabolic rate 30 min before GD. The data was boxcar smoothed with a 5 point moving average to further reduce noise, and replicate chambers were compared and grouped into basal, exposure, and a recovery group, each comprising 30 min (15 stop-flow cycles). To control for the decay in the electrochemical signal, as well as changes to neuronal metabolism due to housing in the microphysiometer, changes in metabolic concentrations of analytes are normalized to control chambers.
Statistical significance was determined by a two-tailed paired Student’s t test with p < 0.05. Statistical significance was tested between exposure and recovery values within the same data set with the assumption that the control chambers values were not different. Comparison between exposure groups of neurons and mixed cultures, as shown in Figure 3, was not performed if the controls were found to be statistically different.
Acknowledgments
The authors would like to thank Drs. Ginger Milne, Rachel Snider, and Jeannette Stankowski for helpful comments and suggestions. We also thank Mrs. Shellie Richards and Ms. Lauren Koenig for editorial assistance. Analysis of eicosanoids was performed at the Vanderbilt University Eicosanoid Core Laboratory.
Glossary
Abbreviations
- CNS
central nervous system
- ATP
adenosine triphosphate
- PC
preconditioning
- MAMP
multianalyte microphysiometry
- OGD
oxygen and glucose deprivation
- GD
glucose deprivation
- LHD
lactate dehydrogenase
- DHA
docosahexaenoic acid
- ASICs
acid sensing ion channels
- GOx
glucose oxidase
- LOx
lactate oxidase
- IrOx
irdium oxide
Author Contributions
J.R.M. performed the research and analyzed the data for MAMP experiments; A.M.P. and J.E.B. conducted cellular analysis; J.E.B. conducted dissection and culture; J.R.M., A.M.P., J.E.B., B.A.M., and D.E.C. helped design the experiments; J.R.M., A.M.P., B.A.M., and D.E.C. wrote the article.
This work was supported by a grant from the JB Marshall Neurovascular Therapeutics Center (BM; AMP) and U01 AI 061223 (DC), as well as the Vanderbilt Institute for Integrative Biosystems Research and Education (JRM) and the Vanderbilt Brain Institute (AMP). Statistical and graphical support was provided by P30HD15052 (Vanderbilt Kennedy Center).
The authors declare no competing financial interest.
References
- Kety S. S. (1957) The general metabolism of the brain in vivo. In Metabolism of the Nervous System (Richter , D.), pp 221–237, Pergamon Press, London. [Google Scholar]
- Kety S. S.; Schmidt C. F. (1948) The nitrous oxide method for the quantitative determination of cerebral blood flow in man: theory, procedure and normal values. J. Clin. Invest. 27, 476–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A.; Langemann D. (2009) Build-ups in the supply chain of the brain: on the neuroenergetic cause of obesity and type 2 diabetes mellitus. Front. Neuroenerg. 2, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gidday J. M. (2006) Cerebral preconditioning and ischaemic tolerance. Nat. Rev. Neurosci. 7, 437–448. [DOI] [PubMed] [Google Scholar]
- Zeiger S. L. H.; McKenzie J. R.; Stankowski J. N.; Martin J. A.; Cliffel D. E.; McLaughlin B. (2010) Neuron specific metabolic adaptations following multi-day exposures to oxygen glucose deprivation. Biochim. Biophys. Acta, Mol. Basis Dis. 1802, 1095–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin B.; Hartnett K. A.; Erhardt J. A.; Legos J. J.; White R. F.; Barone F. C.; Aizenman E. (2003) Caspase 3 activation is essential for neuroprotection in preconditioning. Proc. Natl. Acad. Sci. U.S.A. 100, 715–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Duffy A. E.; Bordelon Y. M.; McLaughlin B. (2006) Killer proteases and little strokes-how the things that do not kill you make you stronger. J. Cereb. Blood Flow Metab. 27, 655–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurr A. (2006) Lactate: the ultimate cerebral oxidative energy substrate?. J. Cereb. Blood Flow Metab. 26, 142–152. [DOI] [PubMed] [Google Scholar]
- Aguilera P.; Vazquez-Contreras E.; Gomez-Martinez C. D.; Cardenas M. E. C. (2009) Hypoxia inducible factor-1 as a therapeutic target in cerebral ischemia. Curr. Signal Transduction Ther. 4, 162–173. [Google Scholar]
- Xiong Z. G.; Chu X. P.; Simon R. P. (2006) Ca2+-Permeable Acid-sensing Ion Channels and Ischemic Brain Injury. J. Membr. Biol. 209, 59. [DOI] [PubMed] [Google Scholar]
- Barone F. C. (2004) Endogenous Brain Protection. Stroke Genomics: Methods and Reviews 105–184. [Google Scholar]
- Vazquez A. L.; Masamoto K.; Fukuda M.; Kim S.-G. (2010) Cerebral oxygen delivery and consumption during evoked neural activity. Front. Neuroenerg. 2, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eklund S.; Thompson R.; Snider R.; Carney C.; Wright D.; Wikswo J.; Cliffel D. (2009) Metabolic Discrimination of Select List Agents by Monitoring Cellular Responses in a Multianalyte Microphysiometer. Sensors 9, 2117–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider R. M.; McKenzie J. R.; Kraft L.; Kozlov E.; Wikswo J. P.; Cliffel D. E. (2010) The Effects of Cholera Toxin on Cellular Energy Metabolism. Toxins 2, 632–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eklund S. E.; Snider R. M.; Wikswo J.; Baudenbacher F.; Prokop A.; Cliffel D. E. (2006) Multianalyte microphysiometry as a tool in metabolomics and systems biology. J. Electroanal. Chem. 587, 333. [Google Scholar]
- Snider R. M.; Ciobanu M.; Rue A. E.; Cliffel D. E. (2008) A multiwalled carbon nanotube/dihydropyran composite film electrode for insulin detection in a microphysiometer chamber. Anal. Chim. Acta 609, 44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sickmann H. M.; Waagepetersen H. S.; Schousboe A.; Benie A. J.; Bouman S. D. (2012) Brain glycogen and its role in supporting glutamate and GABA homeostasis in a type 2 diabetes rat model. Neurochem. Int. 60, 267–275. [DOI] [PubMed] [Google Scholar]
- Zeiger S. L. H.; Musiek E. S.; Zanoni G.; Vidari G.; Morrow J. D.; Milne G. J.; McLaughlin B. (2009) Neurotoxic lipid peroxidation species formed by ischemic stroke increase injury. Free Radical Biol. Med. 47, 1422–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J. E.; Zeiger S. L. H.; Hettinger J. C.; Brooks J. D.; Holt B.; Morrow J. D.; Musiek E. S.; Milne G.; McLaughlin B. (2010) Essential role of the redox-sensitive kinase p66shc in determining energetic and oxidative status and cell fate in neuronal preconditioning. J. Neurosci. 30, 5242–5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busse O.; Hoffmann O. (1983) CSF lactate and CT findings in middle cerebral artery infarction. A comparative study. Stroke 14, 960–963. [DOI] [PubMed] [Google Scholar]
- Jung J. Y.; Lee H. S.; Kang D. G.; Kim N. S.; Cha M. H.; Bang O. S.; Ryu D. H.; Hwang G. S. (2011) (1)H-NMR-Based Metabolomics Study of Cerebral Infarction. Stroke 42, 1282–1288. [DOI] [PubMed] [Google Scholar]
- Simon R.; Xiong Z. (2006) Acidotoxicity in brain ischemia. Biochem. Soc. Trans. 34, 1356–1361. [DOI] [PubMed] [Google Scholar]
- Xiong Z.-G.; Chu X.-P.; Simon R. P. (2007) Acid sensing ion channels - novel therapeutic targets for ischemic brain injury. Front. Biosci. 12, 1376–1386. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Stevens B.; Chang J.; Milbrandt J.; Barres B. A.; Hell J. W. (2008) NS21: Re-defined and modified supplement B27 for neuronal cultures. J. Neurosci. Methods 171, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts L. J. 2nd; Montine T. J.; Markesbery W. R.; Tapper A. R.; Hardy P.; Chemtob S.; Dettbarn W. D.; Morrow J. D. (1998) Formation of isoprostane-like compounds (neuroprostanes) in vivo from docosahexaenoic acid. J. Biol. Chem. 273, 13605–13612. [DOI] [PubMed] [Google Scholar]
- Reich E. E.; Zackert W. E.; Brame C. J.; Chen Y.; Roberts L. J. 2nd; Hachey D. L.; Montine T. J.; Morrow J. D. (2000) Formation of novel D-ring and E-ring isoprostane-like compounds (D4/E4- neuroprostanes) in vivo from docosahexaenoic acid. Biochemistry 39, 2376–2383. [DOI] [PubMed] [Google Scholar]
- Montine T. J.; Quinn J. F.; Milatovic D.; Silbert L. C.; Dang T.; Sanchez S.; Terry E.; Roberts L. J. 2nd; Kaye J. A.; Morrow J. D. (2002) Peripheral F2-isoprostanes and F4-neuroprostanes are not increased in Alzheimer’s disease. Ann. Neurol. 52, 175–179. [DOI] [PubMed] [Google Scholar]
- Roberts L. J.; Fessel J. P.; Davies S. S. (2005) The Biochemistry of the Isoprostane, Neuroprostane, and Isofuran Pathways of Lipid Peroxidation. Brain Pathol. 15, 143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petroni A.; Salami M.; Blasevich M.; Papini N.; Galli C. (1994) Inhibition by n-3 fatty acids of arachidonic acid metabolism in a primary culture of astroglial cells. Neurochem. Res. 19, 1187–1193. [DOI] [PubMed] [Google Scholar]
- Milne G. L.; Sanchez S. C.; Musiek E. S.; Morrow J. D. (2007) Quantification of F2-isoprostanes as a biomarker of oxidative stress.(Report). Nat. Protoc. 2, 221(6). [DOI] [PubMed] [Google Scholar]
- McLaughlin B.; Pal S.; Tran M. P.; Parsons A. A.; Barone F. C.; Erhardt J. A.; Aizenman E. (2001) p38 Activation Is Required Upstream of Potassium Current Enhancement and Caspase Cleavage in Thiol Oxidant-Induced Neuronal Apoptosis. J. Neurosci. 21, 3303–3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eklund S. E.; Taylor D.; Kozlov E.; Prokop A.; Cliffel D. E. (2004) A Microphysiometer for Simultaneous Measurement of Changes in Extracellular Glucose, Lactate, Oxygen, and Acidification Rate. Anal. Chem. 76, 519–527. [DOI] [PubMed] [Google Scholar]
- Eklund S. E.; Cliffel D. E.; Kozlov E.; Prokop A.; Wikswo J.; Baudenbacher F. (2003) Modification of the Cytosensor(TM) microphysiometer to simultaneously measure extracellular acidification and oxygen consumption rates. Anal. Chim. Acta 496, 93–101. [Google Scholar]
- Jin L.; Jin P.; Ye J.; Fang Y. (1992) Determination of dissolved oxygen by catalytic reduction on Nafion-methyl viologen chemically modified electrode. Talanta 39, 145–7. [DOI] [PubMed] [Google Scholar]
- Ohara T. J.; Rajagopalan R.; Heller A. (1994) ″Wired″ Enzyme Electrodes for Amperometric Determination of Glucose or Lactate in the Presence of Interfering Substances. Anal. Chem. 66, 2451–2457. [DOI] [PubMed] [Google Scholar]
- Oldenziel W. H.; Dijkstra G.; Cremers T. I. F. H.; Westerink B. H. C. (2006) Evaluation of Hydrogel-Coated Glutamate Microsensors. Anal. Chem. 78, 3366–3378. [DOI] [PubMed] [Google Scholar]
- Casciari J. J.; Sotirchos S. V.; Sutherland R. M. (1992) Variations in tumor cell growth rates and metabolism with oxygen concentration, glucose concentration, and extracellular pH. J. Cell. Physiol. 151, 386–394. [DOI] [PubMed] [Google Scholar]
- Owicki J. C.; Bousse L. J.; Hafeman D. G.; Kirk G. L.; Olson J. D.; Wada H. G.; Parce J. W. (1994) The light-addressable potentiometric sensor: principles and biological applications. Annu. Rev. Biophys. Biomol. Struct. 23, 87–113. [DOI] [PubMed] [Google Scholar]