

Abstract
ΔFosB protein accumulates in the striatum in response to chronic administration of drugs of abuse, L-DOPA, or stress, triggering long lasting neural and behavioral changes that underlie aspects of drug addiction, abnormal involuntary movements (dyskinesia), and depression. ΔFosB binds AP-1 DNA consensus sequences found in promoters of many genes and can both repress or activate gene transcription. In the striatum, ΔFosB is thought to dimerize with JunD to form a functional transcription factor, though strikingly JunD does not accumulate in parallel. One explanation is that ΔFosB can recruit different partners, including itself, depending on the neuron type in which it is induced and the chronic stimulus, generating protein complexes with different effects on gene transcription. To develop chemical probes to study ΔFosB, a high-throughput screen was carried out to identify small molecules that modulate ΔFosB function. Two compounds with low micromolar activity, termed C2 and C6, disrupt the binding of ΔFosB to DNA via different mechanisms, and in in vitro assays stimulate ΔFosB-mediated transcription. In cocaine-treated mice, C2 significantly elevates mRNA levels of the AMPA glutamate receptor GluR2 subunit with specificity, a known target gene of ΔFosB that plays a role in drug addiction and endogenous resilience mechanisms. C2 and C6 show different activities against ΔFosB homodimers compared to ΔFosB/JunD heterodimers, suggesting that these compounds can be used as probes to study the contribution of different ΔFosB-containing complexes on the regulation of gene transcription in biological systems and to assess the utility of ΔFosB as a therapeutic target.
Keywords: ΔFosB, high throughput screening, transcription factor, drug addiction, dyskinesia, depression
The transcription factor ΔFosB accumulates in specific regions of the brain (in particular the striatum) upon chronic administration of drugs of abuse, L-DOPA (as used in the treatment of Parkinson’s disease), certain antipsychotic drugs, or stress. The elevated levels of ΔFosB protein are thought to mediate long-lasting neural and behavioral changes in response to chronic cocaine1,2 and mediate dyskinesia (abnormal involuntary movements) in response to chronic L-DOPA.3−5 ΔFosB also mediates endogenous resilience or “coping” mechanisms in response to stress.6 The ΔFosB protein is unusually stable and persists for weeks in neurons, even after cessation of drug administration, leading to its accumulation.1,7
ΔFosB is a member of the Fos/Jun family of transcription factors.1 ΔFosB consists of a large N-terminal domain (ca. 142 a.a.) predicted to contain little secondary structure (DisEMBL),8 a bZIP domain comprising a “basic” region (ca. 40 a.a.) that is connected to a leucine zipper (ca. 36 a.a.), and last a short C-terminal region (ca. 19 a.a.). ΔFosB lacks the 101 a.a. C-terminal transactivation domain of FosB. ΔFosB is modular in nature in that the extended N-terminal region recruits different proteins involved in transcriptional regulation, while the bZIP domain interacts with DNA.9 The leucine zipper of ΔFosB dimerizes with other bZIP domain-containing proteins, forming a coiled-coil that brings the basic regions from the two partners together to interact with DNA like a pair of forceps.10,11 ΔFosB binds DNA at AP-1 consensus sites (TGAC/GTCA) within the promoters of specific target genes and regulates their expression.1 Two important genes that can be regulated by ΔFosB in the nucleus accumbens (ventral striatum) in response to cocaine or stress are cyclin-dependent kinase 5 (cdk5) which plays a role in mediating morphological changes to synapses and the AMPA glutamate receptor subunit 2 (GluR2, also known as GluA2) involved in glutamatergic synaptic transmission.1,2,6 However, in vitro and in vivo studies paint a complex picture of ΔFosB function, because it can work both as an activator as well as a repressor, even on the same target gene, depending on the length of time and level of ΔFosB induction, the inducing chronic stimulus (i.e., cocaine versus L-DOPA), and the experimental conditions used.9,12−14 It is thought that ΔFosB partakes in both activating and repressing transcriptional regulatory complexes (containing chromatin remodeling factors, other transcription factors, coactivators, and the basal transcription machinery), integrating their transcriptional impact with the accessibility and conformation of AP-1 sites at target gene promoters.
It is not clear which protein(s) dimerize with ΔFosB rendering it competent to bind DNA, as it accumulates in the striatum in response to chronic stimuli. ΔFosB is thought to heterodimerize with JunD, based largely on the argument that it is relatively highly expressed in the striatum under baseline conditions whereas other Jun proteins, JunB and c-Jun, are not significantly expressed.1 However, accumulation of striatal ΔFosB protein upon chronic cocaine or L-DOPA treatment, or upon transgenic- or virus-mediated overexpression, in animal models does not result in the induction of JunD protein in parallel.5,15 On the other hand, ΔFosB readily forms homodimers in vitro, which specifically bind the cdk5 and GluR2 AP-1 consensus sequences.16 Many bZIP domain-containing transcription factors form multiple dimers, binding the most abundant partners present in the cell at the time, and their choice of partner determines their functional effect on the transcription of target genes.11,17 This has led to the suggestion that in vivo ΔFosB not only forms active heterodimers with JunD, but also heterodimerizes with other bZIP domain-containing proteins and homodimerizes with itself depending on the chronic stimulus and the neuron type, producing different complexes that activate or repress transcription.16
To develop tools to pharmacologically probe ΔFosB function, a chemical genomics campaign was carried out to find small molecules that disrupt the interaction of ΔFosB with DNA in vitro and might have the potential to diminish or potentiate long-term neural adaptations induced by ΔFosB in vivo (Figure 1a). Compounds might work via different mechanisms, that is, by affecting the DNA binding site, dimerization of ΔFosB, or allosterically (Figure 1b). We identified and validated two chemical scaffolds. The compounds employ different mechanisms to disrupt DNA binding and show different selectivities for ΔFosB homodimers compared to ΔFosB/JunD heterodimers. In a cell based-assay, these compounds increased reporter activity. One compound was tested in vivo and selectively elevated mRNA levels of GluR2 in the nucleus accumbens. The compounds we have identified may be useful tools in vivo to tease apart the activating as well as repressing transcriptional effects of ΔFosB at different promoters, and to probe the role of different ΔFosB-containing molecular species in mediating mechanisms that underlie drug addiction, L-DOPA-induced dyskinesia, endogenous resilience mechanisms, and other neuropsychiatric phenomena.
Figure 1.
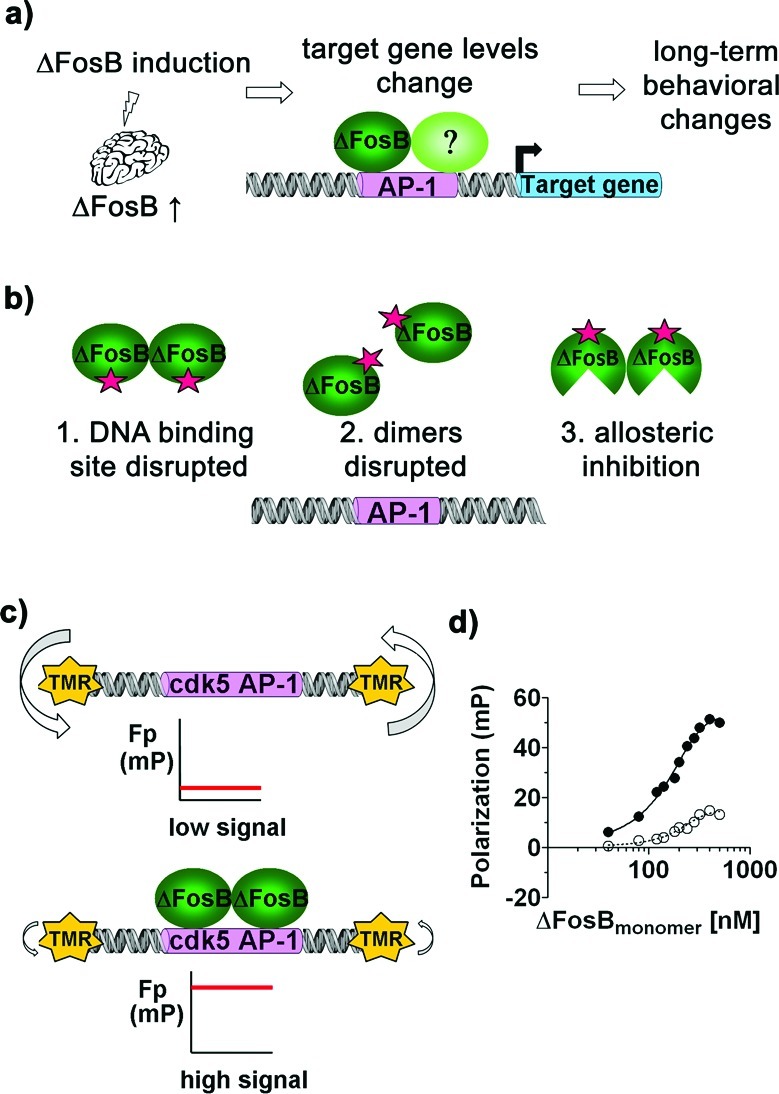
Small molecule modulators of ΔFosB. (a) Chronic administration of stimuli such as drugs of abuse or L-DOPA increase ΔFosB protein levels in specific regions of the brain. ΔFosB binds to promoter regions of numerous target genes, altering their expression. The altered expression of key target genes is thought to mediate long-term neural and behavioral changes. (b) Potential mechanisms of compounds disrupting the binding of ΔFosB to an oligonucleotide containing an AP-1 consensus sequence. (c) Principle of the FP assay used to screen libraries for small molecules that disrupt the ability of ΔFosB to bind DNA. TMR-cdk5 tumbles rapidly in solution, giving a low FP signal. When ΔFosB binds TMR-cdk5, the tumbling of the oligo is slowed down, leading to a dramatic increase in FP signal. Compounds that disrupt ΔFosB binding to DNA will decrease this signal. (d) ΔFosB binds TMR-cdk5 (●) with sequence specificity compared to an oligonucleotide with a random, scrambled sequence (TMR-SCR) (○), as monitored by fluorescence polarization.
Results and Discussion
Screening for Small Molecule Regulators of ΔFosB
To identify small molecule regulators of ΔFosB function, we developed a fluorescence polarization (FP) assay to detect the specific binding of ΔFosB to a TAMRA-labeled cdk5 oligonucleotide carrying the cdk5 AP-1 consensus site (TMR-cdk5) (Figure 1c and d). A 54 498 compound library of commercially available small molecules at the Center for Chemical Genomics (CCG), University of Michigan, was screened. The hits in the primary screen were subject to a series of confirmation and secondary assays to identify active compounds (Supporting Information Table 1). Compounds C1, C2, C4, C5, and C6 disrupted ΔFosB binding to TMR-cdk5 with IC50 values in the low micromolar range (ca. 4–10 μM) in FP-based dose response curves (Figures 2 and 3a). Compound C7 did not require protein to decrease the FP signal in the assay, and neither did the signal saturate at concentrations up to 1 mM, revealing it as a false positive, so C7 was used further as inactive negative control compound (Figures 2 and 3a). NMR spectra and mass spectrometry revealed C1 to be heterogeneous, likely as a result of degradation or modification. C1 was nevertheless taken along in the assays to demonstrate proof-of-principle that multiple, highly different chemical scaffolds can disrupt ΔFosB binding to DNA.
Figure 2.

Small molecule inhibitors of ΔFosB binding to DNA. The structures of five active compounds (C1–C6) and one negative control compound (C7) are indicated together with their commercial source. An additional negative control compound C3 was taken along in the studies, but gave comparable results to C7 and is not further described.
Figure 3.

Validating small molecule modulators of ΔFosB. (a) Compounds were tested in dose response assays by incubating 50 nM TMR-cdk5 with increasing amounts of compound in the presence of 280 nM ΔFosB (▼) or in the absence of ΔFosB (▽), for 15 min at room temperature and monitoring the decrease in FP signal. The positive control oligo alone (○) and the negative control ΔFosB+cdk5 (●) are indicated. (b) Ethidium bromide (EthBr) displacement assays confirm that C1, C2, C4, C5, C6, and C7 are not DNA intercalators and do not efficiently displace EthBr from the cdk5 oligonucleotide unlike the known intercalator NSC311152. Controls for no displacement (cdk5+EthBr, ■) and 100% displacement (EthBr, ◆) are indicated. (c) Compounds were tested using EMSA by incubating 8 pmol of ΔFosB (0.2 μg) with 1 pmol of digoxygenin-labeled cdk5 oligonucleotide (DIG-cdk5) and increasing amounts of compound (1, 5, 10, 25, 50, and 100 μM); see lanes 3–8. DIG-cdk5 alone (lane 1, which also shows a small amount of higher order oligomer) and the starting amount of ΔFosB:DIG-cdk5 complex in the absence of compound (lane 2) are shown as well.
A series of assays were carried out to confirm that C1, C2, C4, C5, and C6 disrupted the binding of ΔFosB to DNA in a protein-dependent manner. An ethidium bromide displacement assay demonstrated that the compounds did not significantly intercalate with DNA (Figure 3b). C1, C2, C4, C5, and C6 disrupted binding of ΔFosB to the cdk5 oligo in electrophoretic mobility shift assays (EMSA), while the negative control compound C7 did not (Figure 3c). Though not quantitative, EMSA confirmed the activity of the compounds in a non-FP-based method.
The compounds were next tested in cell-based assays using Neuro2A cells; these neuronlike cells contain low amounts of endogenous fos/jun proteins. The compounds were tested for their toxicity using an ATP-based cell viability assay (Supporting Information Figure S1), revealing that, at concentrations as high as 50 μM for C1, C2, C4, C5, and C7, and 12.5 μM for C6, cell viability was not negatively impacted in a significant way compared to DMSO (less than 20%). A transactivation assay was used to assess the impact of the compounds on ΔFosB-mediated transcription of a reporter plasmid. Because a cdk5 AP-1 luciferase reporter construct is not substantially activated by ΔFosB (indeed the dynorphin gene promoter is repressed by ΔFosB, and AP-1 sites in the GluR2 promoter influence transcription only in certain cell types),14,18,19 we used a synthetic reporter containing four concatenated AP-1 consensus sites in front of the luciferase gene that is moderately transcriptionally activated by ΔFosB. While the mechanism underlying ΔFosB-mediated activation of this synthetic reporter is not known, it enabled us to test if the compounds could increase or decrease transcription of the reporter in the presence of ΔFosB with or without coexpression of JunD (Figure 4a). C5 was excluded due to poor solubility in the cell medium. Transfection of ΔFosB increased the luciferase signal 3-fold and ΔFosB/JunD increased it 4-fold above basal levels. All of the compounds that had an effect on the luciferase signal in the assay, that is, C1, C2, and C6, increased the luciferase signal 1.5- to 3-fold, suggesting that they resulted in a net activating effect on gene transcription (either through stimulation or derepression of transcription). Compound C4 and the negative control compound C7 were inactive (Figure 4a). Similar results were obtained in cells transfected with full-length FosB in the presence or absence of JunD, as well as JunD alone, although the latter did not activate the luciferase reporter gene above endogenous background levels (Supporting Information Figure S2). The increase in luciferase signal due to addition of C1, C2, or C6 cannot be explained by the compounds working directly on the luciferase enzyme,20 because the compounds clearly require the presence of ΔFosB to exert high levels of reporter gene activation in the assay. It is possible that, in the presence of the compounds, the N-terminal domain of ΔFosB continues to be integrated into the transcriptional regulatory machinery assembling on the reporter, while the binding of the ΔFosB bZIP domain to DNA is disrupted, permitting other more activating, endogenous transcription factors to dominate the transcription of the reporter gene. Alternatively, it is possible that the compounds, by disrupting ΔFosB binding to the concatamer of AP-1 sites, alter the conformation of the DNA or its position with respect to the rest of the transcriptional machinery, relieving DNA bending known to be induced by Fos/Jun complexes,21 for example thereby promoting transcription.
Figure 4.

Cell-based and in vivo effects of small molecule ΔFosB regulators. (a) ΔFosB regulators alter reporter activity in cell-based transactivation assays. Neuro2A cells were cotransfected with a luciferase reporter gene (4xAP-1/RSV-Luc) and one of the following: ΔFosB, ΔFosB, and JunD, or the pcDNA 3.1 vector (to measure endogenous AP-1 transcriptional activity). Transactivation of the reporter gene was monitored after 48 h of incubation with 50 μM C1, C2, or C7, 25 μM C4, 12.5 μM C6, or 0.1% DMSO as a control. The change in luciferase signal is expressed as luciferase units per total cellular protein (LU/μgr). (b) ΔFosB regulators alter transcription of a ΔFosB target gene in mice. Real-time quantitative PCR was performed on triplicate samples of RNA isolated from the nucleus accumbens to evaluate levels of mRNA for ΔFosB target genes, GluR2 and cdk5, following 14 days administration of C2 (7 mice), a structurally related but inactive analogue (Chembridge 5996481, 8 mice) or vehicle (6 mice) in the nucleus accumbens. During the later 7 days of treatment, the animals additionally received an injection of 10 mgr/kg weight cocaine. Data (means ± SEM for each group of mice) were analyzed using an unpaired two-tailed t test with 95% confidence interval to reveal statistically significant differences in mRNA levels compared to the vehicle (p < 0.05).
Because regulation of gene transcription at eukaryotic promoters is often highly cell-type specific, involving a host of transcription factors and chromatin modifying enzymes, as well as the state of the chromatin which controls accessibility to the gene target,22 we assessed the activity of C2 in vivo (C6 was not tested). C2 was chosen because of its lower toxicity in the cell-based toxicity assay and 2-fold higher activity in the FP-assay. C2 was infused directly into the nucleus accumbens (the ventral portion of striatum) of mice treated with cocaine. Alterations in transcript levels of known target genes for ΔFosB were assessed by qPCR on RNA samples isolated from the nucleus accumbens of treated mice (Figure 4b). Administration of C2 resulted in a 3-fold increase of endogenous mRNA levels for GluR2, while administration of a structurally closely related but inactive analogue “Chembridge 5996481” (described next section and Figure 5a) or vehicle did not. Intriguingly, transcriptional regulation appeared gene specific, because the cdk5 gene showed no significant difference in mRNA levels (Figure 4b). This result is consistent with the highly complex chromatin mechanisms observed in vivo in response to cocaine whereby only certain specific genes are primed; that is, their promoter region unwound from the compact chromatin state, rendering them accessible for regulation of their gene transcription.9 Additionally, administration of C2 in vivo revealed that even though transcription of GluR2 is regulated by many transcription factors in addition to ΔFosB including NRF-1, Sp1, and MECP2,19,23,24 regulating ΔFosB pharmacologically was sufficient to dramatically alter GluR2 mRNA levels. These results are intriguing because glutamatergic neurotransmission is emerging as a focus for the development of new drug targets for both drug addiction as well as other neuropsychiatric disorders including depression and autism.25−28 Further study of C2 and other ΔFosB modulators also offers the potential of unraveling the interactions that ΔFosB mediates in transcriptional regulatory macromolecular complexes as it acts at the promoters of specific target genes.
Figure 5.

Commercially available analogues of C2 and C6. The “active” compounds inhibited ΔFosB binding to TMR-cdk5 with an IC50 < 25 μM and activated transcription of the luciferase reporter gene in the cell-based assays similarly to the parent compounds. The “low-active” compounds inhibited ΔFosB binding to TMR-cdk5 with an IC50 between 25 and 300 μM and were not tested in cell-based assays. The “inactive” compounds did not inhibit ΔFosB binding to TMR-cdk5 in the FP assay. Note: CB, CD, AS, and MB represent Chembridge, ChemDiv, Asinex, and Maybridge, respectively.
Mechanism of Action for Compounds C2 and C6
Compounds C2 and C6 were further investigated for their ability to specifically interact with ΔFosB. C1 was excluded because of its heterogeneous composition. While C2 and C6 do not violate Lipinski’s rules,29 they contain α,β-unsaturated carbonyl groups which could potentially undergo Michael addition to the protein. However, these compounds are active under the standard assay conditions which contain 1 mM DTT, suggesting that even if these compounds are thiol-reactive, they are only weakly so, or react in a reversible manner. Furthermore, the compounds do not covalently bind ΔFosB as determined by mass spectrometry (results not shown). C2 and C6 are also active in presence of 0.1% Triton X-100 in FP assays, suggesting they are not aggregator compounds (results not shown). In addition, while C2 has been run in 49 screens and C6 in 46 screens at the CCG to date, these two compounds have demonstrated activity only against ΔFosB (AC50 ≤ 10–6) and low activity only against one additional target each (AC50 ≤ 10–5) in dose response curves, providing evidence that C2 and C6 target ΔFosB with selectivity. To confirm that the compounds represent chemical scaffolds that can regulate ΔFosB binding to DNA specifically, we searched for commercially available analogues. One analogue for C2 and three analogues for C6 were found that disrupted ΔFosB binding to the TMR-cdk5 oligo with an IC50 less than 25 μM in FP-based dose response curves (Figure 5). These compounds again increased the luciferase signal in the cell-based assays (data not shown). A number of low active and nonactive compounds were identified as well, demonstrating that preliminary structure–activity relationships can be constructed for C2 and C6, and that they interact with ΔFosB in a specific manner.
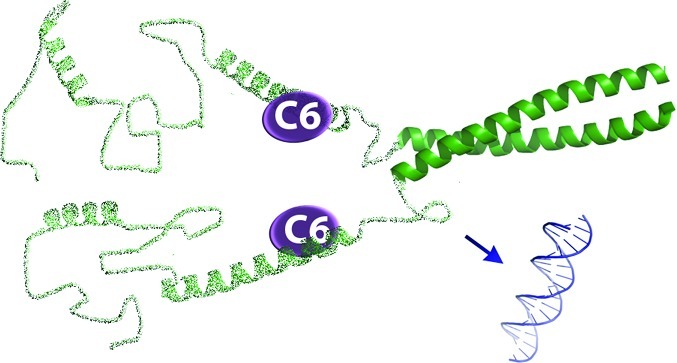
The mechanism of action of C2 and C6 was further investigated by circular dichroism (CD). Purified ΔFosB was incubated with C2 or C6 to test if either compound induced changes to the protein’s secondary structure (Figure 6a and b). Deconvolution of the data showed that ΔFosB is largely unstructured in solution (50% unstructured and 15% helical content) (Figure 6c). Upon addition of the cdk5 oligo, the helical content increased to 45% at the cost of the unstructured component, suggesting that DNA binding introduces significant helical content to the protein. Incubation with C2 did not change the CD spectrum of ΔFosB significantly, suggesting little change to the secondary structure of ΔFosB (Figure 6a). Importantly, subsequent addition of the DNA substrate also no longer induced additional helical content, suggesting that the DNA binding site was blocked (Figure 6c). Strikingly, incubation of ΔFosB with C6 increased the helical content of the protein to 35% at the cost of the unstructured component, triggering rearrangements on a similar scale as addition of DNA (Figure 6b, Supporting Information Figure S3). Like C2, when oligonucleotide was added to ΔFosB preincubated with C6, no additional increase in helical content was seen, suggesting that C6 also prevents ΔFosB binding to DNA (Figure 6c). So while C2 appears to directly block the DNA binding site of ΔFosB, C6 converts unstructured protein regions outside the DNA binding region to helical structure, working allosterically to disrupt DNA binding.
Figure 6.
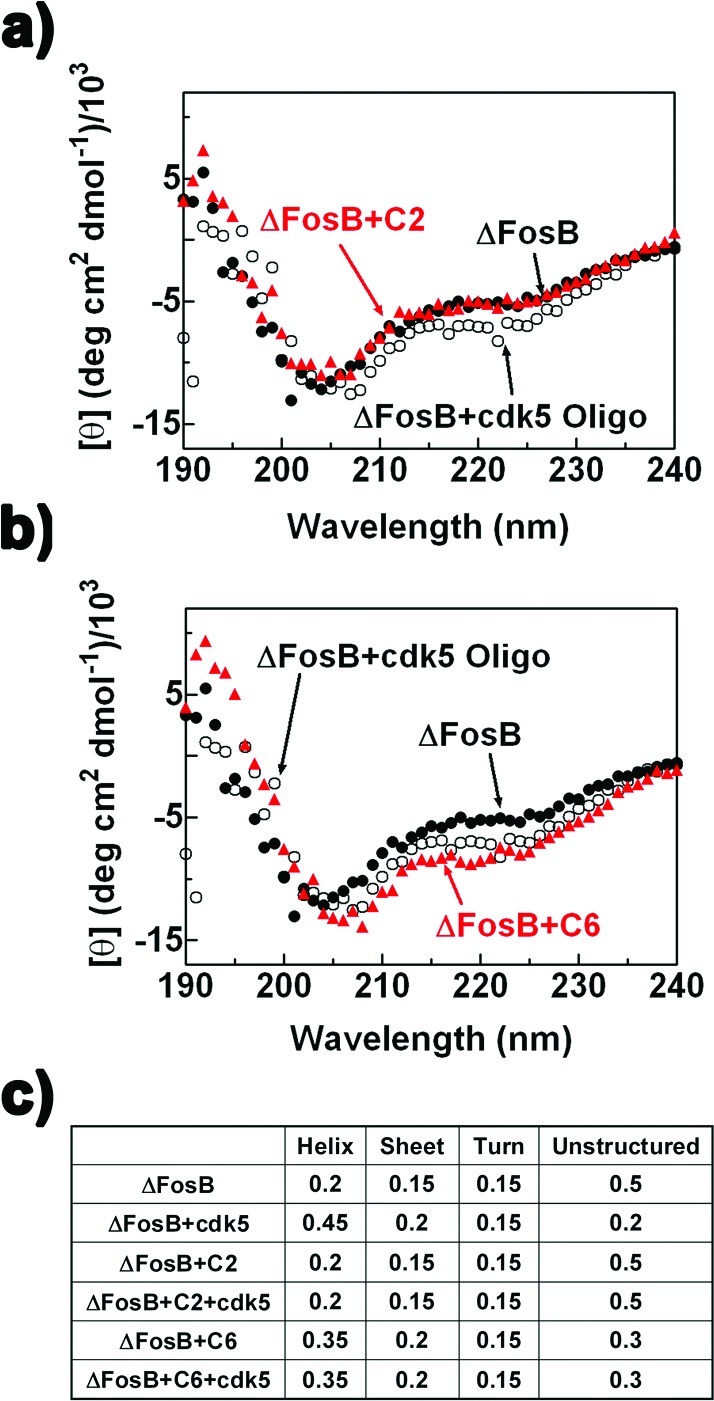
Changes in the secondary structure of ΔFosB monitored by CD. (a) CD spectra of ΔFosB alone, with cdk5, and with 100 μM C2. (b) CD spectra of ΔFosB alone, with cdk5, and with 100 μM C6. (c) Deconvolution of the CD spectra and secondary structural content (in %) of ΔFosB, ΔFosB with cdk5, ΔFosB and C2 with or without cdk5, and ΔFosB and C6 with or without cdk5.
Target Specificity of the Compounds
To investigate if the active compounds were specific for ΔFosB homodimers, compounds C1, C2, and C6 were also tested for their ability to disrupt binding of ΔFosB/JunD heterodimers (Figure 7) and JunD homodimers (Supporting Information Figure S4) to the TMR-cdk5 oligonucleotide. ΔFosB homodimers bind to TMR-cdk5 in a sequence-specific manner at low salt concentration (50 mM), while at high salt concentration (175 mM) DNA binding is lost (Figure 7a). In contrast, ΔFosB/JunD heterodimers (and JunD homodimers) require high salt conditions to efficiently discriminate between TMR-cdk5 and a scrambled oligonucleotide, TMR-SCR (Figure 7b, Supporting Information Figure S4a). Protein/DNA interactions are typically salt-dependent, but salt concentrations can also drastically affect the conformation of proteins. We therefore tested the ability of C1, C2, and C6 to disrupt DNA binding under conditions that support sequence-specific binding to DNA (i.e., ΔFosB under low salt conditions, and ΔFosB/JunD (as well as JunD) under high salt conditions), but also under conditions that promote sequence-aspecific binding to DNA (i.e., ΔFosB/JunD and JunD under low salt conditions), because the two DNA-binding processes might involve different protein conformational states that could be probed by C2 and C6.
Figure 7.

Specificity of small molecule ΔFosB modulators. (a) ΔFosB homodimer binding to TMR-cdk5 under low salt conditions (50 mM NaCl, solid gray circle) and high salt conditions (175 mM NaCl, ▲). Binding is also shown for TMR-SCR (50 mM NaCl, ○) and (175 mM NaCl, △). (b) ΔFosB/JunD binding to TMR-cdk5 under low salt conditions (50 mM NaCl, solid gray circle) and high salt conditions (175 mM NaCl, ▲). Binding is also shown for TMR-SCR (50 mM NaCl, ○) and (175 mM NaCl, △). Under high salt conditions, ΔFosB/JunD binds TMR-cdk5 specifically though not as efficiently, and the binding of ΔFosB/JunD to TMR-SCR (△) is suppressed. (c) Dose response curves of C1, C2, and C6 with ΔFosB and ΔFosB/JunD under low salt conditions (50 mM NaCl, left panel) and high salt conditions (175 mM NaCl, right panel). C1, C2, and C6 were tested in dose response assays as described in Figure 3 without protein (▽), in the presence of 280 nM ΔFosB (◇) and in the presence of 280 nM ΔFosB/JunD (■). The positive control (oligo alone, ○) and two negative controls (ΔFosB+cdk5, * and ΔFosB/JunD+cdk5, □) are shown as well.
Striking differences were observed in the ability of the compounds to disrupt DNA binding to the three transcription factor species as a function of salt concentration. C1 and C2 inhibited ΔFosB homodimers, ΔFosB/JunD heterodimers, and JunD homodimers similarly under low salt conditions (Figure 7c, Supporting Information Figure S4b). However, under high salt conditions, which support sequence-specific binding of ΔFosB/JunD and JunD to DNA (and keep C2 in solution), C2 was more than 10-fold less active compared to low salt conditions. This salt dependence was not seen for compound C1, which disrupted DNA binding to all three species more or less equally independent of the salt conditions. The opposite was seen for C6, which under low salt conditions was 10-fold less effective in disrupting the ΔFosB/JunD heterodimer and JunD homodimer complexes with DNA (compared to ΔFosB), but regained some of its activity under high salt conditions (Figure 7c, Supporting Information Figure S4b). These results suggest that ΔFosB homodimers have significant conformational differences in their DNA-binding sites compared to JunD-containing species, which can be probed by compounds C2 and C6.
Transcription Factors as Targets for Small Molecule Intervention
This study is the first to specifically target ΔFosB with small molecules, revealing compounds that inhibit the binding of ΔFosB to DNA using multiple mechanisms of action and alter transcriptional activity when tested in cell-based assays and in the brain in vivo. The compounds C2 and C6 appear to work using different mechanisms to disrupt DNA binding (Figure 8). Compound C2 does not significantly change the secondary structure of ΔFosB and likely inhibits DNA binding by binding directly to the DNA binding site (Figure 8b). C6 employs a different mechanism to disrupt DNA binding, introducing helical content to ΔFosB in a protein conformation that prevents DNA binding (Figure 8c), though the exact binding site of C6 in ΔFosB must now be determined. Remarkably, these studies suggest that ΔFosB homodimers and ΔFosB/JunD heterodimers have different protein conformations that impact their mechanisms of DNA binding and recognition which can be probed by C2 and C6.
Figure 8.
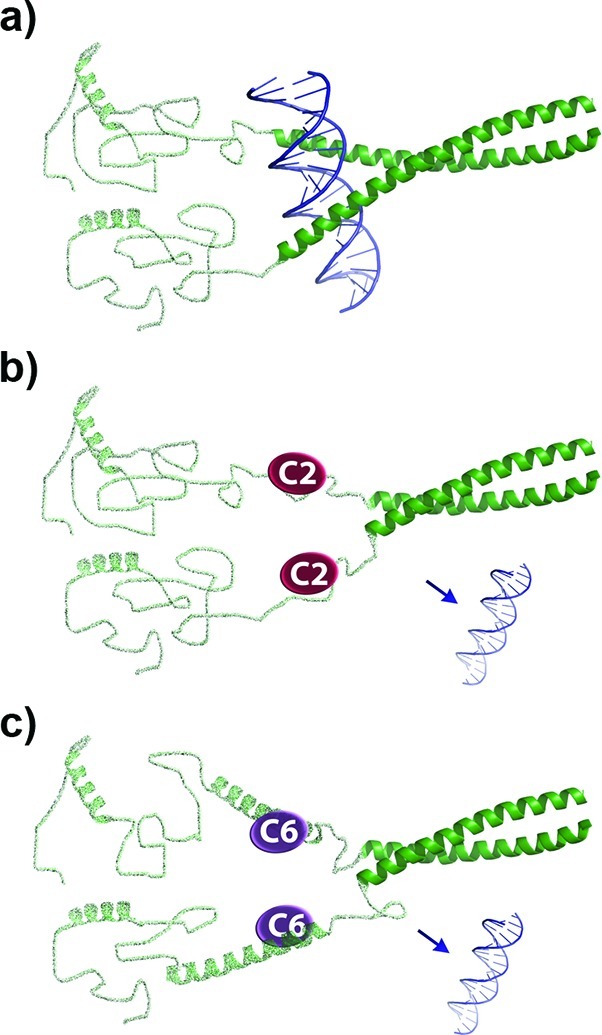
Possible mechanisms of action for C2 and C6. (a) In solution the N-terminus of ΔFosB is mostly unstructured. The C-terminal leucine zipper mediates dimer formation, while the preceding basic region adopts a helical conformation that interacts with DNA. (b) C2 likely binds to the DNA binding region, and thus blocks ΔFosB from binding DNA. (c) C6 likely binds to a region N-terminal to the basic region, inducing helical content to ΔFosB that prevents ΔFosB from undergoing conformational changes required for it to bind to DNA.
Recent studies show that a number of transcription factors can be regulated with small molecules.30,31 In particular, transcription factors which contain regions that undergo disorder-to-order transitions to dimerize or to form binding sites for their ligands such as DNA, coactivators, cofactors, and other transcription factors, appear particularly susceptible to regulation by small molecule compounds.32−34 While small molecule inhibitors of c-Fos-containing AP-1 complexes are known, such as curcumin,35 tanshinone IIA,36 and nordihydroguaiaretic acid,37 these are not active against ΔFosB or JunD-containing species (tested up to 100 μM, results not shown). A benzophenone derivative in preclinical trials to resolve arthritis, T-5224, also inhibits c-Fos and c-Jun-containing AP-1 activity, though the mechanism of action and its activity against ΔFosB are not known.38 Compounds active against bZIP domains of c-Fos/JunD, C/EBPα, and C/EBPβ have been identified, though their mechanism is not known.39,40 Compounds that disrupt c-Myc/Max binding to DNA appear to disrupt dimerization between the bHLH-ZIP domains in the c-Myc/Max heterodimer30,41−43 or disrupt the c-Myc/Max bHLH-ZIP complex with DNA.44,45 Interestingly, the 5-benzylidene-rhodanine group of C6 is also found in 10058-F4, an inhibitor that disrupts c-Myc/Max dimerization by binding to a distinct region of c-Myc and inducing a more rigid and defined conformation,42 though our CD spectra show no evidence that dimerization of ΔFosB is affected by C6.
ΔFosB may be an attractive therapeutic target because the protein accumulates in very specific brain regions in response to several chronic stimuli, and mediates long-term neural and behavioral changes. Our study provides evidence that ΔFosB function can be regulated by small molecules in vitro and in vivo. Beneficial effects of suppressing ΔFosB function in vivo have already been demonstrated using antisense technology or virally expressed dominant negative mutants, and show that reducing ΔFosB protein levels lowers the sensitivity to rewarding effects of cocaine and morphine14,46 as well as the severity of L-DOPA-induced dyskinesia.3,47 However, upregulating ΔFosB activity in a brain region specific way may also prove useful to enhance endogenous resilience mechanisms to combat depression.6 While developing five-membered multiheterocyclics into drugs may be a long-term endeavor,48,49 C2 and C6 can already be used as chemical tools, by administering them directly into the brain to tease apart the role of ΔFosB in repressive as well as activational transcriptional complexes and to assess the epigenetic state of its different target genes during maladaptive neural changes. Such molecular insight could guide assessment of the therapeutic utility of ΔFosB.
Methods
Materials
Compounds C1 (Chembridge 6572652), C2 (Chembridge 5997715), C4 (Chembridge 5375994), C5 (Chembridge 5847737), C6 (Chembridge 5162044 or alternative Chembridge 3018304), and C7 (ChemDiv 5735-0011) were purchased from their respective vendors. Analogues were purchased from Chembridge, ChemDiv, Asinex, or Maybridge. Compounds were dissolved in DMSO (20 mM stocks or 4 mM if poorly soluble) and stored at −20 °C. For the CD experiments, compounds were dissolved in ethanol (2.5 mM stock).
The sense and antisense oligonucleotides of the 19-mer TMR-cdk5 (5′-CGTCGGTGACTCAAAACAC-3′) and TMR-SCR (5′-GTATGCGATACGTCTTTCG-3′) (HPLC purified, TAMRA labeled at 5′-end of both strands) were purchased from SigmaAldrich. For EMSA and CD, unlabeled oligos (sense and antisense, desalted) were purchased as well. Oligos were annealed as described16 and stored as 50 μM stocks (TAMRA-labeled) or 500 μM stocks (unlabeled) in annealing buffer (10 mM Tris pH 8, 50 mM NaCl) at −20 °C.
Protein Expression and Purification
ΔFosB from mouse (a splice form of FosB, accession number P13346) was expressed as an N-terminally His-tagged protein in SF9 cells (Bac-to-Bac system, Invitrogen). The recombinant protein contains the N-terminal tag MGHHHHHHAG followed by residues (F2-E237) from ΔFosB. JunD from mouse (accession number J04509) was expressed as an N-terminally His-tagged protein was well, and contains the N-terminal tag MGHHHHHH followed by residues (E2-Y341). Protein was purified essentially as described,16 and details are given in the Supporting Information. ΔFosB, ΔFosB/JunD, and JunD were stored in 20 mM Tris pH 7.5, 1 M NaCl as flash-frozen aliquots.
High Throughput Screening
A fluorescence polarization assay monitoring the binding of ΔFosB to a TMR-cdk5 was adapted for high throughput screening of small molecules,16 using a library containing the compounds from Microsource, ChemDiv, Chembridge, Maybridge, NCl, and BioFocus. For the primary screen, one dose (∼15 μM) of each compound was transferred from library stock plates using an HDR pintool on a Biomex FX robot (Beckman) to 384-well assay plates (Corning, #3676). Subsequently, 280 nM NHis6-ΔFosB (monomer concentration) in FP buffer (20 mM HEPES pH 7.5, 50 mM NaCl, 1 mM DTT) was added to each well. Finally, 50 nM TMR-cdk5 oligo was added and incubated for 15 min at RT in a total volume of 20 μL. Plates were read using a Pherastar plate reader (BMG Labtech). Wells with TMR-cdk5 alone were used as a positive control (100% inhibition). Wells with TMR-cdk5 incubated with 280 nM ΔFosB, but no compound, were used as a negative control (0% inhibition). Active compounds were selected if they caused >3SD decrease in fluorescent polarization signal compared to negative control. The average Z-score per plate was approximately 0.6.
FP Dose–Response Curves
Active primary screen compounds were retested in a confirmation screen using library stocks in an 8-point dose response assay (in triplicate) to confirm the hit status and obtain a rough estimate of the pIC50. In parallel, concentration series of compound with or without TMR-cdk5 were also incubated in the absence of ΔFosB (in duplicate) to monitor if the compounds interfered with the assay or worked on the oligo directly. Each 384-well plate included 8 wells of “positive control” (TMR-cdk5 alone) and 8 wells of “negative control” (ΔFosB+TMR-cdk5). The FP data was fit using the “log(inhibition) vs response – variable slope” algorithm in Prism (GraphPad). For the reconfirmation assay, hit compounds in the confirmation screen were purchased from vendors and retested in dose–response assays as described above.
Analogue Search
The CCG M-Screen external library search was used to find analogues with an 80% structural similarity cutoff. A commercial database (SciFinder) was used to search for vendors and predict compound characteristics. Compounds with predicted logP > 4 and predicted solubility in water < 10–3 g L–1 were not further considered.
Electrophoretic Mobility (Gel) Shift Assay (EMSA)
EMSA were used to confirm the inhibitory activity of compounds identified through high-throughput screening, essentially as described16 with the DIG Gel Shift kit, second Generation (Roche) according the manufacturer’s instructions.
Ethidium Bromide Displacement Assay (EBDA)
Compounds were tested for their potential to intercalate into DNA and displace prebound ethidium bromide (EthBr) as described.39 As a positive control, the known intercalator, NSC311152 (NCI), which robustly displaces DNA, was included as well.
Cell Toxicity Assay
Cell viability in mouse Neuro2A neuroblastoma cells (ATCC) in the presence of the different compounds was assessed using the CellTiter-Glo luminescent cell viability assay (Promega), which determines the number of viable cells as a function of ATP present, according to manufacturer’s instructions (see Supporting Information).
Transactivation Assay
For transfection, Neuro2A cells were plated onto 10 cm dishes (Corning, #430167, at 1 × 106 cells/dish) in EMEM medium without antibiotics. The next day, each dish was transiently cotransfected with 1 μg of 4×AP-1(metalloproteinase-II promoter)/RSV-Luc (the luciferase reporter plasmid) and 0.5 μg of pcDNA 3.1 ΔFosB using Effectene (Qiagen) according to the manufacturer’s instructions. In parallel, cells were also transfected with 1 μg of 4×AP-1/RSV-Luc in combination with 0.5 μg of FosB-vector or 0.5 μg of JunD-vector. In addition, to produce heterodimers, 1 μg of 4×AP-1/RSV-Luc was transfected together with 0.5 μg of ΔFosB-vector and 0.5 μg of JunD-vector, as well as 0.5 μg of FosB-vector and 0.5 μg of JunD-vector. As a control for endogenous expression of the transcription factors, Neuro2A cells were transfected with 1 μg 4×AP-1/RSV-Luc and 1 μg of the pcDNA3.1 alone.
Based on the toxicity assays, C1, C2, and C7 were tested at 50 μM, C4 at 25 μM, and C6 at 12.5 μM in the luciferase assay as described18 (see Supporting Information). As a control for the effect of DMSO, Neuro2A cells were exposed to 0.1% DMSO (i.e., no compound). The effect of compound on transcription-factor-mediated luciferase activity was expressed as luciferase activity/μg protein in order to take into account any effects of the compound on cell-growth, and is reported relative to wells containing cells exposed to DMSO alone.
In Vivo Studies
Mice were administered C2 (100 μM, 7 mice), a structurally related but inactive analogue (Chembridge 5996481, 100 μM, 8 mice) or vehicle (0.5% DMSO in PBS, 6 mice) bilaterally to the nucleus accumbens for 14 days (see Supporting Information). During the last 7 days of compound administration, all mice received additionally a daily intraperitoneal injection of cocaine at 10 mg/kg weight. The effect of compound administration on mRNA levels of two known target genes for ΔFosB, GluR2, and cdk51 was analyzed by qPCR using bilateral nucleus accumbens punches to isolate RNA (see Supporting Information). The qPCR data were analyzed with the ΔΔCt method using expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which was unaffected by C2, as a normalizing control, as previously described.14
Circular Dichroism (CD)
ΔFosB protein was dialyzed against CD buffer (12.5 mM phosphate buffer, pH 8, 50 mM NaF and 1 mM DTT). Reagents were prepared in CD buffer, that is, the protein sample (ΔFosB at 0.1 mg/mL), compounds (100 μM C2 or C6 prepared from a 2.5 mM stock in ethanol) or ethanol (as a control for no compound), and cdk5 oligo (12.5 μM cdk5 made from a 500 μM stock in annealing buffer), and incubated at RT for 15 min. For each protein sample, a corresponding background sample was prepared that contained the identical sample components including cdk5 oligo and compounds as necessary, but no protein. CD spectra were recorded at RT on an Aviv-202 spectrometer from 190 to 260 nm in a 1.0 mm path length quartz cuvette using an average time of 2 s at the spectral bandwidth of 1.0 nm. Final background cleared averaged spectra were analyzed and deconvoluted using “dichroweb”50,51 (see Supporting Information).
Supporting Information Available
Additional methods describing protein overexpression and purification, cell toxicity assays, transactivation assays, in vivo studies, and circular dichroism (CD); flowchart of the high-throughput screening campaign (Table S1); toxicity studies of small molecule ΔFosB modulators (Figure S1); cell-based transactivation assays testing small molecule ΔFosB modulators (Figure S2); CD studies of ΔFosB in presence of increasing amounts of compound C2 and C6 (Figure S3); and FP-assays testing small molecule ΔFosB modulators against JunD homodimers (Figure S4). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
G.R. and E.J.N. designed the experiments. Y.W., T.I.C., Y.O., R.B., and V.L. performed the experiments. P.D.K., S.D.L., and M.J.L. analyzed the HTS results. Y.W., T.I.C., and G.R. wrote the manuscript, aided by comments from the co-authors.
This work has been supported by a NARSAD Young Investigator Award (G.R.), NIMH (RC MH088477 to E.J.N. and G.R.), and MICHR (UL1RR024986 to G.R.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Nestler E. J. (2008) Review. Transcriptional mechanisms of addiction: role of DeltaFosB. Philos. Trans. R. Soc., B 363, 3245–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo S. J.; Dietz D. M.; Dumitriu D.; Morrison J. H.; Malenka R. C.; Nestler E. J. (2010) The addicted synapse: mechanisms of synaptic and structural plasticity in nucleus accumbens. Trends Neurosci. 33, 267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berton O.; Guigoni C.; Li Q.; Bioulac B. H.; Aubert I.; Gross C. E.; Dileone R. J.; Nestler E. J.; Bezard E. (2009) Striatal overexpression of DeltaJunD resets L-DOPA-induced dyskinesia in a primate model of Parkinson disease. Biol. Psychiatry 66, 554–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenci M. A.; Konradi C. (2010) Maladaptive striatal plasticity in L-DOPA-induced dyskinesia. Prog. Brain Res. 183, 209–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X.; Yasuda T.; Uthayathas S.; Watts R. L.; Mouradian M. M.; Mochizuki H.; Papa S. M. (2010) Striatal overexpression of DeltaFosB reproduces chronic levodopa-induced involuntary movements. J. Neurosci. 30, 7335–7343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vialou V.; Robison A. J.; Laplant Q. C.; Covington H. E. 3rd; Dietz D. M.; Ohnishi Y. N.; Mouzon E.; Rush A. J. 3rd; Watts E. L.; Wallace D. L.; Iniguez S. D.; Ohnishi Y. H.; Steiner M. A.; Warren B. L.; Krishnan V.; Bolanos C. A.; Neve R. L.; Ghose S.; Berton O.; Tamminga C. A.; Nestler E. J. (2010) DeltaFosB in brain reward circuits mediates resilience to stress and antidepressant responses. Nat. Neurosci. 13, 745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulery P. G.; Rudenko G.; Nestler E. J. (2006) Regulation of DeltaFosB stability by phosphorylation. J. Neurosci. 26, 5131–5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linding R.; Jensen L. J.; Diella F.; Bork P.; Gibson T. J.; Russell R. B. (2003) Protein disorder prediction: implications for structural proteomics. Structure 11, 1453–1459. [DOI] [PubMed] [Google Scholar]
- Robison A. J.; Nestler E. J. (2011) Transcriptional and epigenetic mechanisms of addiction. Nat. Rev. Neurosci. 12, 623–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover J. N.; Harrison S. C. (1995) Crystal structure of the heterodimeric bZIP transcription factor c-Fos-c-Jun bound to DNA. Nature 373, 257–261. [DOI] [PubMed] [Google Scholar]
- Hess J.; Angel P.; Schorpp-Kistner M. (2004) AP-1 subunits: quarrel and harmony among siblings. J. Cell Sci. 117, 5965–5973. [DOI] [PubMed] [Google Scholar]
- Andersson M.; Konradi C.; Cenci M. A. (2001) cAMP response element-binding protein is required for dopamine-dependent gene expression in the intact but not the dopamine-denervated striatum. J. Neurosci. 21, 9930–9943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClung C. A.; Nestler E. J. (2003) Regulation of gene expression and cocaine reward by CREB and DeltaFosB. Nat. Neurosci. 6, 1208–1215. [DOI] [PubMed] [Google Scholar]
- Zachariou V.; Bolanos C. A.; Selley D. E.; Theobald D.; Cassidy M. P.; Kelz M. B.; Shaw-Lutchman T.; Berton O.; Sim-Selley L. J.; Dileone R. J.; Kumar A.; Nestler E. J. (2006) An essential role for DeltaFosB in the nucleus accumbens in morphine action. Nat. Neurosci. 9, 205–211. [DOI] [PubMed] [Google Scholar]
- Valastro B.; Andersson M.; Lindgren H. S.; Cenci M. A. (2007) Expression pattern of JunD after acute or chronic L-DOPA treatment: comparison with deltaFosB. Neuroscience 144, 198–207. [DOI] [PubMed] [Google Scholar]
- Jorissen H. J.; Ulery P. G.; Henry L.; Gourneni S.; Nestler E. J.; Rudenko G. (2007) Dimerization and DNA-binding properties of the transcription factor DeltaFosB. Biochemistry 46, 8360–8372. [DOI] [PubMed] [Google Scholar]
- Vinson C.; Acharya A.; Taparowsky E. J. (2006) Deciphering B-ZIP transcription factor interactions in vitro and in vivo. Biochim. Biophys. Acta 1759, 4–12. [DOI] [PubMed] [Google Scholar]
- Ulery P. G.; Nestler E. J. (2007) Regulation of Delta FosB transcriptional activity by Ser27 phosphorylation. Eur. J. Neurosci. 25, 224–230. [DOI] [PubMed] [Google Scholar]
- Brené S.; Messer C.; Okado H.; Hartley M.; Heinemann S. F.; Nestler E. J. (2000) Regulation of GluR2 promoter activity by neurotrophic factors via a neuron-restrictive silencer element. Eur. J. Neurosci. 12, 1525–1533. [DOI] [PubMed] [Google Scholar]
- Auld D. S.; Thorne N.; Nguyen D. T.; Inglese J. (2008) A specific mechanism for nonspecific activation in reporter-gene assays. ACS Chem. Biol. 3, 463–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinenov Y.; Kerppola T. K. (2001) Close encounters of many kinds: Fos-Jun interactions that mediate transcription regulatory specificity. Oncogene 20, 2438–2452. [DOI] [PubMed] [Google Scholar]
- Fuxreiter M.; Tompa P.; Simon I.; Uversky V. N.; Hansen J. C.; Asturias F. J. (2008) Malleable machines take shape in eukaryotic transcriptional regulation. Nat. Chem. Biol. 4, 728–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers S. J.; Peters J.; Huang Y.; Comer M. B.; Barthel F.; Dingledine R. (1998) Transcriptional regulation of the GluR2 gene: neural-specific expression, multiple promoters, and regulatory elements. J. Neurosci. 18, 6723–6739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Z.; Sylwestrak E. L.; Lieberman D. N.; Zhang Y.; Liu X. Y.; Ghosh A. (2012) The Rett Syndrome protein MeCP2 regulates synaptic scaling. J. Neurosci. 32, 989–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitt D. C.; Schoepp D.; Kalivas P. W.; Volkow N. D.; Zarate C.; Merchant K.; Bear M. F.; Umbricht D.; Hajos M.; Potter W. Z.; Lee C. M. (2011) Translating glutamate: from pathophysiology to treatment. Sci. Transl. Med. 3, 102mr2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers M. S.; Chen B. T.; Bonci A. (2010) AMPA receptor synaptic plasticity induced by psychostimulants: the past, present, and therapeutic future. Neuron 67, 11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanacora G.; Zarate C. A.; Krystal J. H.; Manji H. K. (2008) Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat. Rev. Drug Discovery 7, 426–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokita K.; Yamaji T.; Hashimoto K. (2012) Roles of glutamate signaling in preclinical and/or mechanistic models of depression. Pharmacol., Biochem. Behav. 100, 688–704. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 46, 3–26. [DOI] [PubMed] [Google Scholar]
- Berg T. (2008) Inhibition of transcription factors with small organic molecules. Curr. Opin. Chem. Biol. 12, 464–471. [DOI] [PubMed] [Google Scholar]
- Koehler A. N. (2010) A complex task? Direct modulation of transcription factors with small molecules. Curr. Opin. Chem. Biol. 14, 331–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells J. A.; McClendon C. L. (2007) Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 450, 1001–1009. [DOI] [PubMed] [Google Scholar]
- Cheng Y.; LeGall T.; Oldfield C. J.; Mueller J. P.; Van Y. Y.; Romero P.; Cortese M. S.; Uversky V. N.; Dunker A. K. (2006) Rational drug design via intrinsically disordered protein. Trends Biotechnol. 24, 435–442. [DOI] [PubMed] [Google Scholar]
- Metallo S. J. (2010) Intrinsically disordered proteins are potential drug targets. Curr. Opin. Chem. Biol. 14, 481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C. H.; Lee J. H.; Yang C. H. (2005) Curcumin derivatives inhibit the formation of Jun-Fos-DNA complex independently of their conserved cysteine residues. J. Biochem. Mol. Biol. 38, 474–480. [DOI] [PubMed] [Google Scholar]
- Park S.; Song J. S.; Lee D. K.; Yang C. H. (1999) Suppression of AP-1 activity by tanshinone and cancer cell growth inhibition. Bull. Korean Chem. Soc. 20, 925–928. [Google Scholar]
- Park S.; Lee D. K.; Yang C. H. (1998) Inhibition of fos-jun-DNA complex formation by dihydroguaiaretic acid and in vitro cytotoxic effects on cancer cells. Cancer Lett. 127, 23–28. [DOI] [PubMed] [Google Scholar]
- Aikawa Y.; Morimoto K.; Yamamoto T.; Chaki H.; Hashiramoto A.; Narita H.; Hirono S.; Shiozawa S. (2008) Treatment of arthritis with a selective inhibitor of c-Fos/activator protein-1. Nat. Biotechnol. 26, 817–823. [DOI] [PubMed] [Google Scholar]
- Rishi V.; Potter T.; Laudeman J.; Reinhart R.; Silvers T.; Selby M.; Stevenson T.; Krosky P.; Stephen A. G.; Acharya A.; Moll J.; Oh W. J.; Scudiero D.; Shoemaker R. H.; Vinson C. (2005) A high-throughput fluorescence-anisotropy screen that identifies small molecule inhibitors of the DNA binding of B-ZIP transcription factors. Anal. Biochem. 340, 259–271. [DOI] [PubMed] [Google Scholar]
- Rishi V.; Oh W. J.; Heyerdahl S. L.; Zhao J.; Scudiero D.; Shoemaker R. H.; Vinson C. (2010) 12 Arylstibonic acids that inhibit the DNA binding of five B-ZIP dimers. J. Struct. Biol. 170, 216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X.; Giap C.; Lazo J. S.; Prochownik E. V. (2003) Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene 22, 6151–6159. [DOI] [PubMed] [Google Scholar]
- Follis A. V.; Hammoudeh D. I.; Wang H.; Prochownik E. V.; Metallo S. J. (2008) Structural rationale for the coupled binding and unfolding of the c-Myc oncoprotein by small molecules. Chem. Biol. 15, 1149–1155. [DOI] [PubMed] [Google Scholar]
- Hammoudeh D. I.; Follis A. V.; Prochownik E. V.; Metallo S. J. (2009) Multiple independent binding sites for small-molecule inhibitors on the oncoprotein c-Myc. J. Am. Chem. Soc. 131, 7390–7401. [DOI] [PubMed] [Google Scholar]
- Kiessling A.; Sperl B.; Hollis A.; Eick D.; Berg T. (2006) Selective inhibition of c-Myc/Max dimerization and DNA binding by small molecules. Chem. Biol. 13, 745–751. [DOI] [PubMed] [Google Scholar]
- Kiessling A.; Wiesinger R.; Sperl B.; Berg T. (2007) Selective inhibition of c-Myc/Max dimerization by a pyrazolo[1,5-a]pyrimidine. ChemMedChem 2, 627–630. [DOI] [PubMed] [Google Scholar]
- Peakman M. C.; Colby C.; Perrotti L. I.; Tekumalla P.; Carle T.; Ulery P.; Chao J.; Duman C.; Steffen C.; Monteggia L.; Allen M. R.; Stock J. L.; Duman R. S.; McNeish J. D.; Barrot M.; Self D. W.; Nestler E. J.; Schaeffer E. (2003) Inducible, brain region-specific expression of a dominant negative mutant of c-Jun in transgenic mice decreases sensitivity to cocaine. Brain Res. 970, 73–86. [DOI] [PubMed] [Google Scholar]
- Andersson M.; Westin J. E.; Cenci M. A. (2003) Time course of striatal DeltaFosB-like immunoreactivity and prodynorphin mRNA levels after discontinuation of chronic dopaminomimetic treatment. Eur. J. Neurosci. 17, 661–666. [DOI] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. (2010) New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- Mendgen T.; Steuer C.; Klein C. D. (2012) Privileged Scaffolds or Promiscuous Binders: A Comparative Study on Rhodanines and Related Heterocycles in Medicinal Chemistry. J. Med. Chem. 55, 743–753. [DOI] [PubMed] [Google Scholar]
- Whitmore L.; Wallace B. A. (2004) DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 32, W668–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmore L.; Wallace B. A. (2008) Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers 89, 392–400. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.